
Abstract
The Myocyte Enhancer Factor 2C (MEF2C) transcription factor plays a critical role in skeletal muscle differentiation, promoting muscle-specific gene transcription. Here we report that in proliferating cells MEF2C is degraded in mitosis by the Anaphase Promoting Complex/Cyclosome (APC/C) and that this downregulation is necessary for an efficient progression of the cell cycle. We show that this mechanism of degradation requires the presence on MEF2C of a D-box (R-X-X-L) and 2 phospho-motifs, pSer98 and pSer110. Both the D-box and pSer110 motifs are encoded by the ubiquitous alternate α1 exon. These two domains mediate the interaction between MEF2C and CDC20, a co-activator of APC/C. We further report that in myoblasts, MEF2C regulates the expression of G2/M checkpoint genes (14–3–3γ, Gadd45b and p21) and the sub-cellular localization of CYCLIN B1. The importance of controlling MEF2C levels during the cell cycle is reinforced by the observation that modulation of its expression affects the proliferation rate of colon cancer cells. Our findings show that beside the well-established role as pro-myogenic transcription factor, MEF2C can also function as a regulator of cell proliferation.
Abbreviations
| APC/C | = | Anaphase Promoting Complex/Cyclosome |
| CDK | = | Cyclin Dependent Kinase |
| CHX | = | Cycloheximide |
| CRC | = | ColoRectal Cancer |
| Gadd45b | = | Growth Arrest and DNA Damage b |
| degron | = | degradation signal |
| HDAC | = | Histone Deacetylases |
| MADS | = | Minichromosome maintenance, Agamous, Deficiens, Serum response factor |
| MEF2 | = | Myocyte Enhancer Factor 2 |
| MyHC | = | Myosin Heavy Chain |
| UPS | = | Ubiquitin Proteasome System |
Introduction
MEF2 proteins are members of the MADS (MCM1, Agamous, Deficiens and SRF) family of transcription factors that in vertebrates comprises 4 members, MEF2A-D. All MEF2 proteins share an N-terminal MADS box and an adjacent MEF2 domain that are required for MEF2 proteins homo- and hetero-dimerization and high-affinity DNA binding to the consensus sequence (C/T TA(A/T)4TAG/A) within the regulatory regions of target genes. The C-terminal regions of MEF2 factors contain transcription activation domains and are subject to complex patterns of alternative splicing. MEF2s are expressed in many tissues and organs where they control the activation of their differentiation programs.Citation1 MEF2 proteins are highly expressed in skeletal muscle where they play a pivotal role in terminal differentiation but they are unable to activate myogenesis alone; instead, these factors work in concert with the MYOD family of myogenic determinants, synergistically promoting myofiber differentiation.Citation2-Citation4 Genetic studies in mouse and zebrafish have shown that among Mef2 genes, Mef2c plays a unique role in the late phases of myogenesis, where it is essential for myofibers maturation.Citation5,6 Despite the well established role in terminal differentiation, Mef2c transcripts and protein are already detected in primary mouse myoblasts.Citation7,8 To date most models suggest that MEF2 proteins exhibit no specific functions in myoblasts, but exist to “prime” muscle cells for differentiation when the environmental conditions become permissive (for example cessation of cellular proliferation signals). Consequently, we and other have proposed several mechanisms contributing to the silencing of the pro-myogenic activity of MEF2C in proliferating myoblasts. Mechanisms mediating this negative regulation include post-translational modifications leading to increased physical interaction with inhibitors such as class II HDACs and PIN1, reduced DNA binding capabilities and/or protein stability.Citation9-Citation11 Besides, in vertebrates, a fundamental role in MEF2C activity regulation is played by alternative splicing processes.Citation12-Citation14 In mammals 3 major exons are alternatively spliced in MEF2A, MEF2D or MEF2C: the 2 mutually exclusive exons α1 and α2, a short exon β and an exon γ, which is only spliced in some MEF2C gene transcripts. In muscle cells the alternate inclusion in Mef2c transcripts of the ubiquitous α1 or muscle-specific α2 exon has a particular relevance in regulating the pro-myogenic activity of the encoded protein.Citation15,16 The recent observation that MEF2Cα1, present in proliferating myoblasts, is devoid of myogenic activity suggests that this “priming model” cannot be applied to this splice variant and indicate some novel functions of MEF2C in myoblasts. Indeed, in other cell types, Mef2c stimulates proliferation and regulate the expression of growth-related genes.Citation17-Citation22 Furthermore, recently Mef2c has been shown to regulate cell cycle related-genes in muscle cells.Citation23 Therefore, in the attempt to investigate this unexplored activity, we found that the level of MEF2C protein fluctuates during the cell cycle and we describe a novel mechanism by which the Anaphase Promoting Complex/Cyclosome (APC/C) ubiquitin ligase controls MEF2C abundance. The functional relevance of this mechanism during the cell cycle is suggested by the observation that ectopic expression of a MEF2C mutant, resistant to APC/C-dependent degradation, impairs entry into mitosis and cell proliferation. Furthermore, modulation of Mef2c expression in colon cancer cells affects their proliferation rates. We also demonstrate that MEF2C, directly or indirectly, controls the expression of genes that regulate G2/M transition (Gadd45B, p21 and 14.3.3 γ)Citation24-Citation26 and CYCLIN B1 sub-cellular localization. In summary, we present evidence that MEF2C plays a role in the transcriptional control of cell cycle-related genes and its degradation in mitosis contributes to the G2/M checkpoint inactivation in growing myoblasts.
Results
The ubiquitin-proteasome system (UPS) regulates MEF2C protein level during the cell cycle
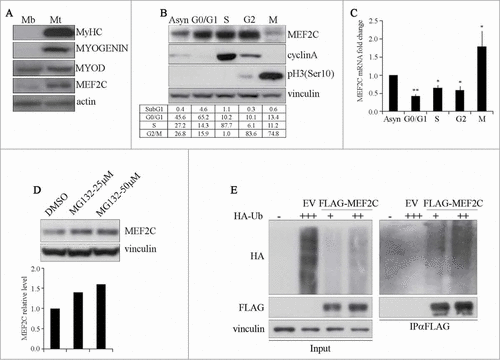
MEF2C function in terminal differentiation of skeletal muscle has been well established, on the contrary little is known about its role in proliferating muscle cells. To investigate this issue, we used the C2 mouse cell line, derived from adult muscle cells, the satellite cells.Citation27 C2 cells proliferate as mononucleated myoblasts in high-serum medium, then terminally differentiate to multinucleated myotubes upon serum withdrawal. As shown in , MEF2C is already present in C2 myoblasts where also MYOD is expressed, as previously described.Citation28 MEF2C level raises upon terminal differentiation, concomitantly with the expression of MYOSIN HEAVY CHAIN (MyHC) and MYOGENIN, a well established MEF2C target.Citation29 To start evaluating a potential role of MEF2C in growing cells, we determined if the cell cycle might have an impact on MEF2C protein level, as it is reported for several genes that influence cellular proliferation. To address this issue, we synchronized C2 muscle cells in G0/G1, S, G2 and M phase. Western blot analysis confirmed that C2 cells were highly synchronized in our experimental conditions, as attested by the appearance of CYCLIN A as DNA synthesis proceeds and of Histone H3 phosphorylated on Serine 10 during mitosis (pH3(Ser10), ). The distribution of cells in the different phases was measured by flow cytometry analysis after propidium iodide staining ( lower table, Fig. S1A). To analyze the expression of Mef2c in the course of the cell cycle, proteins and mRNA extracts were prepared from synchronized myoblasts. Western blot analysis revealed that the level of MEF2C protein is high in G0/G1, S and G2 phases, whereas it is largely decreased and exhibits a lower electrophoretic mobility in M phase cells compared to the other phases of the cell cycle (). To further verify the efficacy of our synchronization protocol of C2 myoblasts, we also examined the protein level of the key myogenic regulators MYOD and MYF5. As shown in Figure S1B, the expression pattern of MEF2C during the cell cycle resembles that already described for MYF5,Citation30-Citation32 whereas, as expected, MYOD protein level was very low in G0/G1 and only slightly reduced in M phase cell extracts.Citation33-Citation35 The mitotic down-regulation profile of endogenous MEF2C was also observed in a non muscle cell line, the mouse NIH-3T3 fibroblasts (Fig. S1C). C2 synchronized extracts were further analyzed by quantitative PCR (RT-qPCR). While the amounts of MEF2C protein significantly decreased during the M phase, the level of Mef2c mRNA was higher if compared to the other phases suggesting that post-transcriptional mechanisms must be involved in the downregulation of MEF2C. Treatment of asynchronous C2 cells with the proteasome inhibitor MG132 prevented MEF2C protein down-regulation in a dose-dependent manner (), suggesting that its decrease in mitosis is at least partially due to proteasome-dependent degradation. Proteasome targets are usually modified by the covalent addition of poly-ubiquitin chains. To test whether MEF2C is poly-ubiquitinated, C2 cells were co-transfected with FLAG-MEF2C and HA-ubiquitin expression plasmids. After inhibition of the proteasome, we immunoprecipitated FLAG-MEF2C proteins and observed by Western blot with the anti-HA antibody the presence of poly-ubiquitinated forms of MEF2C (). When HA-ubiquitin was transfected with an empty vector (EV), no product was detected. Overall these data indicate that in myoblasts MEF2C undergoes an increased protein degradation in mitosis and that this downregulation is due to the UPS system.
Figure 1. MEF2C protein level is downregulated in mitosis and is targeted for degradation by the Ubiquitin-Proteasome Pathway. (A) MEF2C is expressed in proliferating myoblasts. Total protein extracts from C2 growing myoblasts (Mb) or differentiated myotubes (Mt) were analyzed by Western blot with antibodies against myosin heavy chain (MyHC), MYOGENIN, MYOD and MEF2C. Actin was used as loading control. (B) MEF2C protein level decreases in mitotic cells. C2 cells were grown asynchronously in high serum medium (Asyn), arrested in G0/G1 by incubation in methionine-depleted medium, synchronized in S by treatment with aphidicoline or incubated with Nocodazole before shake off treatment to generate a mitotic fraction (Shake off fraction; M) and a non-mitotic fraction (adherent; G2). Cell lysates obtained from synchronized cell populations were analyzed by Western blot assay with antibody against MEF2C. Antibodies specific for CYCLIN A and histone H3 phosphorylated on Ser10 (pH3(Ser10)) were used to verify the level of synchronization in S and M phases obtained with the treatments. Vinculin was used as loading control. DNA content was assessed by flow cytometry analysis, percentage of distribution in G0/G1, S and G2/M phase are reported (lower panel). (C) Analysis of Mef2C transcripts during the cell cycle. Total RNA was isolated from synchronized populations obtained as in A and Mef2c mRNA level was quantified by RT-qPCR. The ratio between Mef2c and Gapdh transcripts was calculated in each synchronized cell population. Value obtained for asynchronous C2 was arbitrarily set equal to 1. Histograms report the mean of 2 independent experiments ± SEM. * and ** represent P-values ≤ 0.05 and ≤ 0.01 respectively. (D) MEF2C is degraded by the proteasome. C2 proliferating myoblasts were incubated with increasing concentrations of the proteasome inhibitor MG132 (25 μM or 50 μM) from a stock in DMSO, the control sample received an equivalent volume of DMSO. After 4 hours of treatment cells were harvested and protein extracts were analyzed by Western blotting with anti-MEF2C specific antibody. Vinculin was used as a loading control. Lower panel shows the results of the densitometric quantification of the signals obtained with the anti-MEF2C antibody normalized for the total amount of protein. The quantity of MEF2C protein in MG132 treated cells is expressed relative to the quantity of MEF2C in the control sample (DMSO) taken as 1. (E) MEF2C is poly-ubiquitinated in proliferating myoblasts. CoIP and Western blot assays of lysates from C2 cells transiently transfected with vectors coding for FLAG-MEF2C and increasing amounts of HA-ubiquitin (HA-Ub) or Empty Vector (EV) as control. 30 hours after transfection cells were treated with MG132 and then harvested. Anti-FLAG antibody was used for immunoprecipitation (IPαFLAG), anti-FLAG and anti-HA for Western blot detection of total Input and of immunoprecipitated proteins (IPαFLAG). Western blot results shown in B, D and E are representative of 2 independent experiments showing similar profiles.
Role of phosphorylation in MEF2C degradation
We next explored the regulatory mechanism that triggers MEF2C degradation. Our data indicate that MEF2C protein decreases when cells pass through mitosis, whereas mRNA remains unaffected, and that the residual mitotic MEF2C protein has a lower electrophoretic mobility. Therefore we wanted to determine if a mitosis-specific post-translational modification might be necessary for MEF2C degradation. We considered that phosphorylation of Ser98 and Ser110 were likely candidates as modifications involved in MEF2C degradation, as phospho-Ser98 and phospho-Ser110 are binding sites for PIN1, a peptidyl prolyl cis-trans isomerase that regulates MEF2C protein stability.Citation10 Notably, Ser110 is located within a domain encoded by the α1 exon, that is included in Mef2c transcripts alternatively to exon α2 (see ). In agreement with previous data,Citation15 our results indicate that proliferating myoblasts express only the transcript encoding the MEF2C α1 isoform, containing both Ser98 and Ser110, as determined by reverse transcription PCR (RT-PCR), using 2 common primers () that give 2 isoform-specific amplicons with different electrophoretic mobilities and that are differently susceptible to enzymatic restriction (Fig. S2). We evaluated the level of phosphorylation of these serines during the cell cycle using 2 antibodies that recognize MEF2C when phosphorylated on Ser98 (pSer98 MEF2C) or Ser110 (pSer110 MEF2C). As shown in , the variations of the signals of phospho-Ser110 reflects the fluctuations of the MEF2C protein during the cell cycle (compare to ), indicating a steady state level of MEF2C phosphorylation on this residue throughout the cell cycle. By contrast, in mitotic cells Ser98 phosphorylation is most abundant although the global level of MEF2C protein is the lowest ( and ). Densitometric quantification revealed a 7-fold increase of pSer98 MEF2C signal normalized to the total amount of MEF2C protein in mitosis compared to asynchronous cells (, lower panel). To determine the protein kinase(s) that regulate Ser98 and Ser110 phosphorylation, we carried out a kinase phosphorylation profiling that revealed a preferential ability of the peptide containing Ser110 to act as a substrate for the CDK5/p35 and CDK6/CycD1 protein kinases (Fig. S3A). On the contrary the peptide containing Ser98 was not efficiently phosphorylated by any tested kinases. The proximity of Ser98 and Ser110 led us to examine their importance as reciprocal “priming” sites. To verify this hypothesis we singularly substituted each of the 2 serines with alanine and assayed the effects of these mutations on the phosphorylation of the wild type residue in proliferating cells. As shown in Figure S3B, mutation on Ser98 did not alter the efficiency of Ser110 phosphorylation whereas Ser98 phosphorylation is strongly impaired (35% less) when Ser110 is substituted with an alanine. This data suggest a mechanism whereby phosphorylation of MEF2C on Ser110 is necessary for the subsequent Ser98 phosphorylation by a yet unknown mitotic kinase. The hypothesis of a M phase-specific phosphorylation of MEF2C on Ser98 is reinforced by the observed co-localization of pSer98 MEF2C in cells that are positive for Histone H3 phosphorylated on Ser10 (Fig. S4).
Figure 2. Ser98 phosphorylation of MEF2C peaks in mitosis, triggers poly-ubiquitination of the protein and reduces its stability. (A) Schematic of the MEF2C isoforms in the exon 3 region, with the location of the primers used to detect the alternate α1 and α2 exons indicated (arrows). The amminoacid sequences of the α exons of the mouse protein are indicated below. The positioning of Ser98 and Ser110 phosphoacceptor sites in α1 exon (and the residue equivalent to Ser98 in α2 exon) are highlighted in red. (B) Exclusive inclusion of the α1 exon in Mef2c transcripts in growing myoblasts. Total RNA was isolated from C2 myoblasts (Mb) or C2 cells that were differentiated for various lengths of time (0–72h). Exon expression was detected by RT-PCR using common primer (indicated in A) located on exons 2 and 4 that give rise to amplification products with different electrophoretyc mobility. Amplification of plasmid vectors containing the murine cDNAs of the MEF2C α1 and α2 splice variants were used as controls of the correct size of expected amplicons (α1 and α2 sample lanes). Rplp0 was used as endogenous control. (C). MEF2C is highly phosphorylated on Ser98 in mitotic cells. Cell lysates of asynchronous (Asyn) or synchronized cells were analyzed by Western blot with polyclonal antibodies that recognize the phosphorylated Serine 98 (pSer98) or Serine 110 (pSer110) of MEF2C. Vinculin was used as loading control. The levels of Ser98 and Ser110 phosphorylation, evaluated by densitometric scanning, were normalized to the quantity of MEF2C protein (see ). In the histogram below are reported the phosphorylation grades of the 2 residues in synchronized cells, expressed relative to the value obtained in asynchronous cells, taken as 1. (D). Ser98 and Ser110 phosphorylation triggers MEF2C poly-ubiquitination. C2 myoblasts were transiently transfected with expression vectors for HA-ubiquitin and FLAG-tagged wild type (WT) or not phosphorylable MEF2C 2SA mutant where Ser98 and Ser110 are substituted with alanines. Anti-FLAG antibody was used for immunoprecipitation (IPαFLAG), anti-HA for Western immunoblot detection (IB). (E) The non-phosphorylable MEF2C mutant proteins on Ser98 and Ser110 are more stable than the wild type protein. FLAG MEF2C wild type or mutated singularly on Ser98 (S98A), Ser110 (S110A) or both phosphoacceptor sites (2SA) were overexpressed in COS1 cells. After 36 h, protein synthesis was blocked with cycloheximide, the amount of FLAG MEF2C remaining at different times was checked by Western blot. Protein loading was controlled by anti- vinculin staining. The intensity of FLAG signal was quantified by densitometric analysis. The graph reports average of the intensity of MEF2C signals, relatively to the not treated sample (CHX 0h), of 2 independent experiments. Bars represent SEM. Western blot results reported in C and D are representative of 2 independent experiments that showed similar profiles.
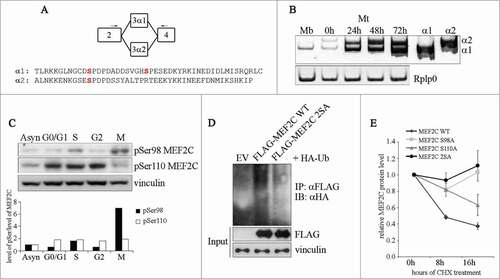
Next we observed that phosphorylation of Ser98 and Ser110 promotes MEF2C poly-ubiquitination, given that the formation of MEF2C-ubiquitin conjugates is strongly impaired with the protein mutated on the phosphoacceptor sites (MEF2C 2SA) (). Coherently with these findings, we found that the lack of MEF2C phosphorylation on Ser98 and Ser110 increases the stability of MEF2C in proliferating cells: in cycloheximide (CHX)-treated COS cells the half-lives of single (S98A, S110A) or double (2SA) not phosphorylable MEF2C mutants are increased if compared to that of the wild type protein (half life 8 hours) (). Altogether our data indicate that a G1 phase kinase, likely CDK6/CycD1, phosphorylates MEF2C on Ser110, priming the protein for the subsequent mitosis-specific phosphorylation on Ser98 that ultimately promotes its degradation through the UPS.
CDH1 or CDC20 mediate the degradation of MEF2C
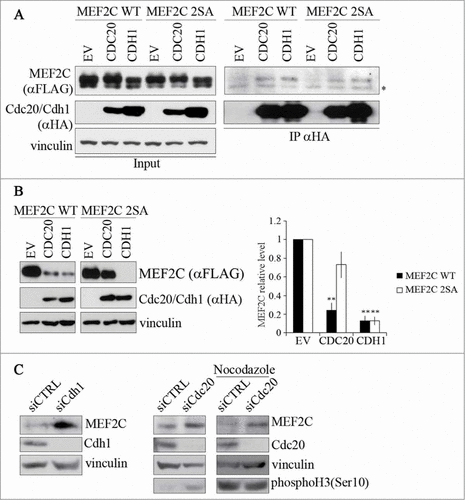
The finding that the level of MEF2C protein decreases during mitosis, suggest that its degradation might be mediated by APC/C, an ubiquitin ligase complex that is responsible for degradation of several protein during this phase of the cell cycle. The activity of APC/C is controlled by 2 related activators, CDC20 and CDH1, that contain a C-terminal WD40 repeat domain, their main function being the recruitment of substrates to APC/C.Citation36 To verify our hypothesis, we first tested if MEF2C physically interacts with CDC20 and CDH1. To this aim, HEK293T cells were transfected with FLAG-MEF2C wild type or the non-phosphorylable FLAG-MEF2C 2SA mutant and HA-tagged CDC20 or CDH1, the whole cell extracts were immunoprecipitated with anti-HA, and the interaction revealed by Western blotting with anti-FLAG (). MEF2C was clearly detected in the purified preparation of both CDC20 and CDH1, but not in the control immunoprecipitation sample (EV). We observed a slight reduction in the efficiency of association between CDC20 and the 2SA mutant, suggesting that the MEF2C/CDC20 interaction involves the phosphorylation of Ser98 and Ser110. We then tested whether the modulation of CDC20 and CDH1 expression could affect the levels of MEF2C protein wild type or mutated. In C2 proliferating cells we observed that the level of exogenous MEF2C decreased when either CDH1 (90% less protein compared to the control, right panel) or CDC20 (80% of decrease) over-expression was induced. Furthermore the accumulation of the MEF2C 2SA mutant (30% of decrease) upon ectopic expression of CDC20 indicates that phosphorylation of Ser98 and Ser110 is required for rapid degradation of MEF2C by APC/CCDC20 but not by APC/CCDH1 (). As a control, we found that SKP2, a known substrates of APC/CCDH1Citation37,38 was down-regulated upon ectopic expression of CDH1 but not CDC20, as shown in Figure S5. To further confirm that APC/CCDC20 and APC/CCDH1 are required for degradation of endogenous MEF2C, we inhibited their activities by siRNA. Silencing of both Cdc20 and Cdh1 transcripts resulted in an increase of the endogenous MEF2C level (). We next tested whether CDC20 is required for MEF2C degradation in mitosis. For this purpose, we silenced Cdc20 in C2 cells arrested in prometaphase with Nocodazole. As expected, MEF2C protein was downregulated in mitosis in the control sample ( siCTRL). In contrast, silencing of Cdc20 in nocodazole treated cells prevents MEF2C mitotic degradation, restoring a protein level comparable to that of asynchronous cells. Overall our results indicate that APC/CCDC20 is responsible for the degradation of phosphoSer98 MEF2C in mitosis.
Figure 3. MEF2C protein level is controlled by CDC20 or CDH1, depending on the phosphorylation of Ser98 and Ser110. (A). MEF2C physically interacts with CDC20 and CDH1. Co-immunoprecipitation analysis of HEK293T cells transiently transfected with empty vector (EV) or HA-tagged vectors coding for CDC20 and CDH1 along with FLAG-MEF2C WT or 2SA. 48 hours after transfection, cells were treated with MG132 and harvested. Total protein extracts (Input) were incubated with protein A beads conjugated with anti-HA antibody. Left: input. Right: immunoprecipitated proteins. FLAG-tagged MEF2C was detected by Western blotting with anti-FLAG antibody. Vinculin was used as a loading control. Asterisk (*) indicates non-specific bands. (B) MEF2C expression is reduced by CDC20 and CDH1 overexpression, CDC20 dependent degradation relies on pSer98/pSer110 phosphorylation. C2 proliferating myoblasts were co-transfected with an empty vector (EV) or HA-tagged CDC20 and CDH1 vectors along with phosphorylable (WT) or not-phosphorylable (2SA) MEF2C. Cell extracts were analyzed by Western blotting with antibodies against FLAG and HA. Vinculin was used as loading control. The histogram (right panel) reports the densitometric quantification of MEF2C protein level (FLAG signal) normalized to the total amount of protein expressed relatively to the quantity of protein in the control sample, taken as 1. Histograms show means ± SEM of 3 independent experiments. ** represent P-values ≤ 0.01. (C) RNAi knockdown of Cdc20 and/or Cdh1 results in an accumulation of MEF2C. Asynchronous C2 cells were transfected with a control siRNA (siCTRL) or a pool of siRNAs targeting Cdh1 (siCdh1, left panel) or Cdc20 (siCdc20, middle panel) mRNAs and analyzed 72 hours later. Alternatively C2 cells transfected with siCdc20 were treated over-night with Nocodazole after 36 hours from transfection (left panel). Protein extracts were analyzed by Western blotting with antibody against MEF2C, CDH1, CDC20 and histone H3 phosphorylated on Ser10, a marker of the M phase. Vinculin was used as loading control. The results of the Western blot shown in A and C are representative of 2 independent experiments that gave similar profiles.
D-box, pSer98 and pSer110 motifs are required for CDC20-dependent degradation of MEF2C
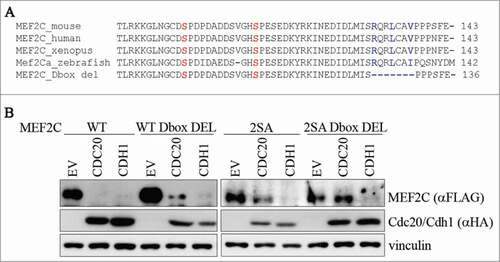
Previous studies revealed that APC/C recognizes sequence recognition motifs including a Destruction-box (RXXL; D-box)Citation39 and/or a KEN-box (KEN).Citation40 Since our results indicate that MEF2C is a target of APC/C, we tried to identify degradation signals in MEF2C. By sequence analysis, no KEN-boxes were found in the MEF2C α1 isoform expressed in proliferating muscle cells. On the contrary we identified one putative conserved D-box motif in the amminoacidic sequence encoded by α1 exon, proximal to the Ser98 and Ser110 residues (). Indeed we found that the CDC20-dependent degradation of the D-box deleted mutant was lower than that of wild-type MEF2C (, lane 5 compared to lane 2). When both D-box and phosphoacceptor sites were mutated, the protein level was almost completely stabilized (, lane 11). Similar results were obtained using the MEF2C 2SA mutant protein where the D-box arginine and leucine were replaced by alanines (2SA D-box MUT) (Fig. S6A). Furthermore we also observed that the MEF2C 2SA D-box deleted protein interacts less efficiently with CDC20 (Fig. S6B). On the contrary either the phosphoacceptor sites or the D-box are not required for the CDH1-dependent degradation (lanes 3, 6, 9 and 12). Taken together, a model emerges in which MEF2C establishes a multivalent interaction with CDC20 as both the D-box and the phospho Ser98 and Ser110 motifs are required for APC/CCDC20-dependent destruction. Our results also suggest that the phospho Ser98 and Ser110 are the major degradation signals (degrons) for CDC20-dependent degradation.
Figure 4. Ser98/Ser110 phosphorylation and a proximal conserved D-box are required for CDC20-dependent degradation of MEF2C. (A) Amminoacid sequence encoded by the α1 isoform. Sequence of a putative D-box near the Ser98 and Ser110 phosphoacceptor sites. Alignment with ClustalW of the amminoacid sequences encompassing the phosphorylable residues of MEF2C proteins from different species revealed the presence of a conservd D-box. Amminoacids forming the putative destruction box (D-box) are highlighted in blue, Ser98 and Ser110 in red. The deletion introduced in the MEF2C D-box mutant (MEF2C D-box del) is indicated. (B) The D-box and Ser98/Ser110 phosphorylation are all necessary for MEF2C degradation. C2 proliferating myoblasts were co-transfected with an empty vector (EV) or HA-tagged CDC20 and CDH1 vectors along with, alternatively the wild type MEF2C protein (WT), the D-box deleted mutant (WT D-box DEL), the not-phosphorylable MEF2C 2SA mutant or the MEF2C 2SA mutant deleted of the D-box (2SA D-box DEL). Cell extracts were analyzed by Western blotting with antibodies against FLAG and HA. Vinculin was used as loading control.
Stabilization of MEF2C impairs progression through mitosis and inhibits CYCLIN B1 nuclear accumulation
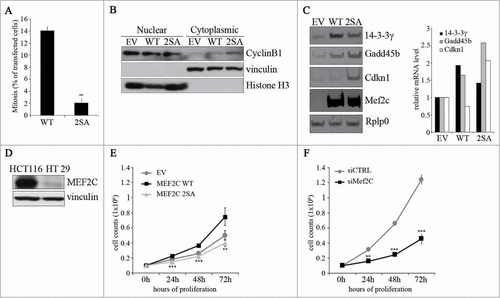
Our previous results indicate that MEF2C protein undergoes a mitotic-specific degradation, suggesting that it might have a role in the regulation of cell cycle progression. To analyze the biological relevance of MEF2C cell cycle-regulated degradation and to verify whether MEF2C stabilization can affect mitosis, we evaluated the mitotic index of COS cells over-expressing the wild type or the CDC20-resistant MEF2C 2SA mutant. Overexpression of MEF2C 2SA determined a 15-fold reduction of the percentage of mitotic cells compared to the wild type protein (). These data indicated that degradation of MEF2C is important for an efficient progression in mitosis. Since CYCLIN B1 expression, nuclear localization and binding to CDK1 are key events for G2/M transition, we investigated the role of MEF2C phosphorylation in the regulation of CYCLIN B1. Intriguingly, MEF2C 2SA over expression in HT29 cells, a colorectal cancer (CRC) cell line that expresses low levels of endogenous MEF2C (), leads to increased cytoplasmic accumulation of CYCLIN B1 (). Furthermore, our data revealed that ectopic expression of MEF2C induces increased expression of the genes encoding GADD45B, the cyclin-dependent kinase inhibitor p21Waf1(Cip1 (Cdkn1) and 14.3.3 γ, all inhibitors of the G2/M transition (), and this transcriptional effect is more evident with the not degradable 2SA mutant. We then evaluated the impact of MEF2C level fluctuation on cell divisions in CRC cell lines. Consistent with the mitotic effect described above, expression of the stable MEF2C 2SA mutant in HT29 cells, caused a proliferative defect compared to cells transfected with an empty vector. In contrast, expression of the wild type MEF2C protein promoted cell proliferation (). Coherently with these results, we found that siRNA-mediated silencing of Mef2c in HCT116 cells, that express high amount of endogenous MEF2C, impairs their proliferation rate compared to cells transfected with a control siRNA (). Taken together our data suggest a model where MEF2C controls, directly or indirectly, the expression of genes of the G2/M checkpoint and its degradation in the early M phase is necessary for an efficient progression of the cell cycle through mitosis and ultimately for cell proliferation.
Figure 5. The ectopic expression of a stable, non phosphorylable mutant of MEF2C can delay cell cycle progression. (A) Overexpression of the MEF2C 2SA mutant is linked to a reduced number of mitotic events. COS-1 cells were transfected with vectors coding for wild type MEF2C (WT) or the not-phosphorylable FLAG-MEF2C 2SA mutant. 48 hours later cells were fixed for immunofluorescence analysis and stained with antibodies for phosphorylated histone H3 and FLAG. Transfected cells were scored for the frequency of mitosis. Histograms show means ± SEM of 2 independent experiments. ** represent P-values ≤ 0.01. (B) MEF2C 2SA impairs nuclear translocation of CYCLIN B1. HT29 cells transiently transfected with an empty vector (EV) or the expression vectors for the wild type or the not-phosphorylable MEF2C 2SA mutant were lysed in order to obtain nuclear and cytoplasmic protein extracts. The different fractions were analyzed by Western blotting with CYCLIN B1 antibody. Histone H3 and Vinculin antibodies were used as loading control respectively for nuclear and cytoplasmic fraction. (C) MEF2C regulates the expression of genes encoding inhibitors of the G2/(M) transition. Left panel, Total RNA was isolated from transfected C2 cells overespressing the wild type or 2SA MEF2C protein. The transcripts of Gadd45b, P21 and 14.3.3 γ genes were amplified by RT-PCR, PCR products were separated in 8% polyacrylamide gels. Rplp0 was used as endogenous control. The histograms in the right panel report the results of the densitometric quantification of the bands normalized to Rplp0 and expressed relatively to the empty vector transfected cells taken as 1. The RT-PCR results are representative of 2 independent experiments. (D) MEF2C is differentially expressed in CRC cell lines. Total protein extracts from HCT116 and HT29 proliferating cells were analyzed by Western blot with antibody against MEF2C. Vinculin was used as loading control. (E) MEF2C modulates cell cycle progression in a phosphorylation-dependent manner. HT29 cells were transfected with an empty vector (EV) or vectors coding for the wild type MEF2C protein or the non-phosphorylable MEF2C 2SA. After 24 hours, 0.1 × 106 cells were plated for each sample. Cells were then harvested every 24 hours and the number of proliferating cells were counted with an hemacytometer after Trypan blue staining. The graphs show means ± SEM of 4 independent experiments. Statistical significance of variation between values obtained at each time points with MEF2C 2SA and MEF2C WT was calculated. Differences between 2SA and WT are statistically significant. ** and *** represent P-values ≤ 0.01 and ≤ 0.001 respectively. (F) RNAi knockdown of Mef2c impairs HCT116 proliferation rate. HCT116 cells were transfected with a control siRNA (siCTRL) or a pool of siRNAs targeting Mef2c (siMef2c) and proliferation rate was evaluated as described in E. The graphs show means ± SEM of 4 independent experiments. ** and *** represent P-values ≤ 0.01 and ≤ 0.001 respectively.
Discussion
The MEF2C transcription factor has a key role in the control of muscle-specific genes during skeletal muscle terminal differentiation.Citation5,6,41 Undifferentiated myoblasts express a splice variant of MEF2C, the ubiquitous MEF2C α1 isoform lacking pro-myogenic activity. The function played by this protein in growing myoblasts is currently unknown. In the present work, we provide evidences that MEF2C is involved in the control of cell cycle progression. First we show that the level of MEF2C fluctuates during the cell cycle in muscle and non-muscle cells, being targeted for degradation early in mitosis by the ubiquitin-proteasome pathway. Second, we observed that ectopic expression of a MEF2C α1 protein mutant, resistant to mitotic degradation, impairs cell cycle progression and results in a lower number of cells in mitosis, suggesting a previous unexplored role for MEF2C α1 in the regulation of cell proliferation. Finally we observed a modulation of genes that regulate G2/M transition upon overexpression of MEF2C.
We present several lines of evidence to indicate that both CDC20 and CDH1, the 2 co-activators of APC/C, promote MEF2C degradation and that mitotic degradation of MEF2C is mediated by APC/CCDC20. Timely downregulation of MEF2C in mitosis by APC/CCDC20 requires MEF2C phosphorylation of Ser98 and Ser110. We found an even level of phosphorylation of the MEF2C α1-specific Ser110 residue during the cell cycle, this phosphoacceptor site is efficiently phosphorylated by CYCLIN D1/CDK6 in vitro. Whereas Ser98 is phosphorylated in mitosis by a yet unknown mitotic kinase. Further, our data indicate that a priming phosphorylation of Ser110 is necessary for achieving an efficient phosphorylation of Ser98. On the contrary we report that APC/CCDH1 mediates the down-regulation of MEF2C independently from the status of Ser98 and Ser110 phosphorylation. The APC/CCDH1 ubiquitin ligase normally regulates exit from mitosis and events in G1. Although overexpression of CDH1 results in a strong decrease of MEF2C, the persistence of some of MEF2C following mitosis exit suggests that a subpopulation of protein is protected from the CDH1-mediated degradation. We speculate that phosphorylation on serine and threonine residues, other than Ser98 and Ser110, might protect MEF2C from APC/CCDH1 mediated degradation as described for other targets of CDH1.Citation42-Citation45 The functional relevance of CDH1-mediated degradation will be subject of future studies. Interestingly, we observed APC/CCDC20 MEF2C degradation in nocodazole-arrested pseudo-prometaphase cells, when the spindle checkpoint (SAC) is actively inhibiting CDC20. The escape from SAC inhibition has been observed also for other APC/C targets such as CYCLIN A, Nek2A and HoxC10.Citation46-Citation49 One proposed model to explain the premature degradation of some APC/CCDC20 targets envisages an increased affinity for CDC20 due to the presence of multiple interaction domains in the substrate. Indeed, we found that APC/CCDC20 -dependent degradation of MEF2C requires a conserved D-box together with the 2 phospho-motifs pSer98 and pSer110, located in close proximity to the D-box. Notably, the D-box and Ser110 residue are located in the protein sequence encoded by the ubiquitous alternative exon α1, that we and other have described to be specifically expressed in proliferating myoblasts (see and reference 15). We previously reported that pSer98 and pSer110 are binding sites for PIN1,Citation10 a peptidyl-prolyl cis-trans isomerase that regulates the activities of several phospho-proteins in mitosis.Citation50 A possible role played by PIN1 in this mechanism warrants future investigations.
Despite the high sequence conservation of the N-terminal region among MEF2s, we found that the MEF2C α1-specific D-box, necessary for CDC20-dependent degrading activity, is not present in any other member of the family. Although we cannot exclude a cell cycle regulated expression and/or degradation also for the other MEF2s, our data prompted us to speculate that the CDC20-dependent mitotic degradation is a specific feature of MEF2C and particularly of the α1 isoform. These observations together with the recent evidence that individual MEF2sCitation23,51 and alternative splicing isoformsCitation12 can induce different subsets of gene during skeletal muscle differentiation and during development, lead us to speculate about specific biological function(s) played by the different members of MEF2 family.
To date, MEF2C protein function in skeletal muscle and in other tissues, correlates with their terminal differentiation programs.Citation5,6,41 The fluctuations of its protein level during the cell cycle suggest a yet unexplored role in the regulation of cell proliferation. A cell cycle-regulated expression has been described in muscle cells also for MYF5 and MYOD, involved in the control of myoblasts proliferation.Citation31 The observation that the overexpression of a non phosphorylable mutant, resistant to CDC20-dependent degradation, impairs mitosis progression means that it is plausible that MEF2C can have a role in mitosis regulation. Although our data do not clarify whether the reduced number of mitotic events is due to a delayed entrance or a faster exit from mitosis, we can speculate that MEF2C mutant overexpression slows down the transition from G2 phase to mitosis. As a matter of fact, upon overexpression of MEF2C, we observed an induction of 14–3–3γ, Gadd45b and p21 transcripts, encoding inhibitors of the CYCLIN B1/CDK1 kinase.Citation24,52-56 These genes have a role in the G2-M checkpoint both in normal cell cycle progression and in response to DNA damage. We also found that failure of MEF2C degradation correlates with an increase in the cytoplasmic inactive fraction of CYCLIN B1. It has been recently reported that MEF2D protein negatively regulates the expression of Gadd45b and p21 genes and binds to MEF2 sites in their promoter sequences in hepatocellular carcinoma (HCC) cell lines.Citation57 Thus it may be argued that MEF2C induces directly the expression of these G2/M transition inhibitors. In contrast, our bioinformatic analysis did not reveal any MEF2 site in the promoter region of 14–3–3γ, suggesting a possible indirect control of MEF2C on this gene. In addition to the mitotic effect, we observed that over-expression of the non-phosphorylable MEF2C mutant in low expressing HT29 CRC cells impairs cell proliferation possibly as a consequence of the delayed progression into mitosis, whereas the ectopic expression of wild type MEF2C increases their proliferation rate. Coherently with these observations, we found that knockdown of Mef2c expression in high expressing HCT116 cells had a detrimental effect on cell proliferation. Hence MEF2C degradation might be a “sine qua non” condition to achieve mitotic progression and, in turn, to allow cell proliferation. We can hypothesize a positive role of MEF2C in directly stimulating cell cycle progression by activating the transcription of proliferation-related genes such as c-jun, a well-known MEF2 target,Citation19 and/or other yet unknown genes likely related to G1/S transition. This has been recently described by a gene expression analysis study in muscle cells.Citation23
In conclusion our work reveal a novel function of MEF2C beyond its well known role in terminal differentiation. According to the classical model MEF2C does not play any specific function in growing myoblasts but is necessary to trigger the differentiation program upon differentiation stimuli. With this work we propose that the ubiquitous MEF2C splice variant plays an active role in promoting, directly or indirectly, the expression of genes that control the G2/M transition thus contributing to the regulation of cell cycle progression.
Materials and Methods
Cell culture and synchronization
C2C12, NIH-3T3, COS-1, HEK293T, HCT116 and HT29 cells were cultured in Dulbecco's Modified Eagle's medium, high glucose, GLUTAMAX (Invitrogen) supplemented with 10% fetal bovine serum (Gibco) and 100 U/ml Penicillin/Streptomycin (Euroclone). C2 cells were induced to differentiate, when approaching confluence, with DMEM and 2% horse serum (Hyclone). Cells were cultured at 37°C under a 5% CO2 atmosphere. The proteasome inhibitor MG132 (Sigma) was added at the indicated concentration 5 hours before harvesting the cells. Transfected COS-1 cells were treated with 25 μM cycloheximide (Sigma) in order to assess protein half-life.
C2 cells were synchronized by culturing them in methionine-depleted DMEM medium (Sigma) supplemented with 1% fetal bovine serum, apo-transferrin and sodium selenite (5 μg/ml, Sigma). C2 cells were treated over-night with aphidicolin (2,5 μg/ml, Sigma) or nocodazole (1 μM, Sigma) to obtain respectively S or G2/M phase synchronization. Nocodazole treated cells were subjected to a gentle shake-off in order to collect separately round mitotic cells and G2 adherent cells. Synchronized cells were harvested and DNA distribution analysis was performed after staining with propidium iodide (PI) (50 mg/ml) with an Epics cytofluorimeter (Beckman Coulter). Cell cycle distribution was analyzed with WinMDI software.
Proliferation assay
HT29 and HCT116 transfected cells grown in 6 cm plates, were harvested by trypsinization 24 hours after transfection and aliquoted in 12-wells plates. For the following 72 hours, cells were harvested every day and the number of proliferating cells was counted with hematocytometer and Trypan Blue staining.
Plasmids and RNA interference
The FLAG tagged MEF2C full-length cDNA (FLAG-MEF2C) and the Ser98 (FLAG-MEF2C S98A), Ser 110 (FLAG-MEF2C S110A) or both the serines (FLAG-MEF2C 2SA) mutants were previously described.Citation10 The deletion or mutation of the Destruction box (D-box) were obtained by PCR-based site-directed mutagenesis using FLAG-MEF2C or FLAG-MEF2C 2SA as a template, mutations were confirmed by sequencing. The cDNAs deleted or mutated of the sequence encoding R130 XXLXXV137 were cloned into HindIII-BglII digested pFLAG-CMV (Sigma) with Gibson Assembly Kit (NEB). Plasmid coding for HA-CDC20, HA-CDH1, HA-Ubiquitin and FLAG-SKP2 were kindly provided by Daniele Guardavaccaro (Hubrecht Institute). Transient transfections were performed using Lipofectamine LTX with PLUS Reagent (Invitrogen) according to manufacturer's instruction. For siRNA experiments C2C12 or HCT116 cells were plated in 6-well plates and transfected with the indicated amounts of a pool of 3 siRNAs using RNAiMAX (Invitrogen) following the manufacturer's instruction. Cells were analyzed 48 h after transfection. Silencer siRNAs targeting Cdh1 (74510, 74322, 165926), Cdc20 (171932, 171933, 171934), Mef2c (157132, 157133, 157134) and Silencer Negative Control #3 siRNA were purchased from Ambion (Life Technologies).
Western blot analysis and Immunoprecipitation
Total cell extracts were prepared with RIPA buffer (50mMTris HCl pH 7.5, 150mMNaCl, 1mMEDTA, 1% Na Deoxycholate, 1% Igepal, 0.1% SDS, 1mM DTT) supplemented with 1mM phenylmethyl sulfonylflouride (PMSF) and Proteases Inhibitor Complete cocktail (PIC, Roche). Equal amounts of protein extracts were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and analyzed by Western blot as previously described.Citation12 The following antibodies were used: mouse monoclonal anti-CYCLIN A (CY-A1), anti-FLAG (M2) and anti-Vinculin (VIN-11–5) from Sigma-Aldrich; mouse monoclonal anti-p55 CDC (E-7) from Santa Cruz Biotechnologies; mouse monoclonal anti-CDH1 (DH01) from Thermo Scientific; mouse monoclonal Phospho-Histone H3 (6G3) from Cell signaling; mouse monoclonal anti-MyHC (MF20) and anti-myogenin (IF5D) from DSHB; rabbit monoclonal anti-MEF2C (D80C1) from Cell Signaling; rabbit polyclonal anti-MEF2C (SBS-002) from Sparrow Biosciences; rabbit polyclonal anti-HA (H6908) from Sigma-Aldrich; rabbit polyclonal anti-MYOD (C-20) and anti-MYF5 (C-20) from Santa Cruz Biotechnologies. Custom polyclonal antibodies against MEF2C phosphorylated on Serine 98 (phosphoSer98) or Serine 110 (phosphoSer110) were developed in rabbit using synthetic phosphorylated MEF2C peptide. The antibodies were affinity-purified on the immobilized immunizing MEF2C peptides.
For immunoprecipitation of FLAG tagged proteins, C2C12 cells were harvested 48 hours after transfection and lysed in RIPA buffer supplemented with 1 mM PMSF, PIC, 2,5 mM sodium fluoride (NaF), 1 mM sodium orthovanadate (Na3VO4), 0,5 mM EDTA, 10 mM Iodoacetamide, 2 μM MG132 and 1 μM G5, Ubiquitin Isopeptidase Inhibitor I. Cell extracts were precleared with Protein G Agarose (KPL) and then incubated with anti-FLAG M2 Affinity Gel (Sigma). Immunoprecipitates were washed 4 times in RIPA buffer with inhibitors, eluted by resuspension in Laemmli sample buffer and boiled for 5 minutes. For co-immunoprecipitation HEK293T were treated with 10 μM MG132 for 5 hours and collected 24 hours after transfection. Total cell extracts were prepared by lysis in Triton Lysis Buffer (TLB, 50 mM Tris-HCl pH 7.4, 0.25 M NaCl, 1 mM EDTA, 0.1% Triton X-100, 50 mM NaF) supplemented with 1 mM PMSF, PIC, 1 mM Na3VO4, 1 mM (dithiothreitol). Protein G-beads were coated with mouse monoclonal anti-HA (HA7, Sigma) and cell extracts were precleared as described above for 1 hours at 4°C. Cell extract were then incubated with protein G-anti HA coated beads for 3 hours at 4°C. Immunocomplex were washed 4 times with TLB with inhibitors and eluted with Laemmli sample buffer and boiling for 5 minutes.
Protein expression in total extracts and immunoprecitated or coimmunoprecipitated proteins were analyzed by SDS-PAGE and Western blot as described above.
Extraction of cytoplasmic and nuclear protein
Nuclear and cytoplasmic fraction of HT29 transfected cells were obtained with 2 distinct lysis buffers. Briefly cells were washed in cold PBS and harvested in Buffer A (10 mM HEPES pH 7.9, 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 0.15% Nonidet P40 (NP40) supplemented with 1 mM DTT and PIC). Cells were homogenized with several passages through a needle 26G, nuclei were sedimented by centrifugation and supernatant containing cytoplasmic protein was conserved. Nuclei were washed quickly with cold PBS, resuspended in Buffer B (20 mM HEPES pH 7.9, 400 mM NaCl, 1 mM EDTA, 1 mM EGTA, 0,5% NP 40 supplemented with 1 mM DTT and PIC) and lysed by sonication. Nuclei lysates were clarified by centrifugation.
Immunofluorescence and mitotic index
Immunostaining of COS-1 cells was performed 48 hours after transfection as previously described.Citation10 Cellular DNA was stained with Hoeascht 33342 (Sigma) and the following primary antibodies were used: mouse monoclonal anti-FLAG M2 (Sigma) and rabbit polyclonal anti-phospho Histone H3 (Ser10) (Millipore). For fluorescent detection anti-mouse and anti-rabbit IgG secondary antibodies conjugated to Alexa Fluor dye (Molecular Probes) were used. All samples were examinated in a Zeiss Axioskop 40 fluorescence microscope equipped with an Axiocam HRC camera for image acquisition. Mitotic index estimation was performed using Cell Counter plugin of Image J (http://imagej.nih.gov/ij/) analyzing at last 10 fields for each sample for a total amount of about 200 nuclei.
Kinase phosphorylation assay
Kinase phosphorylation assay was performed by ProQinase GmbH (Freiburg, Germany). Briefly byotinilated peptides (1 μM) containing phosphorylable Serine 98 (KGLNGCDSPDPDA) or Serine 110 (ADDSVGHSPESED) were assayed for phosphorylation in a radiometric assay (33PanQinase® Activity Assay) on a panel of 190 Ser/Thr kinases, using streptavidin-coated FlashPlate® HTS PLUS plates.
RNA extraction and RT-qPCR
Total RNA was isolated with Total RNA Purification Kit (Norgen Biotek) and digested with DNaseI (Invitrogen). 500 ng of RNA were reverse transcribed using Superscript III reverse transcriptase (Invitrogen) according to manufacturer's instruction. The obtained cDNA was qPCR quantified using TaqMan® Universal Master Mix II (Applied Biosystems) and the PCR ABI PRISM 7900 HT Sequence Detection System (Applied Biosystems). TaqMan Gene Expression assays for Mef2c transcript and for the normalizer Rplp0 were purchased from Invitrogen.
For quantification of Gadd45b, P21, 14.3.3γ and Mef2c α1 of α2 isoforms, total RNA was isolated using TRIzol® Plus RNA Purification System (Ambion) 48 h hours after transfection, cDNA was obtained and their transcripts analyzed with specific primers. Amplicons were separated in 10% polyacrylamide gel and identified by Ethidium Bromide staining.
Student's t-tests were performed for pairwise comparisons to determine significant differences between groups.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
1026519_Supplemental_Material.zip
Download Zip (4.4 MB)Acknowledgments
We are highly indebted with Carol Imbriano and Michele Pelosi for their great advises. We thank Marco Crescenzi, Andrea Martello, Daniele Guardavaccaro, Gian Paolo Rossini, Paul Burger and Domenico D'Arca for very helpful suggestions and discussion. We are grateful to Valentina Basile, Paolo Benatti, Serena Carra, Valentina Salsi and Lucia Montorsi for sharing reagents, protocols and helpful suggestions.
Funding
This research was supported in part by Optistem (European collaborative project HEALTH-2007–1.4–6), AFM (Association Francaise contre le Myopathies) (grant n°16252) and Fondazione di Vignola.
SUPPLEMENTAL MATERIAL
Supplemental data for this article can be accessed on the publisher's website.
Related Research Data
References
- Potthoff MJ, Olson EN. MEF2: a central regulator of diverse developmental programs. Development 2007; 134:4131-40; PMID:17959722; https://doi.org/10.1242/dev.008367
- Molkentin JD, Black BL, Martin JF, Olson EN. Cooperative activation of muscle gene expression by MEF2 and myogenic bHLH proteins. Cell 1995; 83:1125-36; PMID:8548800; https://doi.org/10.1016/0092-8674(95)90139-6
- Ornatsky OI, Andreucci JJ, McDermott JC. A dominant-negative form of transcription factor MEF2 inhibits myogenesis. J Biol Chem 1997; 272:33271-8; PMID:9407117; https://doi.org/10.1074/jbc.272.52.33271
- Yu YT, Breitbart RE, Smoot LB, Lee Y, Mahdavi V, Nadal-Ginard B. Human myocyte-specific enhancer factor 2 comprises a group of tissue-restricted MADS box transcription factors. Genes Dev 1992; 6:1783-98; PMID:1516833; https://doi.org/10.1101/gad.6.9.1783
- Hinits Y, Hughes SM. Mef2s are required for thick filament formation in nascent muscle fibres. Development 2007; 134:2511-9; PMID:17537787; https://doi.org/10.1242/dev.007088
- Potthoff MJ, Arnold MA, McAnally J, Richardson JA, Bassel-Duby R, Olson EN. Regulation of skeletal muscle sarcomere integrity and postnatal muscle function by Mef2c. Mol Cell Biol 2007; 27:8143-51; PMID:17875930; https://doi.org/10.1128/MCB.01187-07
- Liu N, Nelson BR, Bezprozvannaya S, Shelton JM, Richardson JA, Bassel-Duby R, Olson EN. Requirement of MEF2A, C, and D for skeletal muscle regeneration. Proc Natl Acad Sci U S A 2014; 111:4109-14; PMID:24591619; https://doi.org/10.1073/pnas.1401732111
- Mokalled MH, Johnson AN, Creemers EE, Olson EN. MASTR directs MyoD-dependent satellite cell differentiation during skeletal muscle regeneration. Genes Dev 2012; 26:190-202; PMID:22279050; https://doi.org/10.1101/gad.179663.111
- Angelelli C, Magli A, Ferrari D, Ganassi M, Matafora V, Parise F, Razzini G, Bachi A, Ferrari S, Molinari S. Differentiation-dependent lysine 4 acetylation enhances MEF2C binding to DNA in skeletal muscle cells. Nucleic Acids Res 2008; 36:915-28; PMID:18086704; https://doi.org/10.1093/nar/gkm1114
- Magli A, Angelelli C, Ganassi M, Baruffaldi F, Matafora V, Battini R, Bachi A, Messina G, Rustighi A, Del Sal G, et al. Proline isomerase Pin1 represses terminal differentiation and myocyte enhancer factor 2C function in skeletal muscle cells. J Biol Chem 2010; 285:34518-27; PMID:20801874; https://doi.org/10.1074/jbc.M110.104133
- McKinsey TA, Zhang CL, Olson EN. Identification of a signal-responsive nuclear export sequence in class II histone deacetylases. Mol Cell Biol 2001; 21:6312-21; PMID:11509672; https://doi.org/10.1128/MCB.21.18.6312-6321.2001
- Ganassi M, Badodi S, Polacchini A, Baruffaldi F, Battini R, Hughes SM, Hinits Y, Molinari S. Distinct functions of alternatively spliced isoforms encoded by zebrafish mef2ca and mef2cb. Biochim Biophys Acta 2014; 1839:559-70; PMID:24844180; https://doi.org/10.1016/j.bbagrm.2014.05.003
- Zhu B, Gulick T. Phosphorylation and alternative pre-mRNA splicing converge to regulate myocyte enhancer factor 2C activity. Mol Cell Biol 2004; 24:8264-75; PMID:15340086; https://doi.org/10.1128/MCB.24.18.8264-8275.2004
- Zhu B, Ramachandran B, Gulick T. Alternative pre-mRNA splicing governs expression of a conserved acidic transactivation domain in myocyte enhancer factor 2 factors of striated muscle and brain. J Biol Chem 2005; 280:28749-60; PMID:15834131; https://doi.org/10.1074/jbc.M502491200
- Zhang M, Zhu B, Davie J. Alternative splicing of MEF2C controls its activity in normal myogenesis and promotes tumorigenicity in rhabdomyosarcoma cells. J Biol Chem 2014; 290:310-24; PMID:25404735
- McDermott JC, Cardoso MC, Yu YT, Andres V, Leifer D, Krainc D, Lipton SA, Nadal-Ginard B. hMEF2C gene encodes skeletal muscle- and brain-specific transcription factors. Mol Cell Biol 1993; 13:2564-77; PMID:8455629
- Coso OA, Montaner S, Fromm C, Lacal JC, Prywes R, Teramoto H, Gutkind JS. Signaling from G protein-coupled receptors to the c-jun promoter involves the MEF2 transcription factor. Evidence for a novel c-jun amino-terminal kinase-independent pathway. J Biol Chem 1997; 272:20691-7; PMID:9252389; https://doi.org/10.1074/jbc.272.33.20691
- Wilker PR, Kohyama M, Sandau MM, Albring JC, Nakagawa O, Schwarz JJ, Murphy KM. Transcription factor Mef2c is required for B cell proliferation and survival after antigen receptor stimulation. Nat Immunol 2008; 9:603-12; PMID:18438409; https://doi.org/10.1038/ni.1609
- Han TH, Prywes R. Regulatory role of MEF2D in serum induction of the c-jun promoter. Mol Cell Biol 1995; 15:2907-15; PMID:7760790
- Clarke N, Arenzana N, Hai T, Minden A, Prywes R. Epidermal growth factor induction of the c-jun promoter by a Rac pathway. Mol Cell Biol 1998; 18:1065-73; PMID:9448004
- Kato Y, Kravchenko VV, Tapping RI, Han J, Ulevitch RJ, Lee JD. BMK1/ERK5 regulates serum-induced early gene expression through transcription factor MEF2C. EMBO J 1997; 16:7054-66; PMID:9384584; https://doi.org/10.1093/emboj/16.23.7054
- Kato Y, Tapping RI, Huang S, Watson MH, Ulevitch RJ, Lee JD. Bmk1/Erk5 is required for cell proliferation induced by epidermal growth factor. Nature 1998; 395:713-6; PMID:9790194; https://doi.org/10.1038/27234
- Estrella NL, Desjardins CA, Nocco SE, Clark AL, Maksimenko Y, Naya FJ. MEF2 transcription factors regulate distinct gene programs in mammalian skeletal muscle differentiation. J Biol Chem 2014; 290:1256-68; PMID:25416778
- Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW, Vogelstein B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 1998; 282:1497-501; PMID:9822382; https://doi.org/10.1126/science.282.5393.1497
- Hermeking H, Lengauer C, Polyak K, He TC, Zhang L, Thiagalingam S, Kinzler KW, Vogelstein B. 14-3-3 sigma is a p53-regulated inhibitor of G2/M progression. Mol Cell 1997; 1:3-11; PMID:9659898; https://doi.org/10.1016/S1097-2765(00)80002-7
- Jin S, Tong T, Fan W, Fan F, Antinore MJ, Zhu X, Mazzacurati L, Li X, Petrik KL, Rajasekaran B, et al. GADD45-induced cell cycle G2-M arrest associates with altered subcellular distribution of cyclin B1 and is independent of p38 kinase activity. Oncogene 2002; 21:8696-704; PMID:12483522; https://doi.org/10.1038/sj.onc.1206034
- Yaffe D, Saxel O. Serial passaging and differentiation of myogenic cells isolated from dystrophic mouse muscle. Nature 1977; 270:725-7; PMID:563524; https://doi.org/10.1038/270725a0
- Kitzmann M, Fernandez A. Crosstalk between cell cycle regulators and the myogenic factor MyoD in skeletal myoblasts. Cell Mol Life Sci 2001; 58:571-9; PMID:11361092; https://doi.org/10.1007/PL00000882
- Ridgeway AG, Wilton S, Skerjanc IS. Myocyte enhancer factor 2C and myogenin up-regulate each other's expression and induce the development of skeletal muscle in P19 cells. J Biol Chem 2000; 275:41-6; PMID:10617583; https://doi.org/10.1074/jbc.275.1.41
- Doucet C, Gutierrez GJ, Lindon C, Lorca T, Lledo G, Pinset C, Coux O. Multiple phosphorylation events control mitotic degradation of the muscle transcription factor Myf5. BMC Biochem 2005; 6:27; PMID:16321160; https://doi.org/10.1186/1471-2091-6-27
- Kitzmann M, Carnac G, Vandromme M, Primig M, Lamb NJ, Fernandez A. The muscle regulatory factors MyoD and myf-5 undergo distinct cell cycle-specific expression in muscle cells. J Cell Biol 1998; 142:1447-59; PMID:9744876; https://doi.org/10.1083/jcb.142.6.1447
- Lindon C, Montarras D, Pinset C. Cell cycle-regulated expression of the muscle determination factor Myf5 in proliferating myoblasts. J Cell Biol 1998; 140:111-8; PMID:9425159; https://doi.org/10.1083/jcb.140.1.111
- Batonnet-Pichon S, Tintignac LJ, Castro A, Sirri V, Leibovitch MP, Lorca T, Leibovitch SA. MyoD undergoes a distinct G2/M-specific regulation in muscle cells. Exp Cell Res 2006; 312:3999-4010; PMID:17014844; https://doi.org/10.1016/j.yexcr.2006.09.001
- Song A, Wang Q, Goebl MG, Harrington MA. Phosphorylation of nuclear MyoD is required for its rapid degradation. Mol Cell Biol 1998; 18:4994-9; PMID:9710583
- Tintignac LA, Sirri V, Leibovitch MP, Lecluse Y, Castedo M, Metivier D, Kroemer G, Leibovitch SA. Mutant MyoD lacking Cdc2 phosphorylation sites delays M-phase entry. Mol Cell Biol 2004; 24:1809-21; PMID:14749395; https://doi.org/10.1128/MCB.24.4.1809-1821.2004
- Yu H. Regulation of APC-Cdc20 by the spindle checkpoint. Curr Opin Cell Biol 2002; 14:706-14; PMID:12473343; https://doi.org/10.1016/S0955-0674(02)00382-4
- Bashir T, Dorrello NV, Amador V, Guardavaccaro D, Pagano M. Control of the SCF(Skp2-Cks1) ubiquitin ligase by the APC/C(Cdh1) ubiquitin ligase. Nature 2004; 428:190-3; PMID:15014502; https://doi.org/10.1038/nature02330
- Wei W, Ayad NG, Wan Y, Zhang GJ, Kirschner MW, Kaelin WG Jr. Degradation of the SCF component Skp2 in cell-cycle phase G1 by the anaphase-promoting complex. Nature 2004; 428:194-8; PMID:15014503; https://doi.org/10.1038/nature02381
- Glotzer M, Murray AW, Kirschner MW. Cyclin is degraded by the ubiquitin pathway. Nature 1991; 349:132-8; PMID:1846030; https://doi.org/10.1038/349132a0
- Pfleger CM, Kirschner MW. The KEN box: an APC recognition signal distinct from the D box targeted by Cdh1. Genes Dev 2000; 14:655-65; PMID:10733526
- Hinits Y, Pan L, Walker C, Dowd J, Moens CB, Hughes SM. Zebrafish Mef2ca and Mef2cb are essential for both first and second heart field cardiomyocyte differentiation. Dev Biol 2012; 369:199-210; PMID:22750409; https://doi.org/10.1016/j.ydbio.2012.06.019
- Gao D, Inuzuka H, Tseng A, Chin RY, Toker A, Wei W. Phosphorylation by Akt1 promotes cytoplasmic localization of Skp2 and impairs APCCdh1-mediated Skp2 destruction. Nat Cell Biol 2009; 11:397-408; PMID:19270695; https://doi.org/10.1038/ncb1847
- Littlepage LE, Ruderman JV. Identification of a new APC/C recognition domain, the A box, which is required for the Cdh1-dependent destruction of the kinase Aurora-A during mitotic exit. Genes Dev 2002; 16:2274-85; PMID:12208850; https://doi.org/10.1101/gad.1007302
- Mailand N, Diffley JF. CDKs promote DNA replication origin licensing in human cells by protecting Cdc6 from APC/C-dependent proteolysis. Cell 2005; 122:915-26; PMID:16153703; https://doi.org/10.1016/j.cell.2005.08.013
- Rodier G, Coulombe P, Tanguay PL, Boutonnet C, Meloche S. Phosphorylation of Skp2 regulated by CDK2 and Cdc14B protects it from degradation by APC(Cdh1) in G1 phase. EMBO J 2008; 27:679-91; PMID:18239684; https://doi.org/10.1038/emboj.2008.6
- den Elzen N, Pines J. Cyclin A is destroyed in prometaphase and can delay chromosome alignment and anaphase. J Cell Biol 2001; 153:121-36; PMID:11285279; https://doi.org/10.1083/jcb.153.1.121
- Gabellini D, Colaluca IN, Vodermaier HC, Biamonti G, Giacca M, Falaschi A, Riva S, Peverali FA. Early mitotic degradation of the homeoprotein HOXC10 is potentially linked to cell cycle progression. EMBO J 2003; 22:3715-24; PMID:12853486; https://doi.org/10.1093/emboj/cdg340
- Geley S, Kramer E, Gieffers C, Gannon J, Peters JM, Hunt T. Anaphase-promoting complex/cyclosome-dependent proteolysis of human cyclin A starts at the beginning of mitosis and is not subject to the spindle assembly checkpoint. J Cell Biol 2001; 153:137-48; PMID:11285280; https://doi.org/10.1083/jcb.153.1.137
- Hames RS, Wattam SL, Yamano H, Bacchieri R, Fry AM. APC/C-mediated destruction of the centrosomal kinase Nek2A occurs in early mitosis and depends upon a cyclin A-type D-box. EMBO J 2001; 20:7117-27; PMID:11742988; https://doi.org/10.1093/emboj/20.24.7117
- Shen M, Stukenberg PT, Kirschner MW, Lu KP. The essential mitotic peptidyl-prolyl isomerase Pin1 binds and regulates mitosis-specific phosphoproteins. Genes Dev 1998; 12:706-20; PMID:9499405; https://doi.org/10.1101/gad.12.5.706
- Wales S, Hashemi S, Blais A, McDermott JC. Global MEF2 target gene analysis in cardiac and skeletal muscle reveals novel regulation of DUSP6 by p38MAPK-MEF2 signaling. Nucleic Acids Res 2015; 42:11349-62; https://doi.org/10.1093/nar/gku813
- Hollander MC, Sheikh MS, Bulavin DV, Lundgren K, Augeri-Henmueller L, Shehee R, Molinaro TA, Kim KE, Tolosa E, Ashwell JD, et al. Genomic instability in Gadd45a-deficient mice. Nat Genet 1999; 23:176-84; PMID:10508513; https://doi.org/10.1038/13802
- Hosing AS, Kundu ST, Dalal SN. 14-3-3 Gamma is required to enforce both the incomplete S phase and G2 DNA damage checkpoints. Cell Cycle 2008; 7:3171-9; PMID:18843201; https://doi.org/10.4161/cc.7.20.6812
- Kasahara K, Goto H, Enomoto M, Tomono Y, Kiyono T, Inagaki M. 14-3-3gamma mediates Cdc25A proteolysis to block premature mitotic entry after DNA damage. EMBO J 2010; 29:2802-12; PMID:20639859; https://doi.org/10.1038/emboj.2010.157
- Lee JH, Lu H. 14-3-3Gamma inhibition of MDMX-mediated p21 turnover independent of p53. J Biol Chem 2011; 286:5136-42; PMID:21148311; https://doi.org/10.1074/jbc.M110.190470
- Vairapandi M, Balliet AG, Hoffman B, Liebermann DA. GADD45b and GADD45g are cdc2/cyclinB1 kinase inhibitors with a role in S and G2/M cell cycle checkpoints induced by genotoxic stress. J Cell Physiol 2002; 192:327-38; PMID:12124778; https://doi.org/10.1002/jcp.10140
- Ma L, Liu J, Liu L, Duan G, Wang Q, Xu Y, Xia F, Shan J, Shen J, Yang Z, et al. Overexpression of the transcription factor MEF2D in hepatocellular carcinoma sustains malignant character by suppressing G2-M transition genes. Cancer Res 2014; 74:1452-62; PMID:24390737; https://doi.org/10.1158/0008-5472.CAN-13-2171