
Abstract
The DNA-dependent protein kinase catalytic subunit (DNA-PKcs) plays a major role in DNA damage signaling and repair and is also frequently overexpressed in tumor metastasis. We used isogenic cell lines expressing different levels of DNA-PKcs to investigate the role of DNA-PKcs in metastatic development. We found that DNA-PKcs participates in melanoma primary tumor and metastasis development by stimulating angiogenesis, migration and invasion. Comparison of conditioned medium content from DNA-PKcs-proficient and deficient cells reveals that DNA-PKcs controls secretion of at least 103 proteins (including 44 metastasis-associated with FBLN1, SERPINA3, MMP-8, HSPG2 and the inhibitors of matrix metalloproteinases, such as α-2M and TIMP-2). High throughput analysis of secretomes, proteomes and transcriptomes, indicate that DNA-PKcs regulates the secretion of 85 proteins without affecting their gene expression. Our data demonstrate that DNA-PKcs has a pro-metastatic activity via the modification of the tumor microenvironment. This study shows for the first time a direct link between DNA damage repair and cancer metastasis and highlights the importance of DNA-PKcs as a potential target for anti-metastatic treatment.
Abbreviations
| α-2M | = | α-2-macroglobulin |
| CM | = | conditioned media |
| DNA-PK | = | DNA-dependent protein kinase |
| DNA-PKcs | = | DNA-PK catalytic subunit |
| DSB | = | double-strand break |
| ECM | = | extracellular matrix |
| MMP | = | matrix metalloproteinase |
| MS | = | mass spectrometry |
| NHEJ | = | non-homologous end joining |
| SILAC | = | stable isotope labeling by amino acids in cell culture |
| TIMP | = | tissue inhibitor of metalloproteinase. |
Introduction
The dysregulation of DNA repair genes affects the response of cells to DNA-damaging anti-cancer treatment. Several studies have reported a strong relationship between the metastatic capacity of tumor cells and their resistance to treatments such as radiotherapy and chemotherapy.Citation1,2 As resistance to chemo/radiotherapy may result from the upregulation of DNA repair pathwaysCitation3,4 we investigated the possible correlation between metastasis and one of the key proteins involved in DNA repair, the DNA-dependent protein kinase (DNA-PK).
DNA double-strand breaks (DSBs) pose the greatest threat to cell viability and, if left unrepaired, they result in a massive loss of genetic information, chromosomal aberrations, or cell death. Two major pathways exist for the repair of DSBs: non-homologous end-joining (NHEJ) and homologous recombination (HR). NHEJ is more rapid and efficient than HR in human cells.Citation5 A DNA-PK complex formed at DNA breaks by the catalytic subunit (DNA-PKcs) and a heterodimer of Ku70/Ku80 proteins plays a critical role in the repair of DSBs by NHEJ. Cells lacking DNA-PK are extremely sensitive to radiation and to many DNA-damaging therapeutic agents.Citation6 DNA-PKcs is strongly expressed in human cells.Citation7 The other factors involved in NHEJ are less abundant than the DNA-PK complex,Citation8,9 suggesting that the repair activity alone is not sufficient to account for the increased expression of this complex in human cells. Indeed, many authors have reported that DNA-PKcs could be involved in cell survival through Akt activation,Citation10 inflammatory responses (NFκB activation),Citation11 the regulation of metabolic genes,Citation12 cancer cell radioresistance through EGFR signaling,Citation13,14 mRNA metabolismCitation15 and cell cycle control, through its roles in mitosis, microtubule dynamics, and correct chromosomal segregation.Citation16 How these functions are related to DNA-PKcs role in DNA repair remains controversial.
Since some targets of DNA-PKcs play an important role in the development of cancer metastasisCitation17-19 we designed experiments to estimate the requirement in DNA-PKcs for migration and invasion. Metastasis, defined as the spread of malignant tumor cells from the primary tumor mass to distant sites, involves a complex series of interconnected events. Two of these events, migration and invasion, are critical processes requiring proteolysis of the basement membrane and the surrounding extracellular matrix (ECM). Cancer cells interact with their surrounding microenvironment by secreting proteolytic enzymes called matrix metalloproteases (MMPs).Citation20,21 Improvements in our understanding of the regulation of the tumor microenvironment are required to elucidate the processes at work during cancer metastasis and to limit the risk of metastatic spread during treatment.
In this work, we demonstrate that inhibition or depletion of DNA-PKcs impairs cell migration and invasion in vitro. DNA-PKcs silencing inhibits the formation of primary melanomas and delays lymph node metastatic invasion in vivo. We demonstrate that secretion of regulators of MMPs and other metastasis-associated factors, which influence the tumor microenvironment is regulated by DNA-PKcs.
Results
DNA-PKcs is critical for melanoma tumor implantation but does not affect primary tumor growth
Very few human tumor cells have been shown to be deficient in DNA-PKcs. Glioma-derived M059J cells are the DNA-PKcs-deficient cells most widely studied. These cells and their DNA-PKcs-proficient counterpart (MO59K) were isolated from the same tumor.Citation22 We attempted to graft these tumor cells into SCID mice. We found that MO59J cells did not form tumors, whereas MO59K cells gave rise to tumors in 100% of the animals tested (Fig. S1A). The role of DNA-PKcs in tumor formation was further confirmed in SK28 human melanoma cells depleted of DNA-PKcs by stable transfection with a shRNA. The abundance of DNA-PKcs in the shDNA-PK-treated cells was only 19% that in shCTL-treated cells (). The SK28 cells and shCTL-treated cells displayed efficient subcutaneous tumor formation in Nude mice and at least 60% of the animals receiving cell grafts developed metastases in proximal lymph nodes (Fig. S1B, C). The DNA-PKcs-depleted cells had a slightly slower proliferation rate than control cells, consistent with previous reportsCitation23 (). Tumor implantation was strongly impaired following the transplantation of shDNA-PK-treated cells at low cell concentrations, as demonstrated by comparison with shCTL-treated cells (). By increasing the number of shDNA-PK-treated cells used for transplantation, we were able to achieve successful engraftment, but tumor growth was significantly delayed (). Once the tumors had reached a size of 100 mm3, both tumor types were found to be growing at the same speed (Fig. S1D, E). Histological analyses of large shDNA-PK and shCTL tumors (size ≥ 1500 mm3) showed no difference in proliferation, apoptosis or angiogenesis (). By contrast, the examination of small tumors (size ≤ 150 mm3) revealed the presence of smaller numbers of blood vessels () and a lower proliferation index () in shDNA-PK tumors, potentially accounting for their slower growth after grafting. DNA-PKcs depletion was weaker (52% residual protein) in shDNA PK tumors than in cell culture, but remained constant during tumor growth (; Fig. S2A, B).
Figure 1. DNA-PKcs plays a role in primary tumor formation. The downregulation of DNA-PKcs protein levels in cells and tumors (volume of 1500 mm3) after infection with lentiviral particles carrying a shRNA directed against DNA-PKcs (shDNA-PK) or a nontargeting shRNA (shCTL) was checked by (A) protein gel blotting with an antibody against DNA-PKcs (β-actin was used as an internal control) and by (B) immunostaining for DNA-PKcs (green). DNA was stained with DAPI (blue). Scale bar = 20 μm. (C) Proliferation of SK28 cells stably transduced with shCTL and shDNA-PK. Results are expressed as mean cell number ± standard error of mean (SEM) for 2 independent experiments. (D) The percentage of mice displaying primary tumor formation 21 d after the injection of different number of cells transduced with shDNA-PK and shCTL (n ≤ 6 mice/each injection condition). (E) Growth of primary tumors from cells transduced with shCTL and shDNA-PK, after the injection of 4 × 106 cells into Nude mice. (F) Comparison of angiogenesis in shCTL and shDNA-PK tumors of different sizes. Results show the mean number of vessels stained with anti-CD31 antibodies ± SEM per microscopic field for each group (shCTL, n=3 ; shDNA-PK, nequals;4 ). (G) Comparison of proliferation index at different tumor sizes, for shCTL and shDNA-PK tumors. Results are expressed as the mean percentage of proliferative cells stained with anti-Ki67 antibodies ± SEM per microscopic field, for each group (shCTL, nequals;3; shDNA-PK, nequals;4 ). The significance of differences was assessed in Mann-Whitney tests. *P < 0.05, **P < 0.005.
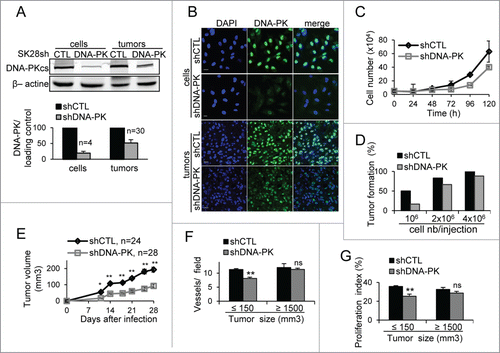
These results suggest that a minimal basal DNA-PKcs content is required for tumor formation, particularly for the promotion of neoangiogenesis and early-stage proliferation.
DNA-PKcs gene silencing impairs melanoma metastasis in lymph nodes
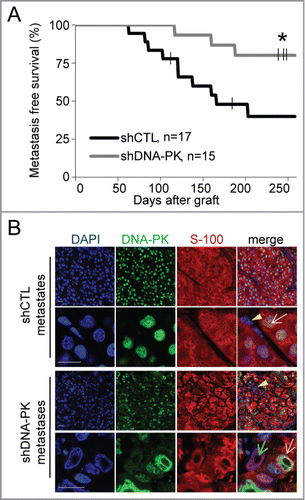
We investigated whether DNA-PKcs affected the metastatic properties of primary tumors, by surgically removing all primary tumors when their volume reached 1500 mm3 and monitoring the occurrence of metastasis in the proximal axillary and/or inguinal lymph node in the flank into which the cells had been grafted. Metastatic cells were identified on the basis of the presence of S100 protein, which is specific to human melanoma cellsCitation24 (). Metastases appeared 2 months earlier in animals with DNA-PKcs-proficient tumors than in animals with DNA-PKcs-deficient tumors (). This difference is much greater than the delay in early-stage tumor development, which did not exceed 14 d ( and ). Moreover, 258 d after tumor cell injection, metastasis-free survival was only 39.8% (7/17) for the shCTL group, whereas it was as high as 80% (12/15) for the shDNA-PK group (P = 0.018, ). All shDNA-PK metastases contained a mixed population of DNA-PKcs-proficient and -deficient cells, present in proportions similar to those in the primary tumor (, Fig. S3) suggesting a collective invasion and migration by cells with and without DNA-PKcs.
Figure 2. DNA-PKcs depletion impairs the formation of melanoma metastases. We grafted 4×106 cells of shCTL- and shDNA-PK-treated SK28 cells into Nude mice and then surgically removed the resulting primary tumors when they attained a volume of 1500 mm3. Animals were monitored for 258 d after grafting, to check for the occurrence of metastases in the proximal lymph node. (A) Lymph-node metastasis-free survival curves for shCTL and shDNA-PK tumors (*P=0 .018, Kaplan-Meier test). (B) Representative images of double-immunostaining for DNA-PK (green) and human melanoma marker S-100 (red) in shCTL and shDNA-PK metastases. Yellow arrows indicate infiltrating mouse cells, white arrowheads indicate human DNA-PK-positive cells and green arrowheads indicate human DNA-PK-deficient cells. DNA was stained with DAPI (blue). Scale bar = 20 μm.
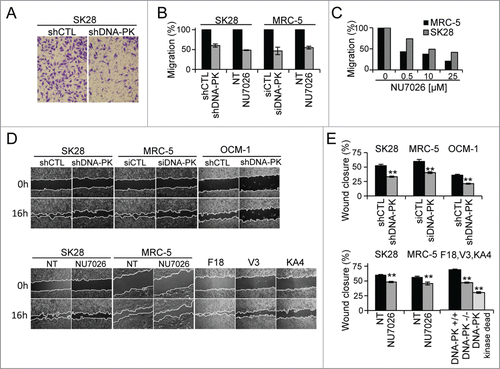
The inhibition or depletion of DNA-PKcs impairs cell migration and invasion
The defect of the capacity of shDNA-PK-treated cells to initiate primary tumor or metastatic growth led us to analyze the capacity of these cells to migrate and invade the extracellular matrix. The depletion of DNA-PKcs with shDNA-PK in SK28 and OCM-1 cells or siRNA in MRC5 cells, or the inhibition of its activity with NU7026Citation25 resulted in a strong inhibition of cell migration in Transwell migration assay and wound closure assays (), whereas cell proliferation and viability during this time remained similar in all the conditions ( and Fig. S4B, C). The migration-inhibiting effect of NU7026 was dose-dependent in all cell lines tested (). V3 hamster cell lines lacking DNA-PKcs or complemented with the kinase-dead DNA-PKcs mutant (KA4 cells) displayed slower wound closure () than V3 cells complemented with functional DNA-PKcs (F18 cells), demonstrating the requirement of DNA-PKcs kinase activity for migration in rodent cells. The migration defect was more pronounced in V3-KA4 (kinase dead) cells than in the DNA-PKcs deficient V3 cells. A similar observation was reported by Kurimasa et alCitation26 who observed a higher repair defect in KA4 mutants than in V3 deficient cells. This difference was explained by a competition with other proteins of the inactive kinase dead enzyme on common targets.
Figure 3. The depletion or inhibition of DNA-PKcs impairs cell migration. Cell migration was measured in a Transwell assay (A–C) and in a wound healing assay (D, E), in cells depleted of DNA-PKcs by siDNA-PK (MRC-5) or shDNA-PK (SK28 and OCM-1) or after the inhibition of DNA-PKcs with a specific inhibitor (10 μM NU7026). Wound healing was assessed in DNA-PK-deficient V3 rodent cells complemented with wild-type DNA-PK (F18 cells) or the kinase-dead DNA-PKcs mutant (KA4 cells). (A) Representative image of shCTL-treated and shDNA-PK-treated migratory cells, fixed on the bottom of a Transwell filter. (B) The percentage cell migration was calculated from the number of cells migrating to the bottom of the filter and was normalized with respect to the corresponding control. The values shown are the means of at least 3 independent experiments (each conducted in duplicate) for each set of experimental conditions. (C) Dose effect of the inhibitor, NU7026, on the migration of MRC-5 and SK28 cell lines. (D) Example of wound healing microscopy measurements in the various conditions. (E) Percentage wound closure, estimated as the difference in cell-free surface area between the images obtained at 0 h and 16 h (24 h for OCM-1 cells) in the various conditions (n=2 independent experiments per set of conditions). The significance of differences was assessed in Mann-Whitney tests. *P < 0.05, **P < 0.005.
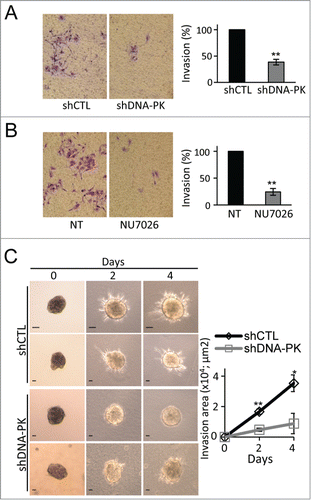
We further investigated the role of DNA-PKcs in tumor invasion. The invasive capacity of DNA-PKcs-depleted or NU7026-treated cells was significantly impaired in the 2D Matrigel Transwell assay () and the 3D collagen-embedded spheroid invasion assay (). DNA-PKcs is thus clearly important for cell migration and invasion, 2 critical processes in cancer metastasis.
Figure 4. The depletion or inhibition of DNA-PKcs impairs melanoma cell invasion. Matrigel invasion by SK28 human melanoma cells (A) transformed with shCTL or shDNA-PK, or (B) incubated in the presence of DNA-PK inhibitor (10 μM NU7026). The graphs show the mean percentage invasion ± SEM for each set of conditions, normalized with respect to control conditions (n=3 , each experiment conducted in duplicate or triplicate). Representative fields from the bottom of the filter are shown and the percentage invasive cells is shown on the right. (C) Inhibition of SK28 spheroid invasion after DNA-PKcs-depletion. Spheroids from shCTL-treated or shDNA-PK-treated SK28 cells were embedded into collagen I for 3-dimensional invasion assays. Representative images of SK28 spheroid invasion obtained just after embedding in collagen (0 days), and 2 and 4 d after embedding, from left to right. Scale bar: 20 μm. The graph shows the mean invasion area ± SEM (shCTL, n= 5 and shDNA-PK, n=6 ), as measured by time-lapse microscopy and calculated with ImageJ software. The significance of differences was assessed in Mann-Whitney tests. *P < 0.05, **P < 0.005.
Inhibition of cell migration and invasion by conditioned media from DNA-PKcs-deficient cells
Secreted proteins play a key role in cell motility, migration and invasiveness. We monitored the migration of cells incubated in different conditioned media (CM). Cells (shCTL or shDNA-PK) were re-suspended in the 4-times concentrated CM from shCTL or shDNA-PK cells and added to the upper chamber of migration inserts. By this approach we limit the effects of the proteins secreted during the experimental time and analyze essentially the effects of the proteins present in the concentrated CM. The addition of CM from shCTL-treated cells restored the migration of shDNA-PK-treated cells (). By contrast, CM from shDNA-PK-treated cells did not increase the rate of migration of shDNA-PK-treated cells and significantly impaired the migration of shCTL-treated cells. These results suggest that factors required for migration are limiting in CM from shDNA-PK-treated cells but can be provided in trans by CM from shCTL-treated cells.
Figure 5. Inhibition of melanoma cell migration and invasion by conditioned media (CM) from DNA-PKcs-deficient melanoma cells. (A) Cell migration, assessed in a Transwell assay of SK28shCTL and SK28shDNA-PK cells with or without fold4- concentrated CM from either SK28shCTL or SK28shDNA-PK cells. Representative images of migratory cells in the indicated conditions are shown. The mean percentage migration ± SEM for each set of conditions, normalized with respect to control conditions (n=3 , each conducted in duplicate) is next to the images. (B) Cell invasion, assessed by Matrigel Transwell assays of SK28shCTL and SK28shDNA-PK cells with and without fold4- concentrated CM from either SK28shCTL or SK28shDNA-PK cells. Representative images of invasive cells in the indicated conditions are shown. The mean percentage invasion ± SEM for each set of conditions, normalized with respect to control conditions (n=3 , each conducted in duplicate) is shown below the images. The significance of differences was determined in Mann-Whitney tests. *P < 0.05, **P < 0.005.
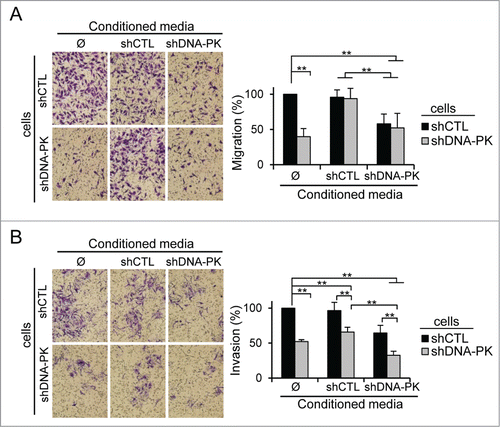
The effect of CM on cell invasion differed from that on cell migration. CM from shCTL-treated cells had almost no effect on invasion, whereas that from shDNA-PK-treated cells strongly inhibited the invasion of both control and shDNA-PK-treated cells (). The additive effect of concentrated CM from shDNA-PK-treated cells on shDNA-PK-treated cells, which was not observed in the migration study, suggests that factors concentrated in CM from shDNA-PK-treated cells are inhibiting the invasion.
These results demonstrate that the control of cell migration and invasion by DNA-PKcs is mostly due to the secretion of promigratory compounds (by DNA-PKcs-proficient cells) and inhibitors of invasion (by DNA-PKcs-deficient cells).
Secretome analysis reveals that DNA-PKcs regulates matrix metalloproteinases
We used SILAC technology coupled with LC-MS/MS mass spectrometryCitation1 to compare the secretome and proteome of DNA-PKcs-deficient and -proficient cells. We detected 1153 proteins in the secretome of these cells, 151 of which were present at different abundances in DNA-PKcs-deficient and –proficient cells. Only 103 proteins (68%) were experimentally shown to be extracellular or containing predicted cleavable signal peptides targeting them to the secretory pathway (SecretomeP databases and SignalP database) were considered to display differential secretion between shDNA-PK-treated and shCTL-treated cells and were retained for further analysis (Table S1). Ingenuity pathway analysis (IPA) highlighted cellular movement as the function for which the largest number of proteins was differentially regulated (44 proteins; Table S1). The pathway most altered by DNA-PKcs silencing was the “Inhibition of Matrix Metalloproteases,” for which 4 proteins (TIMP-2, α-2M, MMP-8, HSPG2) were upregulated, and 2 (MMP-1 and MMP-14) were downregulated ( and Table S1). Moreover, the proteins most strongly overrepresented (10 times higher levels) in CM from shDNA-PK-treated cells included the anti-metastatic protein FBLN-1 (fibulin-1) (). Western blot analysis confirmed the SILAC data ( and Fig. S4A). The full-length MMP-14 (66 kDa) was not present at a lower abundance, but its inactive truncated form (43 kDa), which is released into CM in microvesicular exosomes,Citation27 was significantly less abundant in the CM of shDNA-PK-treated cells (). In agreement with these results we observed an increase of FBLN-1, α-2M and TIMP-2 secretion after DNA-PKcs depletion by siRNA or inhibition by NU7026 ().
Figure 6 (See previous page). SILAC + LC-MS/MS secretome analysis showing the DNA-PKcs-dependent regulation of MMPs and other metastasis-associated proteins. (A) The SILAC ratio (shDNA-PK/shCTL) is shown for proteins from the "Inhibition of Matrix Metalloproteases" pathway: α-2M, MMP-8, HSPG-2, TIMP-2, MMP-14 and MMP-1 and metastasis-associated protein FBLN-1. No difference in HSP70 secretion was observed between SK28shCTL and SK28shDNA-PK cells, so this protein was used as a control. Criteria for a significant change: fold change ≥ 1.5; P ≤ 0.05; peptides used for quantification ≥ 3. (B, C) The differential secretion of proteins was confirmed by western blotting. Conditioned media (CM) from SK28shCTL and SK28shDNA-PK cells was normalized according to the number of cells in the culture from which they were prepared. CM concentrated by a factor of 50 was denatured and FBLN-1, α-2M, MMP-8, TIMP-2, MMP-14 and MMP-1 were detected by immunoblotting. HSP70 served as a loading control. (D) DNA-PKcs silencing increases the expression of FBLN-1 and α-2M but does not change expression of MMP-8, HSPG2, TIMP-2, MMP-14 and MMP-1. The bar graph shows the microarray results as a relative fold-change for secreted proteins of interest after DNA-PKcs silencing in SK28 melanoma cells. Negative values indicate repression. Values highlighted in light gray are significant (FBLN-1, α-2M). Statistical significance: fold-change ≥ 1.5 and P < 0.05. (E) Comparison of the proteome and secretome after the DNA-PKcs-depletion in SK28 cells. The Venn diagrams show the overlap between proteins (with numbers indicated) from the proteome and secretome. A–Proteins differentially secreted and not present in the proteome (n=67 ); B–Proteins differentially secreted but not differentially present in the proteome (n=24 ); C–Proteins both differentially secreted and differentially present in the proteome (n=12 ); D–Proteins differentially present in the proteome, but not differentially secreted (n=14 ); E- Proteins differentially present in the proteome and not detected in the secretome (n=93 ). (F) Quantification of secreted proteins abundance from SK28 cells treatment with siCTL and siDNA-PKcs or DMSO and DNA-PKcs inhibitor (NU7026, 10μM). CM concentrated by a factor of 50 (siCTL, siDNA-PK) or 25 (DMSO, NU7026) was denatured and FBLN-1, α-2M, TIMP-2 and HSP70 were detected by immunoblotting (not shown). The bar charts give the average abundance of indicated proteins normalized to the loading control (HSP70).
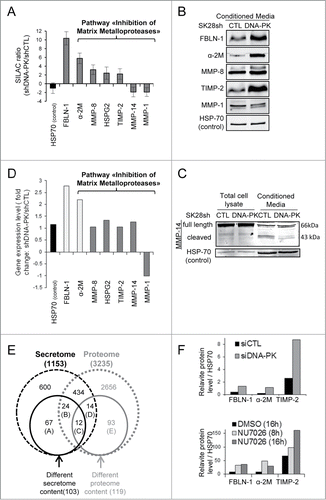
We performed LC-MS/MS to analyze the intracellular protein content of shDNA-PK-treated and shCTL-treated cells. Only 3.7% (119/3235) of the proteins detected in the proteome were affected by DNA-PKcs deficiency ( and Table S2), versus 9% (103/1153) of those in the secretome. For 24 proteins displaying differential secretion, intracellular abundance was not modified by DNA-PKcs depletion, indicating that either the secretion of these proteins or their extracellular stability was modified in DNA-PKcs-deficient cells.
Transcriptional regulation of secreted proteins in shDNA-PK-treated melanoma cells
We analyzed mRNA content to determine whether DNA-PKcs regulates secreted proteins at the translational level or posttranslational level. In total, 380 genes were found to be differentially expressed between the shDNA-PK and shCTL cell lines (data not shown). As expected, the PRKDC mRNA, the target of shDNA-PK, was significantly less abundant (5-fold lower levels) in the silenced cells (data not shown). Only 18 (17.5%) of the 103 secreted proteins affected by DNA-PKcs depletion were differentially expressed (Table S3). For example, SERPINA3, FBLN-1 and α-2M, which code for proteins displaying the highest ratio changes in the secretome, were upregulated in shDNA-PK-treated cells. As the 85 other genes encoding proteins that were differently secreted, HSPG2, TIMP2, MMP8, MMP-14 and MMP-1 displayed no change in expression ().
Discussion
Various authors have suggested that there is a cause-effect relationship between most DNA repair genes and cancer metastasis. Most of these genes were identified in systemic analyses as overexpressed in metastases or primary tumors that develop metastases. DNA damage repair proteins, such as the Ku heterodimer,Citation19 MRE11,Citation28 FANC/BRCA-1,Citation29,30 XRCC3Citation31 and PARP-1,Citation32 were found significantly deregulated in the metastasis of various types of cancer. How other activities than DNA repair are involved in metastasis development remains unclear. However, the KU80 protein, one of the partners of DNA-PKcs in the DNA-PK repair complex, has been shown to be involved in cell-cell and cell-matrix adhesionCitation19 and to colocalize with MMP-9 on the cell outer membrane, where it could play an important role in regulating ECM remodeling in highly invasive cells.Citation33,34 Even if the exact mechanism remains to be elucidated they are clear evidence that DNA repair proteins could participate to the process of cancer metastasis by tumor microenvironment moludation ((MRE-11; Citation35,36, XRCC 1,Citation31 Ku heterodimerCitation19).
Our results demonstrate that, in addition to its key role in the survival of tumor cells,Citation2 DNA-PKcs plays a direct role in their microenvironment regulation and thereby their ability to migrate and invade. Animal studies have shown that low levels of DNA-PKcs are associated with a delay in the initiation of neoangiogenesis and a lower ability of tumors to develop and to metastasize. Endothelial cell migration is essential for angiogenesis. This motile process is directionally regulated by chemotactic stimuli and involves degradation of the ECM to enable progression of the migrating cells. Early in their growth, tumors with DNA-PKcs depletion have few vessels, suggestive of a defect in the signaling pathway promoting cell migration. This defect was overcome in large tumors, in which the number of cells compensated for the limiting amounts of pro-angiogenic factors produced by individual cells. SILAC analysis indicated that DNA-PKcs controlled the secretion of many proteins in the tumor microenvironment, including the MMPs and their inhibitors. These secreted factors contribute to many steps in carcinogenesis, including the establishment of a microenvironment for the primary tumor, angiogenesis, and the tumor cell migration, intravasation, extravasation and invasion.Citation37 The delay in blood vessel formation and tumor proliferation in nascent shDNA-PK tumors may be due to the secretion of the 2 main inhibitors of MMPs, α-2M and TIMP-2,Citation38 and/or the oversecretion of FBLN-1,Citation39 together with the low abundance of MMP-1 and MMP-14Citation37,40 in the microenvironment of the shDNA-PK tumors. A-2M inhibits most proteinases by trapping them in the macroglobulin, leading to rapid clearance by receptor-mediated endocytosis,Citation41 it is possible that the low abundance of MMP-1 and MMP-14 in the secretome of shDNA-PK-treated cells results directly from α-2M overexpression. FBLN-1 and HSPG2 are glycoproteins associated with the basement membraneCitation42,43 that interact with many ECM proteins, such as fibronectin, laminin and nidogen.Citation44-46 Interestingly, laminin α, β and gamma, and fibronectin, were also overrepresented in the secretome of DNA-PKcs-deficient cells, indicating that the balance between the ECM and membrane proteins is altered in the microenvironment of shDNA-PK-treated cells. However, many other proteins (44/105 secreted proteins) oversecreted by DNA-PKcs-deficient cells are involved in cell migration and/or cell invasion (Supplemental Table 1) and may also contribute to the role of DNA-PKcs in cancer metastasis.
How could DNA-PKcs regulate protein secretion? The first possibility is that DNA-PKcs could control proteins secretion at transcriptional level as it has been demonstrated that DNA-PKcs has a direct role in the transcription of some genes.Citation47,48 Actually, DNA-PKcs phosphorylates transcription factors such as Sp1, Oct-1, c-Myc, c-Jun, p53.Citation49 Moreover, DNA-PKcs is involved in transcriptional initiation by interacting with a TopoIIβ and PARP‑1.Citation50,51 DNA-PKcs has been shown to be implicated in the regulation of metabolic gene expression in response to insulin, when transient DSBs occur.Citation12 Though most studies describe a role of DNA-PKcs in transcription after DSB induction,Citation47,48, its role in the regulation of transcription of the extracellular matrix gene laminin α 4 (Lama4) was demonstrated in absence of its activating treatment.Citation48 In our study, transcriptome analysis revealed only 2% of mRNAs (380/19929) varying between shDNA-PK and control SK28 cells. However, only 17,5% of the differently secreted proteins were found over-represented in the mRNAs regulated by DNA-PKcs, eliminating the possibility that their transcription control by DNA-PKcs could account for the observed change in all secreted proteins. Another hypothesis, to explain the DNA-PKcs role in protein secretion comes from the recent finding that DNA-PKcs is involved in the dispersal of the Golgi apparatus after irradiation.Citation52 This result was obtained by inducing DNA damage, and raises the possibility that secretion is controlled by DNA-PKcs in the Golgi apparatus, which plays a key role in exosome production. These results remained to be confirmed but exosomes contain very specific types of proteins involved in cancer development and metastasis.Citation53 The role of DNA-PKcs in regulating secretion by controlling the subclasses of exosomes produced by cancer cells requires further investigation.
The major migration defect of the K4A mutant indicates that DNA-PKcs kinase activity is involved in regulation of migration. The activation of this kinase may promote protein modification, increasing tumor cell migration or invasion. Several studies have reported that irradiation, a treatment likely to induce extensive DNA-PKcs activation, promotes the movement of cells out of the irradiated area and invasive behavior by many types of cancer cells.Citation54,55 Moreover, DNA-PKcs controls pro-angiogenic VEGF secretion in irradiated cells.Citation56 These results illustrate the role of DNA-PKcs in tumor microenvironment. We show here that DNA-PKcs controls the secretion of inhibitors of cell migration and invasion. DNA-PKcs activation at sites of DNA damage leads to the autophosphorylation of this protein, resulting in its inactivation. Thus, DNA-PKcs may act as an "alarm system" that is activated when the cell is endangered by DNA damage, promoting the movement of the cell to enable it to escape the aggressive environment.
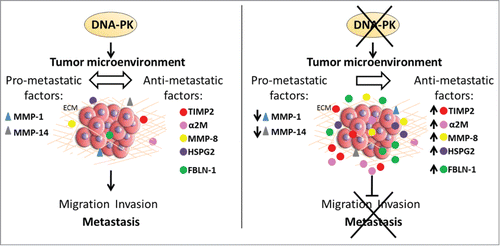
In summary, our findings reveal that DNA-PKcs regulates the tumor microenvironment by controlling the secretion of factors involved in metastasis (). They provide the first direct link between the main DNA repair enzyme, DNA-PKcs, and cancer metastasis, and highlight the importance of DNA-PKcs as a potential target for anti-metastatic treatment.
Figure 7. Model of the role of DNA-PKcs in regulating the tumor microenvironment. In the presence of DNA-PKcs, tumor cells maintain the balance between pro- and anti-metastatic secreted factors (left). In the absence of DNA-PKcs, this balance is modified. DNA-PKcs knockdown leads to the secretion of anti-metastatic factors, such as TIMP-2, α-2M, MMP-8, HSPG2 and FBLN-1, and to downregulation of the secretion of pro-metastatic factors, such as MMP-1 and MMP-14 (right). The absence of DNA-PKcs therefore inhibits cell migration and invasion, thereby preventing tumor metastasis.
Materials and Methods
Cell culture, chemicals, siRNA, shRNA
Cell culture studies were performed with SV40-transformed MRC-5 cells (human fibroblasts, ATCC: CCL-171), OCM-1 cells (human uveal melanomaCitation57), DNA-PKcs-/- CHO cells (V3) and V3 cells expressing wild-type (F18) or a kinase-dead DNA-PKcs mutant (KA4) cellsCitation26 (hamster cells were kindly provided by David J. Chen, Department of Radiation Oncology, University of Texas Southwestern Medical Center, Dallas, TX). Cells were grown as monolayers, at 37°C, in complete DMEM (Gibco, Cergy Pontoise, France) supplemented with 10% fetal calf serum (FCS), 110 mg/l sodium pyruvate and antibiotics (100 μg/ml streptomycin and 100 μg/ml penicillin). SK28 (human melanomaCitation58) cells were grown in RPMI 1640 medium supplemented with 10% FCS and antibiotics. Cells were grown under conditions of 100% humidity, under an atmosphere of 95% air/5% CO2. The DNA-PKcs inhibitor, NU7026, was purchased from Sigma Aldrich (St. Louis, MO, USA). A small interfering RNA (siRNA) specific for DNA-PKcs (ON-TARGETplus SMART Human PRKDC siRNA, ref L-005030–00, Dharmacon, Lafayette, CO, USA) and control siRNA (ON-TARGETplus Non-targeting pool, Dharmacon) were used to transfect the cells in the presence of DharmaFECT (Dharmacon), according to the manufacturer's instructions.
For stable silencing of the DNA-PKcs gene, SK28 cells were used to seed 6-well plates at a concentration of 1.5 × 10Citation5 cells per well on the day before shRNA transduction, to allow them to reach 50% of confluence before viral infection. Subconfluent cells were transduced with lentiviruses expressing a control, non-targeting, shRNA (shCTL; sc-108080, Santa Cruz Biotechnology, Inc.. Germany) or a shRNA targeting DNA-PKcs (sc-35200-V, Santa Cruz Biotechnology), at a multiplicity of infection (MOI) of 3, in the presence of polybrene (5 μg/ml). They were incubated for 24 h and the medium was then replaced with fresh RPMI medium. Then, 72 h after infection, stably transduced cells were selected with puromycin (1 μg/ml). The efficacy of DNA-PKcs knockdown was checked by western blotting and immunofluorescence analysis. Cells from the clone displaying the most efficient DNA-PKcs depletion (80% knockdown) were used for experiments in vitro and in vivo. DNA-PKcs knockdown was also verified by protein gel blotting after several cell passages and before each experiment.
Animal experiments
Animal experiments were approved by the local ethics committee for animal experimentation (Comité Ethique en Expérimentation Animale de l'Institut Curie, CEEA-IC, national registration number: #59). Detailed procedures can be found in the Supplementary Methods.
Transwell migration and invasion assay and wound healing
Cell migration was assessed in the QCM 24-well colorimetric cell migration assay (pore size: 8 μm; ECM 508, Millipore, Billerica, MA, USA), according to the manufacturer's protocol. We evaluated cell invasion with the Cell Invasion Assay Kit (ECM550, Millipore), using ECM-coated Transwell inserts (pore size: 8 μm) according to the manufacturer's protocol. The wound healing assay was performed as previously described.Citation59 Detailed procedures for the cell migration and invasion and wound healing assays are provided in the Supplementary Methods.
3D melanoma spheroid invasion assay
A three-dimensional (3D) multicellular tumor spheroid culture was performed with DNA-PKcs-deficient SK28shDNA-PK and control SK28shCTL melanoma cells. Cells were encapsulated, with a microfluidic device, in alginate microcapsules, to form 3D spheroids. Additional details of the procedure can be found elsewhere.Citation60 Collagen matrices were prepared at a final concentration of 2 mg/ml with rat tail collagen type I (3.71 mg/ml, BD Biosciences, USA), 1 x PBS, RPMI 1640 medium and 1M NaOH (final pH=7 .4). Spheroids about 200 μm in diameter were embedded in collagen drops such that they remained precisely in the center of the drop (in 3D). Invasion was monitored over a period of 4 days, by bright-field microscopy, at ×4 magnification, and was quantified by measuring the area invaded with ImageJ software.
Preparation of conditioned media for cell migration/invasion and secretome analysis
We allowed SK28shCTL and SK28shDNA-PK cells to grow to 80–90% confluence in 60 mm-diameter Petri dishes for the preparation of conditioned media (CM). The medium was discarded, and the cells were washed 3 times with 1 × PBS and twice with serum-free medium. They were then incubated for 24 h with 2 ml of serum-free medium per dish. The CM was collected and centrifuged (200 × g, 5 min) to remove debris, and then filter-sterilized by passage through a Minisart 0.20 μm-pore syringe-driven filter (Sartorius Stedim Biotech S.A., France). For the migration and invasion assay, the collected CM was used fresh and was 4X concentrated with Amicon Ultra-15 (10K) centrifugal filter tubes (Millipore). Cells (SK28shCTL or SK28shDNA-PK) were resuspended in the 4-times concentrated CM from shCTL or shDNA-PK cells and added to the upper chamber of migration or invasion inserts. By this approach we could asses impact of secreted proteins from both cell lines on theeir migratory and invasive potential. The possible continues secretion of factors important for migration and invasion was overcome by concentration over 4-times of CM before to be added to the cells in transwell chamber were migration and invasion were immediately assayed during 16h. For western blotting, trypsin digestion, and LS/MS analysis, the CM was kept at +4°C until use.
Immunofluorescence, immunohistochemistry and immunoblotting
For cultured cells and tumor samples, we used the following antibodies: mouse monoclonal anti-DNA-PKcs (Ab-4, Cocktail, Thermo Scientific, USA), mouse monoclonal anti-β-actin (Clone AC-15, Sigma Aldrich), goat polyclonal anti-HSP70 (K-20, sc-1060, Santa Cruz Biotechnology), rabbit polyclonal anti-S-100 (Z0311, Dako, France). The tumor samples were stained with rabbit polyclonal anti-Ki67 (Ab15580, Abcam, Cambridge, MA, USA) and rabbit polyclonal anti-CD31 (Ab28364, Abcam) antibodies. The secondary antibody was a biotin-conjugated goat anti-rabbit antibody (BA-1000, Vector Laboratories, USA). For immunofluorescence staining, cells were processed as previously described.Citation61 We immunostained 7 μm histological sections from paraffin-embedded tumor samples as previously described.Citation62 The detailed immunofluorescence and immunohistochemistry procedures are described in the Supplementary Methods.
For immunoblotting, the signal was quenched by incubation with 15% trichloroacetic acid (TCA, Sigma Aldrich) for 30 minutes on ice. Alternatively, tumor samples were weighed (frozen samples: 120 mg) and lysed in a Precellys 24 tissue homogenizer (Bertin Technologies, Montigny-le-Bretonneux, France) for 3 × 60 s at 6000 rpm, or until all visible tissue was dissolved, with Precellys CK28 beads (Bertin Technologies), in ice-cold 15% TCA. Further details of the immunoblotting procedures used can be found in the Supplementary Methods.
For immunoblotting with conditioned media (CM), we used the following antibodies: mouse monoclonal anti-α-2-macroglobulin [2D9] (ab36995, Abcam), mouse monoclonal anti-TIMP-2 [3A4] (ab1828, Abcam), mouse monoclonal anti-fibulin 1 (ab54652, Abcam), rabbit monoclonal anti-MMP-1 [EP1247Y] (ab52631, Abcam), mouse monoclonal anti-MMP8 [115–13D2] (ab77964, Abcam) and rabbit monoclonal anti-MMP-14 [EP1264Y] (ab51047, Abcam) antibodies. For the analysis of secreted proteins, cells were incubated for 24 h without serum; the supernatant was then recovered and concentrated 100-, 50-, 25- or fold10- with Amicon Ultra-0.5 or -15 (10 K) centrifugal filter tubes (Millipore) before processing for protein extraction. Approximately 20 μl of concentrated CM was mixed 1:1 with Laemmli buffer and boiled for 10 min. We then separated 30–40 μl of the protein extract from the CM by SDS-PAGE on 12% or 7.5% polyacrylamide gels.
SILAC labeling, trypsin digestion, LC/MS-MS analysis, protein identification and bioinformatics analyses
SK28shDNA-PK and SK28shCTL human melanoma cells were grown in RMPI (arginine- and lysine-deficient, Thermo Scientific) medium for SILAC, with 10% (vol:vol) dialyzed FBS (Thermo Scientific), 5,000 U/l penicillin/streptomycin and 1 μg/ml puromycin, supplemented with either 13C6,15N4 L-arginine-HCl and 13C6,15N2 L-lysine-2HCl (“Heavy Medium”) or with normal 13C6 L-arginine-HCl and 4,4,5,5-D4, L-lysine-2HCl (“Medium Medium”) (all at 0.1 g/l, Thermo Scientific). Further details of the protocols used are provided in the Supplementary Methods.
Microarray assay and data analysis
Total RNA was extracted from SK28shCTL and SK28shDNA-PK cell monolayers with the RNeasy mini kit (Qiagen, USA), according to the manufacturer's instructions. Human Transcriptome Array 2.0 ST arrays were hybridized according Affymetrix recommendations using the WT PLUS protocol and labeling and hybridization kits from Affymetrix. The assay was performed at the Institut Curie microarray core facility. The detailed microarray protocol is described in the Supplementary Experimental Procedures. Affymetrix HTA2 dataset analysis was performed with GenoSplice technology (http://genosplice.com). Genes were considered to display significant regulation if a fold-change in expression of at least 1.5 was observed and if the P value obtained was 0.05 or below (unadjusted P-value). The analysis of the microarray data is described in more detail in the Supplementary Methods.
Statistical analysis
Data are expressed as means ± SEM. We analyzed the results of in vitro and in vivo experiments, using Mann-Whitney U tests to compare the 2 groups. StatEL software was used for this analysis (ad Science, Paris, France). Median metastasis-free survival was estimated by the Kaplan–Meier method. All tests were considered significant for P values ≤ 0.05*, ≤0 .005**, ≤0 .0005***.
Disclosure of Potential Conflicts of Interest
The authors declare no conflict of interest.
1026522_Supplemental_File.pdf
Download PDF (1.3 MB)Acknowledgments
We thank Sophie Dodier-Leboucher from the immunohistology platform and Fabrice Cordelières and Pauline Chabosseau from the imaging platform of Institut Curie. We also thank Jacky Bonaventure for providing the S-100 antibody. We thank to David Gentien and his group from microarray core facility at Institut Curie. We also thank to the group of GenoSplice technology for bioinformatics analysis.
Funding
Ewa Kotula would like to thank the Association Nationale de la Recherche et de la Technologie for funding (CIFRE 1461/2010). This work was supported by the Institut Curie; the Centre National de la Recherche; the Institut National de la Sante et de la Recherche Medicale.
Supplemental Material
Supplemental data for this article can be accessed on publisher's website
Related Research Data
References
- Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, Mann M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics 2002; 1:376-86; PMID:12118079; http://dx.doi.org/10.1074/mcp.M200025-MCP200
- Shintani S, Mihara M, Li C, Nakahara Y, Hino S, Nakashiro K, Hamakawa H. Up-regulation of DNA-dependent protein kinase correlates with radiation resistance in oral squamous cell carcinoma. Cancer Sci 2003; 94:894-900; PMID:14556663; http://dx.doi.org/10.1111/j.1349-7006.2003.tb01372.x
- Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA. DNA repair pathways as targets for cancer therapy. Nat Rev Cancer 2008; 8:193-204; PMID:18256616; http://dx.doi.org/10.1038/nrc2342
- Lieberman HB. DNA damage repair and response proteins as targets for cancer therapy. Curr Med Chem 2008; 15:360-7; PMID:18288990; http://dx.doi.org/10.2174/092986708783497328
- Mao Z, Bozzella M, Seluanov A, Gorbunova V. Comparison of nonhomologous end joining and homologous recombination in human cells. DNA Repair 2008; 7:1765-71; PMID:18675941; http://dx.doi.org/10.1016/j.dnarep.2008.06.018
- Thompson LH. Recognition, signaling, and repair of DNA double-strand breaks produced by ionizing radiation in mammalian cells: the molecular choreography. Mutat Res 2012; 751:158-246; PMID:22743550; http://dx.doi.org/10.1016/j.mrrev.2012.06.002
- Meek K, Kienker L, Dallas C, Wang W, Dark MJ, Venta PJ, Huie ML, Hirschhorn R, Bell T. SCID in Jack Russell terriers: a new animal model of DNA-PKcs deficiency. J Immunol 2001; 167:2142-50; PMID:11489998; http://dx.doi.org/10.4049/jimmunol.167.4.2142
- Anderson CW, Lees-Miller SP. The nuclear serine/threonine protein kinase DNA-PK. Crit Rev Eukaryot Gene Expr 1992; 2:283-314; PMID:1486241
- Mimori T, Hardin JA, Steitz JA. Characterization of the DNA-binding protein antigen Ku recognized by autoantibodies from patients with rheumatic disorders. J Biol Chem 1986; 261:2274-8; PMID:3511059
- Feng JH, Park J, Cron P, Hess D, Hemmings BA. Identification of a PKB/Akt hydrophobic motif Ser-473 kinase as DNA-dependent protein kinase. J Biol Chem 2004; 279:41189-96; PMID:15262962; http://dx.doi.org/10.1074/jbc.M406731200
- Basu S, Rosenzweig KR, Youmell M, Price BD. The DNA-dependent protein kinase participates in the activation of NF kappa B following DNA damage. Biochem Biophys Res Commun 1998; 247:79-83; PMID:9636658; http://dx.doi.org/10.1006/bbrc.1998.8741
- Wong RH, Chang I, Hudak CS, Hyun S, Kwan HY, Sul HS. A role of DNA-PK for the metabolic gene regulation in response to insulin. Cell 2009; 136:1056-72; PMID:19303849; http://dx.doi.org/10.1016/j.cell.2008.12.040
- Dittmann K, Mayer C, Fehrenbacher B, Schaller M, Raju U, Milas L, Chen DJ, Kehlbach R, Rodemann HP. Radiation-induced epidermal growth factor receptor nuclear import is linked to activation of DNA-dependent protein kinase. J Biol Chem 2005; 280:31182-9; PMID:16000298; http://dx.doi.org/10.1074/jbc.M506591200
- Szumiel I. Epidermal growth factor receptor and DNA double strand break repair: the cell's self-defence. Cell Signal 2006; 18:1537-48; PMID:16713182; http://dx.doi.org/10.1016/j.cellsig.2006.03.010
- Ting NS, Pohorelic B, Yu Y, Lees-Miller SP, Beattie TL. The human telomerase RNA component, hTR, activates the DNA-dependent protein kinase to phosphorylate heterogeneous nuclear ribonucleoprotein A1. Nucleic Acids Res 2009; 37:6105-15; PMID:19656952; http://dx.doi.org/10.1093/nar/gkp636
- Lee KJ, Lin YF, Chou HY, Yajima H, Fattah KR, Lee SC, Chen BP. Involvement of DNA-dependent protein kinase in normal cell cycle progression through mitosis. J Biol Chem 2011; 286:12796-802; PMID:21330363; http://dx.doi.org/10.1074/jbc.M110.212969
- Kang GY, Pyun BJ, Seo HR, Jin YB, Lee HJ, Lee YJ, Lee YS. Inhibition of Snail1-DNA-PKcs Protein-Protein Interface Sensitizes Cancer Cells and Inhibits Tumor Metastasis. J Biol Chem 2013; 288:32506-16; PMID:24085291; http://dx.doi.org/10.1074/jbc.M113.479840
- Um JH, Kang CD, Bae JH, Shin GG, Kim DW, Chung BS, Kim SH. Association of DNA-dependent protein kinase with hypoxia inducible factor-1 and its implication in resistance to anticancer drugs in hypoxic tumor cells. Exp Mol Med 2004; 36:233-42; PMID:15272235; http://dx.doi.org/10.1038/emm.2004.32
- Muller C, Paupert J, Monferran S, Salles B. The double life of the Ku protein: facing the DNA breaks and the extracellular environment. Cell Cycle 2005; 4:438-41; PMID:15738653; http://dx.doi.org/10.4161/cc.4.3.1565
- Coussens LM, Werb Z. Matrix metalloproteinases and the development of cancer. Chem Biol 1996; 3:895-904; PMID:8939708; http://dx.doi.org/10.1016/S1074-5521(96)90178-7
- Nagase H, Visse R, Murphy G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc Res 2006; 69:562-73; PMID:16405877; http://dx.doi.org/10.1016/j.cardiores.2005.12.002
- Allalunis-Turner MJ, Barron GM, Day RS, 3rd, Dobler KD, Mirzayans R. Isolation of two cell lines from a human malignant glioma specimen differing in sensitivity to radiation and chemotherapeutic drugs. Radiat Res 1993; 134:349-54; PMID:8316628; http://dx.doi.org/10.2307/3578196
- Ruis BL, Fattah KR, Hendrickson EA. The catalytic subunit of DNA-dependent protein kinase regulates proliferation, telomere length, and genomic stability in human somatic cells. Mol Cellular Biol 2008; 28:6182-95; PMID:18710952; http://dx.doi.org/10.1128/MCB.00355-08
- Nonaka D, Chiriboga L, Rubin BP. Differential expression of S100 protein subtypes in malignant melanoma, and benign and malignant peripheral nerve sheath tumors. J Cutan Pathol 2008; 35:1014-9; PMID:18547346; http://dx.doi.org/10.1111/j.1600-0560.2007.00953.x
- Biau J, Devun F, Jdey W, Kotula E, Quanz M, Chautard E, Sayarath M, Sun JS, Verrelle P, Dutreix M. A preclinical study combining the DNA repair inhibitor Dbait with radiotherapy for the treatment of melanoma. Neoplasia 2014; 16:835-44; PMID:25379020; http://dx.doi.org/10.1016/j.neo.2014.08.008
- Kurimasa A, Kumano S, Boubnov NV, Story MD, Tung CS, Peterson SR, Chen DJ. Requirement for the kinase activity of human DNA-dependent protein kinase catalytic subunit in DNA strand break rejoining. Mol Cell Biol 1999; 19:3877-84; PMID:10207111
- Hakulinen J, Sankkila L, Sugiyama N, Lehti K, Keski-Oja J. Secretion of active membrane type 1 matrix metalloproteinase (MMP-14) into extracellular space in microvesicular exosomes. J Cell Biochem 2008; 105:1211-8; PMID:18802920; http://dx.doi.org/10.1002/jcb.21923
- Yuan SS, Hou MF, Hsieh YC, Huang CY, Lee YC, Chen YJ, Lo S. Role of MRE11 in cell proliferation, tumor invasion, and DNA repair in breast cancer. J Natl Cancer Inst 2012; 104:1485-502; PMID:22914783; http://dx.doi.org/10.1093/jnci/djs355
- Coene ED, Gadelha C, White N, Malhas A, Thomas B, Shaw M, Vaux DJ. A novel role for BRCA1 in regulating breast cancer cell spreading and motility. J Cell Biol 2011; 192:497-512; PMID:21282464; http://dx.doi.org/10.1083/jcb.201004136
- Sarasin A, Kauffmann A. Overexpression of DNA repair genes is associated with metastasis: a new hypothesis. Mutat Res 2008; 659:49-55; PMID:18308619; http://dx.doi.org/10.1016/j.mrrev.2007.12.002
- Martinez-Marignac VL, Rodrigue A, Davidson D, Couillard M, Al-Moustafa AE, Abramovitz M, Foulkes WD, Masson JY, Aloyz R. The effect of a DNA repair gene on cellular invasiveness: XRCC3 over-expression in breast cancer cells. PloS One 2011; 6:e16394; PMID:21283680; http://dx.doi.org/10.1371/journal.pone.0016394
- Rodriguez MI, Gonzalez-Flores A, Dantzer F, Collard J, de Herreros AG, Oliver FJ. Poly(ADP-ribose)-dependent regulation of Snail1 protein stability. Oncogene 2011; 30:4365-72; PMID:21577210; http://dx.doi.org/10.1038/onc.2011.153
- Monferran S, Paupert J, Dauvillier S, Salles B, Muller C. The membrane form of the DNA repair protein Ku interacts at the cell surface with metalloproteinase 9. Embo J 2004; 23:3758-68; PMID:15385961; http://dx.doi.org/10.1038/sj.emboj.7600403
- Paupert J, Dauvillier S, Salles B, Muller C. Transport of the leaderless protein Ku on the cell surface of activated monocytes regulates their migratory abilities. EMBO Rep 2007; 8:583-8; PMID:17496833; http://dx.doi.org/10.1038/sj.embor.7400976
- Huang C, Cao J, Huang KJ, Zhang F, Jiang T, Zhu L, Qiu ZJ. Inhibition of STAT3 activity with AG490 decreases the invasion of human pancreatic cancer cells in vitro. CancerSci 2006; 97:1417-23; PMID:17054436; http://dx.doi.org/10.1111/j.1349-7006.2006.00340.x
- Xie TX, Wei D, Liu M, Gao AC, Ali-Osman F, Sawaya R, Huang S. Stat3 activation regulates the expression of matrix metalloproteinase-2 and tumor invasion and metastasis. Oncogene 2004; 23:3550-60; PMID:15116091; http://dx.doi.org/10.1038/sj.onc.1207383
- Rundhaug JE. Matrix metalloproteinases, angiogenesis, and cancer: commentary re: A. C. Lockhart et al., Reduction of wound angiogenesis in patients treated with BMS-275291, a broad spectrum matrix metalloproteinase inhibitor. Clin Cancer Res, 9: 00-00, 2003. Clin Cancer Res 2003; 9:551-4.
- Seo DW, Li H, Guedez L, Wingfield PT, Diaz T, Salloum R, Wei BY, Stetler-Stevenson WG. TIMP-2 mediated inhibition of angiogenesis: an MMP-independent mechanism. Cell 2003; 114:171-80; PMID:12887919; http://dx.doi.org/10.1016/S0092-8674(03)00551-8
- Xie L, Palmsten K, MacDonald B, Kieran MW, Potenta S, Vong S, Kalluri R. Basement membrane derived fibulin-1 and fibulin-5 function as angiogenesis inhibitors and suppress tumor growth. Exp Biol Med (Maywood) 2008; 233:155-62; PMID:18222970; http://dx.doi.org/10.3181/0706-RM-167
- Foda HD, Zucker S. Matrix metalloproteinases in cancer invasion, metastasis and angiogenesis. Drug Discov Today 2001; 6:478-82; PMID:11344033; http://dx.doi.org/10.1016/S1359-6446(01)01752-4
- Strickland DK, Ashcom JD, Williams S, Burgess WH, Migliorini M, Argraves WS. Sequence identity between the α 2-macroglobulin receptor and low density lipoprotein receptor-related protein suggests that this molecule is a multifunctional receptor. J Biol Chem 1990; 265:17401-4; PMID:1698775
- Bengtsson E, Morgelin M, Sasaki T, Timpl R, Heinegard D, Aspberg A. The leucine-rich repeat protein PRELP binds perlecan and collagens and may function as a basement membrane anchor. J Biol Chem 2002; 277:15061-8; PMID:11847210; http://dx.doi.org/10.1074/jbc.M108285200
- Hopf M, Gohring W, Mann K, Timpl R. Mapping of binding sites for nidogens, fibulin-2, fibronectin and heparin to different IG modules of perlecan. J Mol Biol 2001; 311:529-41; PMID:11493006; http://dx.doi.org/10.1006/jmbi.2001.4878
- Bix G, Fu J, Gonzalez EM, Macro L, Barker A, Campbell S, Zutter MM, Santoro SA, Kim JK, Hook M, et al. Endorepellin causes endothelial cell disassembly of actin cytoskeleton and focal adhesions through alpha2beta1 integrin. The Journal of cell biology 2004; 166:97-109; PMID:15240572; http://dx.doi.org/10.1083/jcb.200401150
- Miner JH, Yurchenco PD. Laminin functions in tissue morphogenesis. Annu Rev Cell Dev Biol 2004; 20:255-84; PMID:15473841; http://dx.doi.org/10.1146/annurev.cellbio.20.010403.094555
- Sasaki T, Kostka G, Gohring W, Wiedemann H, Mann K, Chu ML, Timpl R. Structural characterization of two variants of fibulin-1 that differ in nidogen affinity. J Mol Biol 1995; 245:241-50; PMID:7844816; http://dx.doi.org/10.1006/jmbi.1994.0020
- Pankotai T, Bonhomme C, Chen D, Soutoglou E. DNAPKcs-dependent arrest of RNA polymerase II transcription in the presence of DNA breaks. Nat Struct Mol Biol 2012; 19:276-82; PMID:22343725; http://dx.doi.org/10.1038/nsmb.2224
- Bryntesson F, Regan JC, Jeggo PA, Taccioli GE, Hubank M. Analysis of gene transcription in cells lacking DNA-PK activity. Radiat Res 2001; 156:167-76; PMID:11448237; http://dx.doi.org/10.1667/0033-7587(2001)156%5b0167:AOGTIC%5d2.0.CO;2
- Lees-Miller SP. The DNA-dependent protein kinase, DNA-PK: 10 years and no ends in sight. Biochem Cell Biol 1996; 74:503-12; PMID:8960356; http://dx.doi.org/10.1139/o96-054
- Ju BG, Lunyak VV, Perissi V, Garcia-Bassets I, Rose DW, Glass CK, Rosenfeld MG. A topoisomerase IIbeta-mediated dsDNA break required for regulated transcription. Science 2006; 312:1798-802; PMID:16794079; http://dx.doi.org/10.1126/science.1127196
- Ju BG, Rosenfeld MG. A breaking strategy for topoisomerase IIbeta/PARP-1-dependent regulated transcription. Cell Cycle 2006; 5:2557-60; PMID:17106262; http://dx.doi.org/10.4161/cc.5.22.3497
- Farber-Katz SE, Dippold HC, Buschman MD, Peterman MC, Xing M, Noakes CJ, Tat J, Ng MM, Rahajeng J, Cowan DM, et al. DNA Damage Triggers Golgi Dispersal via DNA-PK and GOLPH3. Cell 2014; 156:413-27; PMID:24485452; http://dx.doi.org/10.1016/j.cell.2013.12.023
- Yang C, Robbins PD. The roles of tumor-derived exosomes in cancer pathogenesis. Clin Dev Immunol 2011; 2011:842849; PMID:22190973
- Zhang X, Li X, Zhang N, Yang Q, Moran MS. Low doses ionizing radiation enhances the invasiveness of breast cancer cells by inducing epithelial-mesenchymal transition. Biochem Biophys Res Commun 2011; 412:188-92; PMID:21810413; http://dx.doi.org/10.1016/j.bbrc.2011.07.074
- Zhou YC, Liu JY, Li J, Zhang J, Xu YQ, Zhang HW, Qiu LB, Ding GR, Su XM, Mei S, et al. Ionizing radiation promotes migration and invasion of cancer cells through transforming growth factor-β-mediated epithelial-mesenchymal transition. Int J Radiat Oncol Biol Phys 2011; 81:1530-7; PMID:22115555; http://dx.doi.org/10.1016/j.ijrobp.2011.06.1956
- Liu Y, Zhang L, Sun C, Zhang H, Miao G, Xiao Di C, Zhou X, Zhou R, Wang Z. DNA-PKcs deficiency inhibits glioblastoma cell-derived angiogenesis after ionizing radiation. J Cell Physiol 2015; 230:1094-103.
- Kan-Mitchell J, Mitchell MS, Rao N, Liggett PE. Characterization of uveal melanoma cell lines that grow as xenografts in rabbit eyes. Invest Ophthalmol Visual Sci 1989; 30:829-34; PMID:2722439
- Quanz M, Berthault N, Roulin C, Roy M, Herbette A, Agrario C, Alberti C, Josserand V, Coll JL, Sastre-Garau X, et al. Small-molecule drugs mimicking DNA damage: a new strategy for sensitizing tumors to radiotherapy. Clin Cancer Res 2009; 15:1308-16; PMID:19190126; http://dx.doi.org/10.1158/1078-0432.CCR-08-2108
- Kotula E, Faigle W, Berthault N, Dingli F, Loew D, Sun JS, Dutreix M, Quanz M. DNA-PK target identification reveals novel links between DNA repair signaling and cytoskeletal regulation. PloS One 2013; 8:e80313; PMID:24282534; http://dx.doi.org/10.1371/journal.pone.0080313
- Alessandrino EP, Della Porta MG, Pascutto C, Bacigalupo A, Rambaldi A. Should cytoreductive treatment be performed before transplantation in patients with high-risk myelodysplastic syndrome? J Clin Oncol 2013; 31:2761-2; PMID:23775971; http://dx.doi.org/10.1200/JCO.2012.48.0525
- Quanz M, Chassoux D, Berthault N, Agrario C, Sun JS, Dutreix M. Hyperactivation of DNA-PK by double-strand break mimicking molecules disorganizes DNA damage response. PLoS One 2009; 4:e6298; PMID:19621083; http://dx.doi.org/10.1371/journal.pone.0006298
- Solass W, Herbette A, Schwarz T, Hetzel A, Sun JS, Dutreix M, Reymond MA. Therapeutic approach of human peritoneal carcinomatosis with Dbait in combination with capnoperitoneum: proof of concept. Surg Endosc 2012; 26:847-52; PMID:22042585; http://dx.doi.org/10.1007/s00464-011-1964-y