
Intracellular calcium (Ca2+) governs several aspects of the homeostasis of living organisms. The levels of Ca2+ in the cytosol are maintained at a concentration of about 0.1 μM by the continuous action of ATP-powered Ca2+ pumps, which transport Ca2+ ions across the plasma membrane to the outside of the cell or into the lumen of the endoplasmic reticulum (ER) and other vesicles.Citation1 In response to a variety of stimuli, Ca2+ is released from these storage sites leading to the activation of several Ca2+ responsive proteins. Intracellular Ca2+ fluxes play fundamental roles in regulating a wide variety of cellular responses, among which programmed cell death has been one of the most studied biological process in the last decade. Several reports have demonstrated that the Ca2+ content within the ER determines cell sensitivity to apoptotic stress and that changes in the Ca2+ flux from the ER to mitochondria represent a signal capable of triggering the rupture of the mitochondrial membrane as well as the consequent release of several pro-apoptotic factors into the cytosol.Citation1
This pathway is inactivated or impaired in cancer progression, with important consequences. Indeed, several tumor suppressor genes, such as PML, PTEN and TP53, have been unexpectedly found enriched at the ER where they regulate the Ca2+ crosstalk between the ER and mitochondria thus favoring Ca2+-dependent apoptosis after stress stimuli.Citation2 Particularly intriguing is the role of p53, a stress induced tumor suppressor, able to orchestrate mitochondrial apoptosis acting both in the nucleus via transcriptional regulation of several proteins (e.g. Bcl-xL, Bcl-2, PUMA and others) as well as in the cytoplasm.Citation3
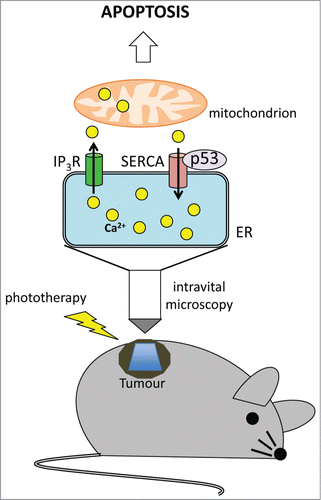
Despite its biological importance, the mechanism of Ca2+ flux between the ER and mitochondria and its regulation by tumor suppressors is not completely understood. Contributing in filling this gap, Pinton and colleagues found that p53 localizes at ER and at mitochondria-associated membranes (MAMs), and upon activation by a variety of stimuli, is able to bind the sarco/ER Ca2+-ATPase (SERCA) pump, leading to an increase of Ca2+ release from the ER and a consequent mitochondrial Ca2+ overload that profoundly impacts on the ability of the cells to undergo apoptosis.Citation4 The same group has also recently overcome the technical limitations that slowered so far the acquisition of evidences on the in vivo impact of this process, by using a novel strategy that, for the first time, allows the direct monitoring of intracellular Ca2+ fluxes within a tumor mass.Citation5,6 For the first time Giorgi and colleagues, starting from the techniques already used to measure Ca2+ in vivo,Citation7 undoubtedly demonstrated a role for p53 in modulating Ca2+ homeostasis in tumors. The authors injected murine p53−/− embryo fibroblasts (MEF) transduced with the oncogene H-RASV12 alone or with wild type (wt) p53 into a skinfold chamber, that had been implanted in the dorsal skin of female athymic mice. Upon tumor growth, the masses originated from these cells have been treated with phthalocyanine, a light-activatable agent used in cancer photodynamic therapy. Photo-activation of the drug with a LED light caused ROS formation in the ER and consequent Ca2+ release. The monitoring of pro-apoptotic Ca2+ signal with the ratiometric Ca2+ indicator Fura-2 by spinning disc confocal microscopy unveiled a massive release of Ca2+ ions into the cytoplasm of p53+/+ tumors after photo-activation. Instead, Ca2+ release was almost absent in the p53–/– counterpart. Of note, the higher Ca2+ response evoked by p53 was instrumental for an efficient induction of apoptosis and reduction of tumor mass. More interestingly, restoration of Ca2+ fluxes in p53–/– cells by overexpression of the mitochondrial Ca2+ uniporter (MCU) or of the sarco/ER Ca2+-ATPase (SERCA) pump, was able to rescue the apoptotic sensitivity of the cells to activated phthalocyanine. On the other hand, pharmacological manipulation of Ca2+ signaling in p53+/+ tumors efficiently prevented apoptosis in vitro and in vivo, meaning that a functional p53 is required to trigger an efficient stress-induced Ca2+ release able to sustain mitochondrial swelling and apoptosis ().
The potential impact of this new technique is wide opening the possibility of studying protein and RNA-based modulators of the Ca2+-mediated apoptotic cascade in vivo and also allowing the screening of molecules that could change cancer cell sensitivity to apoptotic signals.
Figure 1. Intravital imaging of ER-mitochondrial Ca2+ transfer and its regulation by p53. Ca2+ is transferred to the ER by the SERCA pumps and is released by inositol 1,4,5-triphosphate (IP3)-gated channels (IP3R). p53 binds and activates SERCA pumps thus increasing the amount of Ca2+ storage into the ER. The Ca2+ released to the mitochondria after phototherapy leads to mitochondrial swelling with consequent apoptosis induction in p53+/+ tumors.
References
- Orrenius S, et al. Nat Rev Mol Cell Biol 2003; 4:552–565; PMID:12838338; http://dx.doi.org/10.1038/nrm1150
- Bononi A, et al. Cell Death Differ 2013; 20:1631–1643; PMID:23811847; http://dx.doi.org/10.1038/cdd.2013.77
- Sorrentino G, et al. Mitochondrion 2014; 19(Pt A):88–96; PMID:25132079; http://dx.doi.org/10.1016/j.mito.2014.08.003
- Giorgi C, et al. Proc Natl Acad Sci U S A 2015; 112:1779–1784; PMID:25624484; http://dx.doi.org/10.1073/pnas.1410723112
- Giorgi C, et al. Oncotarget 2014; 6:1435–1445; PMID:25544762
- Ellenbroek SI, et al. Nat Rev Cancer 2014; 14:406–418; PMID:24854083; http://dx.doi.org/10.1038/nrc3742
- Pozzan T, et al. Biochim Biophys Acta 2009; 1787:1317–1323; PMID:19100709; http://dx.doi.org/10.1016/j.bbabio.2008.11.012