
Defective regulation of the NFκΒ signaling pathway has been implicated in the pathogenesis of a variety of human diseases including automimmune and neurodegenerative disorders, and cancer. A pivotal regulator of NFκΒ signaling is the inhibitor of κΒ kinase (IκΒ) complex (IKK), which drives NFκΒ signaling in response to inflammation and cellular stress by phosphorylating and targeting IκΒ for degradation, subsequently releasing and activating NFκΒ . The IKK complex itself comprises 3 components, namely the protein kinases IKKα and IKKβ, and the NFκB essential modulator (NEMO, also known as IKKγ). Given the central role of IKK in the NFκΒ signaling pathway, it is no surprise that mutations to the IKK subunits have been associated with several human diseases. Recently, somatic mutations altering lysine 171 (K171) of the IKBKB gene that encodes IKKβ have been described in splenic marginal zone lymphomas, mantle cell lymphoma, and multiple myeloma. This mutation has been shown to keep IKKβ in an active state resulting in constitutive activation of NFκB signaling;Citation1 yet, how the K171 mutation alters IKKβ activity had not been defined.
In a recent study published in Cell Cycle,Citation2 Donoghue and colleagues investigated the mechanism of K171-mediated IKKβ activation by constructing and expressing various IKKβ K171 mutants observed in human cancers. They first examined downstream effects of this mutation and were able to demonstrate that K171 mutation not only leads to prolonged IKKβ activation, but also constitutive activation of STAT3, another transcription factor linked to cell transformation and tumorigenesis. STAT3 has been shown to maintain constitutive activation of NFκB by retaining NFκB in the nucleus.Citation3 The author's results suggest that this mechanism might occur with K171 mutation.
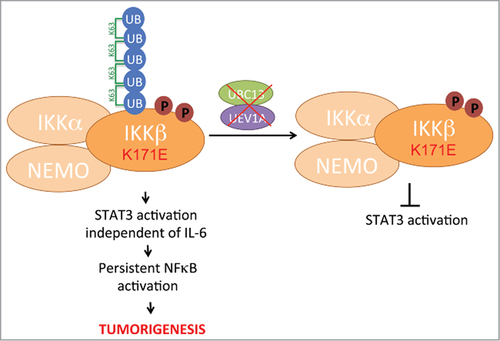
IKK/NFκB signaling has been linked to the ubiquitin pathway, specifically K63-linked polyubiquitylation-mediated IKK activation. K63-linked polyubiquitin chains act as scaffolds to assemble protein kinase complexes and several proteins in the NFκB signaling pathway have been shown to associate with these chains, including RIP1, TAK1, and NEMO.Citation4 As K63-linked ubiquitylation is important for IKK activation, Donoghue and colleagues sought out to determine if IKKβ itself is modified by K63-linked chains. Indeed, the authors found that IKKβ undergoes K63-linked ubiquitylation that is enhanced and stabilized with K171 mutation. This is a significant finding as this is the first report of IKKβ modification by K63-linked ubiquitin chains. Utilizing mass spectrometryapproaches, the authors identified 4 predominant sites of ubiquitylation in IKKβ, only one of which, K147, to be the principal site of K63-linked ubiquitylation. Finally, in a very elegant experiment, the authors used a chemical inhibitor of the ubiquitin-conjugating enzyme (E2) complex Ubc13-Uev1A, which selectively inhibits K63-linked ubiquitylation, and found that this inhibitor blocked constitutive STAT3 activation mediated by the IKKβ K171 mutant. Together, the authors show that deregulated STAT3 activation resulting from IKKβ mutation requires K63-ubiquitin chain assembly on IKKβ (Fig. 1). It is interesting to note that the K147 site of K63-ubiquitylation lies within a highly conserved sequence (DLKPEN) in IKKβ that is also found in other related kinases, suggesting that this modification and mechanism of activation may extend to these kinases. In fact, corresponding K63 ubiquitin sites on TAK1 and BRAF have been shown to be required for activation of their respective downstream signaling pathways.Citation5,6
IKKβ is an attractive target for intervention with small molecule kinase inhibitors; specific IKKβ inhibitors have been found by a number of pharmaceutical companies. Very few of these inhibitors have reached clinical trials, all of them ATP-competitive IKKβ selective inhibitors that exhibit off-target/side effects such as cellular toxicity and immunosuppression.Citation7 As such, there is a need for novel strategies to target and inhibit aberrant IKKβ activity. By inhibiting the catalytic activity of the Ubc13/Uev1A E2 enzyme, this paper demonstrates that pharmacological blockage of IKKβ K63-linked ubiquitylation might be a valid approach to inhibiting STAT3 activation in vivo. Determining the E3 ubiquitin ligase that specifically synthesizes the IKKβ K63-linked ubiquitin chain might offer a more specific component of the pathway for pharmacological intervention. Furthermore, identification of interacting proteins that associate with the K63-ubiquitin scaffold assembled on IKKβ will not only help uncover molecular mechanisms underlying K171 mutation-mediated STAT3 activation, but also reveal novel therapeutic opportunities to inhibit IKK signaling.
Figure 1. K63-linked ubiquitylation of IKKβ activates STAT3 signaling. IKKβ mutation at K171 associated with multiple myeloma and other cancers leads to increased IKKβ activation, indicated by phosphorylation on 2 serine residues (S177 and S181) and K63-linked polyubiquitylation on K147. IKKβ mutation also constitutively activates STAT3 signaling which likely causes tumorigenisis by maintaining elevated NFκΒ activity. Inhibition of the E2 ubiquitin-conjugating complex UBC13/UEV1A responsible for IKKβ K63-linked ubiquitylation blocks this constitutive STAT3 signaling while not affecting IKKβ kinase activity.
References
- Kai X, et al. J Biol Chem 2014; 289(39):26960–72; PMID:25107905; http://dx.doi.org/10.1074/jbc.M114.598763
- Gallo LH, et al. Cell Cycle 2014; 13(24):3964–76; PMID:25486864; http://dx.doi.org/10.4161/15384101.2014.988026
- Lee H, et al. Cancer Cell 2009; 15(4):283–93; PMID:19345327; http://dx.doi.org/10.1016/j.ccr.2009.02.015
- Chen ZJ. Immunol Rev 2012; 246(1):95–106; PMID:22435549; http://dx.doi.org/10.1111/j.1600-065X.2012.01108.x
- An L, et al. Sci Rep 2013; 3:2344; PMID:23907581; http://dx.doi.org/10.1038/srep02344
- Fan Y, et al. J Biol Chem 2010; 285(8):5347–60; PMID:20038579; http://dx.doi.org/10.1074/jbc.M109.076976
- Gamble C, et al. Br J Pharmacol 2012; 165(4):802–19; PMID:21797846; http://dx.doi.org/10.1111/j.1476-5381.2011.01608.x