
Abstract
p31comet plays an important role in spindle assembly checkpoint (SAC) silencing. However, how p31comet's activity is regulated remains unclear. Here we show that the timing of M-phase exit in Xenopus egg extracts (XEEs) depends upon SAC activity, even under conditions that are permissive for spindle assembly. p31comet antagonizes the SAC, promoting XEE progression into anaphase after spindles are fully formed. We further show that mitotic p31comet phosphorylation by Inhibitor of nuclear factor κ-B kinase-β (IKK-β) enhances this role in SAC silencing. Together, our findings implicate IKK-β in the control of anaphase timing in XEE through p31comet activation and SAC downregulation.
Abbreviations
| SAC | = | spindle assembly checkpoint |
| XEEs | = | Xenopus egg extracts |
| IKK- | = | β Inhibitor of nuclear factor κ-B kinase-β |
| KT | = | kinetochore |
| MT | = | microtubule |
| NEBD | = | nuclear envelope breakdown |
| APC/C | = | anaphase promoting complex/cyclosome |
| CSF | = | cytostatic factor |
| DSN | = | demembranated sperm nuclei |
| TPCA | = | [5-(p-fluorophenyl)-2-ureido] thiophene-3-carboxamide |
| MCC | = | itotic checkpoint complex. |
Introduction
The spindle assembly checkpoint (SAC) ensures faithful separation of mitotic sister chromatids by monitoring interactions between kinetochores (KTs) and microtubules (MTs) (reviewed inCitation1–3). The SAC controls the anaphase promoting complex/cyclosome (APC/C), a ubiquitin ligase that is crucial for mitotic exit. SAC activation causes sequestration of an APC/C activator, Cdc20, within an inhibitory complex called the mitotic checkpoint complex (MCC). The MCC is composed of Cdc20 and SAC proteins Mad2, BubR1, Bub3. The Mad2 protein exists in 2 states, the inactive open state (o-Mad2) and the active closed state (c-Mad2); c-Mad2 is incorporated into the MCC complex. Conversion of o-Mad2 to c-Mad2 occurs at a “catalytic platform” complex on unattached KTs, comprised of c-Mad2 and Mad1.
Mad1 and Mad2 are not essential in budding yeast unless KT MTs are disrupted,Citation4 suggesting that the SAC is not required for spindle structure or chromosome segregation per se. However, Mad1 and Mad2 are essential for correct chromosome segregation and organismal viability in metazoans,Citation5,6 indicating that the SAC is required in higher eukaryotes even without external perturbation of spindle assembly. This difference reflects the fact that yeast KTs are constitutively MT bound throughout the cell cycle, whereas organisms that undergo open mitosis re-establish KT-MT attachments after nuclear envelope breakdown (NEBD) in each cell division. Unattached KTs transiently invoke the SAC during the interval between NEBD and MT attachment, and subsequent SAC silencing helps to determine the timing of anaphase onset.Citation6
SAC silencing is not fully understood, although it clearly involves a number of crucial events, including both inactivation of the catalytic platform at KTs and disassembly of existing MCC.Citation1 p31comet is a protein found in higher eukaryotes that plays an important role in SAC silencing.Citation7 p31comet over-expression in tissue culture cells overrides the SAC, while p31comet knockdown delays anaphase onset.Citation8,9 p31comet is structurally related to Mad2Citation10 and it binds c-Mad2 in a manner that is essential for its role in SAC silencing.Citation8,11 Two mechanisms have been suggested for p31comet activity: First, p31comet has been proposed to disrupt soluble MCC. This model is supported by the capacity of p31comet to bind c-Mad2 within the MCC in vitro and thereby disrupt that complex.Citation12 Second, p31comet associates with unattached KTs through direct binding to c-Mad2.Citation8,11 p31comet binding to c-Mad2 occludes o-Mad2 binding,Citation10 and a “capping” model has been proposed wherein p31comet blocks MCC generation by inhibition of the Mad1/c-Mad2 catalytic platform.Citation13
Xenopus egg extracts (XEEs) are a convenient system for ex vivo studies on mitosis. CSF-XEEs preserve the meiotic arrest of the frog egg by cytostatic factor (CSF).Citation14 CSF-XEEs form spindles after addition of demembranated sperm nuclei (DSN). Upon the addition of CaCl2, which mimics fertilization of intact eggs, CSF is lost, spindles are disassembled and CSF-XEEs proceeds into interphase. XEE can recapitulate cell cycle checkpoints, including the SAC. For example, the SAC becomes activated by unattached KTs in CSF-XEEs containing DSN and the MT-depolymerizing agent nocodazole, preventing anaphase onset even after CaCl2 addition and CSF degradation. Cycling XEE are produced through an alternative protocol, so that they mimic the cell cycle of the fertilized egg, spontaneously alternating between interphase and mitosis.Citation14 Like CSF-XEE, cycling XEE activate the SAC in response to unattached KTs. Purified p31comet antagonizes Mad2 inhibition of APC/Ccdc20 in XEE.Citation15
We have used XEEs for investigation of the mitotic role(s) and regulation of p31comet. Here we show that p31comet depletion from XEE caused a SAC-dependent delay in anaphase onset, suggesting that endogenous p31comet is important for mitotic timing in this system. p31comet was mitotically phosphorylated in XEE. While a number of well-established mitotic kinases did not efficiently modify p31comet in vitro, IKK-β (Inhibitor of nuclear factor κ-B kinase-β) was effective for p31comet phosphorylation. Depletion or inhibition of IKK-β delayed mitotic exit of XEEs, and a phosphomimetic p31comet mutant showed increased activity in dissociation of soluble MCC and particularly in disruption of KT association of SAC components. Together, our results suggest that p31comet contributes to the timing of anaphase onset in XEE through antagonism of the SAC and that IKK-β modifies p31comet to enhance its activity.
Results
Xenopus p31comet promotes SAC silencing in XEE
To test whether p31comet modulates mitotic exit timing in XEE under circumstances permissive for spindle assembly, we incubated control and p31comet-depleted CSF-XEE reactions containing DSN for 15 min. at 23°C, followed by CaCl2 addition to initiate mitotic exit. We examined the progression of each reaction into interphase through Western blotting for Cyclin B. Cyclin B degradation was significantly slower in p31comet-depleted samples () in a manner that could be rescued by addition of recombinant p31comet. These observations indicate that p31comet facilitates mitotic exit in CSF-XEE containing DSN.
Figure 1. p31comet depletion causes an SAC-dependent mitotic exit delay. (A) p31comet-depleted XEE reactions containing DSN, with or without 15 nM His6-p31comet were incubated at 23°C for 15 min., followed by CaCl2 addition. At intervals after CaCl2 addition, samples were collected for Western blotting with the indicated antibodies. Mock lanes show samples from a mock-depleted reaction, without added His6-p31comet. (B) XEEs depleted of p31comet, Mad2 or both proteins were subjected to analysis as in (A). (C) Cyclin B levels were quantified for reactions as described in Panel A and B, and normalized relative to the initial level at time = 0. Values represent the mean ± SD derived from 3 independent assays.
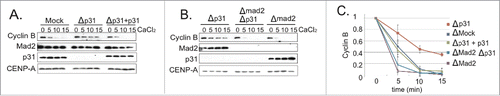
We postulated that transiently unattached KTs temporarily activate the SAC in XEEs, and that p31comet promotes subsequent SAC silencing after MT-KT attachments were formed. To test this idea, we examined whether the delay in p31comet-depleted XEEs requires Mad2 (). Mad2 depletion alone accelerated the rate of Cyclin B degradation, consistent with the notion that SAC activation modulates mitotic exit timing. The presence or absence of p31comet did not significantly alter Cyclin B degradation in Mad2-depleted XEEs, indicating that p31comet modulates mitotic exit through Mad2. Spindle morphology and MT density were indistinguishable in the presence and absence of p31comet (Fig. S1A), arguing that p31comet depletion did not delay mitotic exit by disrupting spindle assembly, and p31comet-depleted XEEs could activate their SAC in response to fully unattached KTs. (Fig. S1B).
Together, these observations suggest that the SAC slows mitotic exit in CSF-XEE containing DSN, and that p31comet promotes anaphase by antagonizing the SAC. This observation is consistent with reports that p31comet-depleted HeLa cells show mitotic exit delays because they are unable to mediate SAC silencing.Citation8,11,16
p31comet phosphorylation by IKK-β during mitotic exit
It has been reported that mammalian p31comet is phosphorylated during mitosis,Citation17,18 and that phosphorylation of human p31comet on Serine-102 weakens its interaction with Mad2 and its capacity to silence the SAC.Citation19 We wished to determine whether p31comet might be phosphorylated in XEE, and, if so, what role this modification might play in its activity. To address this question, we took samples from a cycling XEE reaction at different times, and examined the activity of kinases that can phosphorylate p31comet by adding GST-p31comet and [γ−32P]ATP to each sample. After 30 min., we re-isolated GST-p31comet and subjected it to SDS-PAGE and autoradiography (). We observed that kinase activity directed against GST-p31comet increased progressively, peaking near the onset of Cyclin B degradation and mitotic exit in the intact cycling XEE (≈60 min.) and declining abruptly as the cycling XEE returned to interphase.
Figure 2. IKK-β phosphorylates Xenopus p31comet. (A) Samples from cycling XEEs with 1,000 DSN/µl were collected at intervals after shift to 23°C. GST-p31comet beads and [γ−32P]ATP were added to an aliquot of the sample for analysis of kinase activity as described in Methods (top panel, bottom histogram). In parallel, another aliquot was directly analyzed by Western blotting for Cyclin B (second panel). The arrow indicates32P-labeled GST-p31comet. The stage of the cycling XEE is shown above (I: interphase, M: mitosis). (B) GST-p31comet or GST were incubated with ATP-γ-S and Aurora B, Mps1 or IKK-β. Samples from each reaction were subject to Western blotting using an anti-thiophosphate ester specific antibody (upper panel) and to CBB staining (lower panel). Black arrows within the upper panel indicate bands resultant from kinase autophosphorylation. Arrow to the right of the upper panel indicates phosphorylated GST-p31comet. (C) CSF-XEE containing [γ−32P]ATP and either DMSO or TCPA (final concentration 300 µM) were incubated with GST-p31comet or Histone H1. Each sample was subjected to SDS-PAGE and autoradiography (upper panels. Arrows indicate phosphorylated p31comet or Histone H1) and to Western blotting to assure equal loading (lower panels). (D) Samples as in panel A were subjected to Western blotting using phospho-specific antibody (anti-p31comet-S4p,T6p). Positive control (+ve) lane contains in vitro phosphorylated GST-p31comet as in Figure S3A, except ATP was used instead of [γ−32P]ATP. Negative control (-ve) lane includes untreated purified GST-p31comet.
![Figure 2. IKK-β phosphorylates Xenopus p31comet. (A) Samples from cycling XEEs with 1,000 DSN/µl were collected at intervals after shift to 23°C. GST-p31comet beads and [γ−32P]ATP were added to an aliquot of the sample for analysis of kinase activity as described in Methods (top panel, bottom histogram). In parallel, another aliquot was directly analyzed by Western blotting for Cyclin B (second panel). The arrow indicates32P-labeled GST-p31comet. The stage of the cycling XEE is shown above (I: interphase, M: mitosis). (B) GST-p31comet or GST were incubated with ATP-γ-S and Aurora B, Mps1 or IKK-β. Samples from each reaction were subject to Western blotting using an anti-thiophosphate ester specific antibody (upper panel) and to CBB staining (lower panel). Black arrows within the upper panel indicate bands resultant from kinase autophosphorylation. Arrow to the right of the upper panel indicates phosphorylated GST-p31comet. (C) CSF-XEE containing [γ−32P]ATP and either DMSO or TCPA (final concentration 300 µM) were incubated with GST-p31comet or Histone H1. Each sample was subjected to SDS-PAGE and autoradiography (upper panels. Arrows indicate phosphorylated p31comet or Histone H1) and to Western blotting to assure equal loading (lower panels). (D) Samples as in panel A were subjected to Western blotting using phospho-specific antibody (anti-p31comet-S4p,T6p). Positive control (+ve) lane contains in vitro phosphorylated GST-p31comet as in Figure S3A, except ATP was used instead of [γ−32P]ATP. Negative control (-ve) lane includes untreated purified GST-p31comet.](/cms/asset/3ab71b59-9fa0-4f98-a494-8063c09dca27/kccy_a_1033590_f0002_c.gif)
To determine which kinase mediates p31comet phosphorylation, we examined the capacity of purified Mps1, Aurora B, Cyclin B/Cdk1 and IKK-β to phosphorylate GST-p31comet (, Fig. S2). Mps1, Aurora B and Cyclin B/Cdk1 are well-studied mitotic kinases. IKK-β has been implicated in spindle assemblyCitation20 and SAC regulation.Citation21 The only kinase that efficiently phosphorylated GST-p31comet in vitro was IKK-β. To test whether IKK-β has a major role in p31comet phosphorylation, we performed kinase assays using CSF-XEE treated with IKK-β inhibitor TPCA ([5-(p-fluorophenyl)-2-ureido] thiophene-3-carboxamide).Citation22 TPCA both inhibited phosphorylation of GST-p31comet in CSF-XEE () and abolished IKK-β kinase activity toward GST-p31comet reactions containing purified proteins (Fig. S3A). TPCA did not inhibit phosphorylation of the Cyclin B/Cdk1 substrate Histone H1 within CSF-XEE (), arguing that it specifically blocked mitotic p31comet phosphorylation through IKK-β inhibition.
We performed mass spectrometry to identify the sites on p31comet phosphorylated by IKK-β in vitro, and identified Serine-4 (S4), Threonine-6 (T6) and Threonine-179 (T179) of p31comet as candidate IKK-β sites (Fig. S4A). Mutation of either S4 and T6 to alanine or S179 to alanine reduced in vitro p31comet phosphorylation, while mutation of all 3 residues to alanines nearly abolished p31comet modification (Fig. S4B). We raised rabbit antibodies that specifically recognized p31comet phosphorylated on S4 and T6 (p31comet-S4p,T6p): Anti-p31comet-S4p,T6p antibodies did not recognize bacterially expressed GST-p31comet on Western blots, but showed a prominent band of the appropriate molecular weight after GST-p31comet was treated with IKK-β (Fig. S3B). The appearance of this band was suppressed by the addition of TPCA to the reaction. In a similar fashion, the anti-p31comet-S4p,T6p antibodies recognized GST-p31comet that had been incubated in XEE alone or in XEE plus DMSO, but not GST-p31comet incubated in XEE reactions containing TPCA (Fig. S3C). We also raised rabbit antibodies against Xenopus IKK-β (anti-xIKK-β), which were used to immunodeplete IKK-β from XEE. We examined the capacity of these depleted XEE to phosphorylate GST-p31comet by Western blotting with anti-p31comet-S4p,T6p antibodies (Fig. S3D). We observed that phosphorylation of p31comet was abolished after IKK-β depletion, but could be restored through the addition of purified human IKK-β at a similar concentration. These experiments collectively suggest that IKK-β phosphorylates p31comet on S4 and T6, and that its kinase activity is essential for the modification of these residues in CSF-XEE.
In a manner similar to the experiment in , we analyzed kinase activity against S4 and T6 of p31comet in cycling XEE by Western blotting with anti-p31comet-S4p,T6p antibodies (). We observed that kinase activity against these residues peaked in mitosis, near the onset of Cyclin B degradation. TPCA abolished this peak of kinase activity toward p31comet-S4p,T6p (Fig. S3E). Interestingly, the peak of anti-p31comet-S4p,T6p kinase activity was somewhat sharper than overall kinase activity against p31comet (compare ), potentially suggesting that p31comet may be phosphorylated by other kinases during mitosis in addition to IKK-β. Our observations collectively indicate that p31comet becomes phosphorylated near the onset of anaphase in XEE. Our data implicate IKK-β as a major kinase for at least a subset of these modifications, particularly S4 and T6.
Phosphomimetic p31comet antagonizes the SAC
To examine whether p31comet phosphorylation changes its activity, we depleted endogenous p31comet from CSF-XEE, followed by addition of physiological levels (15 nM) of recombinant wild type p31comet (p31comet-wt), a phosphomimetic form of p31comet, in which S4, T6 and T179 were mutated to glutamic acid (p31comet-EEE), or a non-phosphoryated form, in which those residues were mutated to alanine (p31comet-AAA). We added DSN to each reaction and incubated them at 23°C. After 15 min., we added CaCl2 and took periodic samples for Western blotting analysis of Cyclin B levels (). p31comet depletion delayed mitotic exit in a manner that was reversed by GST-p31comet-wt, but not by GST. GST-p31comet-AAA was less effective than GST-p31comet-wt at relieving this mitotic delay, while GST-p31comet-EEE was more effective in accelerating Cyclin B degradation. Collectively, these findings are consistent with the idea that p31comet phosphorylation enhances its capacity to promote mitotic exit.
Figure 3. Phosphomimetic p31comet accelerates M-phase exit. (A) 15 nM GST-tagged p31comet-wt, p31comet-AAA or p31comet-EEE were added to p31comet-depleted CSF-XEE containing DSN. The reactions were incubated for 20 min. at 23°C, followed by CaCl2 addition. Samples were taken at the indicated times after CaCl2 addition (in minutes), and subjected to Western blotting with anti-Cyclin B (top panel), anti-GST (second panel), anti-p31comet (third panel) or anti-CENP-A (bottom panel, loading control). Graph shows Cyclin B levels, which were quantified at each point, and normalized relative to initial levels at time = 0. Values represent the mean ± SD derived from 3 independent assays. (B) 75 nM His-tagged p31comet-wt, p31comet-AAA or p31comet-EEE were added to p31comet-depleted CSF-XEE containing DSN, and, where indicated, 60 µM nocodazole (Noc.). The reactions were incubated for 30 min. at 23°C, followed by CaCl2 addition. Cyclin B destruction was monitored as in Panel A.
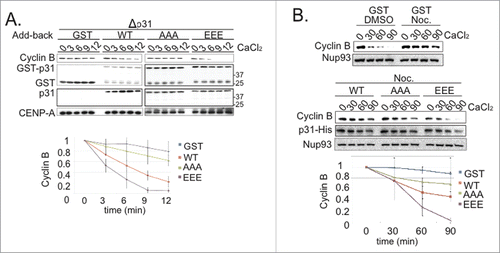
To test the role of p31comet phosphorylation in controlling its antagonism of the SAC per se, 75 nM p31comet-wt, p31comet-AAA and p31comet-EEE were added to p31comet-depleted CSF-XEE that also contained DSN and nocodazole. The reactions were incubated for 30 min. to allow SAC activation. CaCl2 was then added to cause CSF degradation, and mitotic exit was monitored by Western blotting with antibodies against Cyclin B (). As expected, a control sample to which DMSO had been added in place of nocodazole exited from mitosis within 30 min. of CaCl2 addition. Cyclin B remained stable in a nocodazole-treated sample to which GST had been added, indicating the establishment of SAC arrest. The addition of p31comet-AAA or p31comet-wt weakly accelerated Cyclin B degradation in comparison to the nocodazole-treated control, while p31comet-EEE promoted a faster rate of Cyclin B degradation. Our results are consistent with previous reports that excess p31comet can block Mad2 function in XEEs,Citation15 and indicate that p31comet phosphorylation increases its effectiveness in disruption of the SAC.
p31comet phosphorylation alters Mad2 binding and KT interactions
The ability of human p31comet to disrupt the MCC requires c-Mad2 binding.Citation8,10,11 To examine how p31comet phosphorylation alters Mad2 binding, we incubated recombinant Xenopus Mad2L12A with recombinant p31comet-wt, p31comet-EEE or p31comet-AAA. Mad2L12A is a Mad2 mutant that mimics c-Mad2.Citation10 Mad2L12A was immunoprecipitated from each reaction, and the bead-associated fractions were subjected to SDS-PAGE and Coomassie brilliant blue (CBB) staining (). While the levels of p31comet-wt and p31comet-AAA precipitated were similar, p31comet-EEE was more abundant, suggesting that it bound to Mad2L12A more tightly.
Figure 4. p31comet phosphorylation by IKK-β enhances Mad2 association. (A) 50 nM recombinant His-tagged p31comet-wt, p31comet-AAA or p31comet-EEE were incubated with 100 nM recombinant Xenopus Mad2L12A. Mad2L12A was immunoprecipitated using anti-Mad2 antibodies coupled to protein A Agarose beads. Mad2L12A and co-precipitating p31comet were detected by CBB staining. The level of each p31comet variant was quantified and normalized to the amount of Mad2L12A. The relative yield of p31comet-AAA or p31comet-EEE was compared to the yield of p31comet-wt; the ratio of each mutant to the wild type protein is shown (lower graph). Values represent the mean ± SD from three independent assays. (B) His-tagged p31comet-wt, p31comet-AAA or p31comet-EEE were added at increasing concentrations (3× = 45 nM, 6× = 90 nM, 12× = 180 nM) to XEEs containing DSN and nocodazole. After 30 min. at 23°C, chromatin was removed by pelleting, and MCC was isolated from the soluble fraction by immunoprecipitation using anti-Cdc20 antibodies. Co-precipitating Mad2 was assayed by Western blotting. After quantification, Mad2 signals at each concentration for every p31comet variant were normalized to the samples without p31comet addition, as shown in the graph below. Values represent the mean ± SD from three independent assays. (C) Reactions were assembled as in (B), with increasing concentrations of His-tagged p31comet-wt, p31comet-AAA or p31comet-EEE (1× = 15 nM, 5× = 75 nM, 10× = 150 nM). After 30 m in. at 23°C, chromosomes were removed by pelleting and washed. The chromatin fraction was subjected to SDS-PAGE and Western blotting with the indicated antibodies. After quantification, Mad2 signals at each concentration for every p31comet variant were normalized to the samples with 1× p31comet (lower graph). Values represent the mean ± SD from three independent assays.
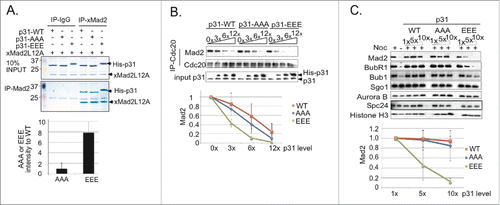
We compared the capacity of recombinant p31comet-wt, p31comet-AAA and p31comet-EEE to release Mad2 from Cdc20 when added to CSF-XEE containing DSN and nocodazole. In each case, p31comet was added at increasing concentrations and incubated for 30 min. at 23°C. Cdc20 was then immunoprecipitated from each sample, co-precipitating Mad2 was detected by immunoblotting (). While all 3 forms of p31comet disrupted the association of Cdc20 with Mad2, p31comet-EEE was noticeably more effective. When we examined the capacity of p31comet variants to release Mad2 from isolated MCC complexes (Fig. S5A), p31comet-EEE was similarly more efficient than p31comet-wt or p31comet-AAA. Together, these results suggest that p31comet phosphorylation by IKK-β enhances its capacity for MCC dissociation, thus improving its capacity for SAC silencing.
To determine if IKK-β phosphorylation controls p31comet activity at unattached KTs, we isolated chromatin from XEE incubated with nocodazole and p31comet-wt, p31comet-AAA and p31comet-EEE. We analyzed the levels of KT-bound SAC components (Mad2, BubR1, Bub1), KT structural components (Spc24) and chromosome-associated inner centromeric proteins (Shugoshin1 and Aurora B).Citation23 p31comet-wt and p31comet-AAA had little effect on the chromosome-bound levels of these components (, Fig. S5B). By contrast, chromosomes showed a progressive loss of MCC components (Mad2, BubR1) and non-MCC SAC proteins (Bub1) with increasing p31comet-EEE concentrations (, Fig. S5B). p31comet-EEE was not effective in releasing SAC components from KT in Mad2-depleted XEEs (Fig. S5C), consistent with the idea that p31comet-EEE must associate with KTCitation8,11 in order to act in this context. These data suggest Mad2-dependent p31comet recruitment to KT allows it to play a positive role in dispersion of SAC proteins from KT, and that this function is enhanced by IKK-β phosphorylation. We did not observe appreciably higher levels of SAC proteins on KTs in p31comet-depleted XEEs than in control samples, so we do not believe that p31comet normally determines the maximal amount of their recruitment.
Depletion or inhibition of IKK-β compromises SAC silencing
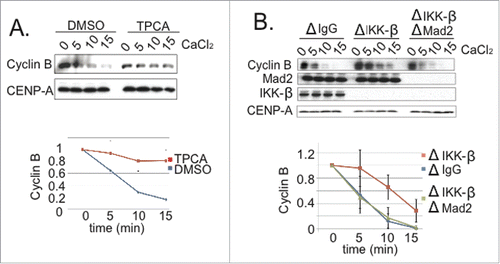
Our findings indicated that p31comet plays an important role in anaphase onset in XEE, and that its activity is significantly enhanced through phosphorylation by IKK-β. Together, these observations predicted that IKK-β should promote anaphase in XEE. To directly test this prediction, we examined the consequences of IKK-β inhibition or depletion upon mitotic exit in CSF-XEE. First, we added TPCA to CSF-XEE containing DSN. After 20 min. at 23°C, we added CaCl2 and followed mitotic exit (). We observed slower Cyclin B degradation in the TPCA-treated reaction, indicating that IKK-β inhibition delays mitotic exit. Second, we examined mitotic exit in IKK-β-depleted CSF-XEE containing DSN. Spindles appeared normal in the absence of IKK-β before CaCl2 addition (Fig. S6B). However, mitotic exit was significantly delayed after CaCl2 addition, with longer persistence of both high Cyclin B levels () and spindle MTs (Fig. S6B). This delay was reversed through the addition of purified human IKK-β (Fig. S6A). As with the delay caused by p31comet depletion (), co-depletion of Mad2 restored anaphase progression in IKK-β-depleted XEEs (), consistent with the idea that IKK-β accelerates mitotic exit by antagonizing the SAC through p31comet phosphorylation.
Figure 5. IKK-β loss compromises SAC silencing. (A) DMSO or TPCA (final concentration 300 µM) were added to CSF-XEE with DSN. The reactions were incubated for 10 min. at 23°C before CaCl2 addition. Samples were taken at indicated times after CaCl2 addition, and subjected to SDS-PAGE and Western blotting with anti-Cyclin B and -CENP-A (loading control) antibodies. The graph shows Cyclin B levels, which were quantified in each reaction and normalized relative to the initial level at time = 0. Values represent the mean ± SD derived from 3 independent assays. (B) XEEs were depleted of IKK-β, or both IKK-β and Mad2. Reactions were carried out as in Panel A, and samples were subjected to SDS-PAGE and Western blotting with the indicated antibodies. The graph shows Cyclin B levels, which were quantified in each reaction and normalized relative to the initial level at time = 0. Values represent the mean ± SD derived from 3 independent assays. ΔIgG lanes show samples from a mock-depleted reaction.
Discussion
SAC activation has been shown to occur transiently during each cell cycle in other metazoan systems,Citation5,6 even in the absence of external perturbations, so that subsequent SAC silencing is pivotal in determining the timing of anaphase. Our data indicate that similar regulation can be recapitulated within the XEE in vitro system (): Under the conditions that we have used, Mad2 depletion accelerated Cyclin B degradation in XEE, indicating that the SAC acts as a brake to anaphase progression. Moreover, p31comet, a protein required for SAC silencing, promoted anaphase in this system. Since the presence or absence of p31comet did not modulate anaphase timing in the absence of Mad2, our data suggest that p31comet acts primarily by antagonizing the SAC. Furthermore, we found that kinase activity directed against p31comet increased near anaphase onset in XEE, and that IKK-β was among the principal kinases that modify p31comet in this interval (). Analysis of phosphomimetic p31comet mutants showed that p31comet phosphorylation promotes its activity in SAC silencing (), while the depletion or inhibition of IKK-β reduced p31comet's capacity to act in this context ().
p31comet has been reported to act both on soluble MCCCitation11 and at KTs,Citation10,13 and we find evidence consistent with both modes of action in XEE (, Fig. S5). Notably, while p31comet-EEE was somewhat more efficient than p31comet-wt at mediating the dissociation of Mad2 from Cdc20 within the soluble faction (), its release of SAC proteins from unattached KTs was dramatically better than p31comet-wt (), suggesting that p31comet phosphorylation by IKK-β particularly enhances its capacity to act at KT. We estimate that the concentration of p31comet (15 nM) is much lower than the concentration of Mad2 (300 nM) within XEE (Fig. S7). Because of this difference, endogenous p31comet may only be sufficient to neutralize a small fraction of the soluble Mad2 through stoichiometric binding.Citation13 It is interesting to speculate that p31comet phosphorylation may promote its activity specifically against the smaller pool of Mad2 associated to KT, and that this might be its primary site of action within XEE.
Our findings are interesting in light of the underappreciated role of IKK-β within mitosis, which is distinct from its function in cellular stress and inflammatory signaling pathways.Citation24 IKK-β depletion causes mitotic delays in mammalian cells, accompanied by spindle assembly defects and stabilization of the Aurora A kinase.Citation20 On the other hand, the IKK-β inhibitor BMS-345541 causes precocious APC/C activation in tissue culture cells, resulting in early mitotic exit, defective chromosome separation and improper cytokinesis, and BMS-345541 can override the SAC in nocodazole-arrested cells.Citation21 It is notable in this context that the transcription factor NF-κB, a critical IKK-β target, controls the expression of numerous cell cycle regulators.Citation24 NF-κB's activity is stimulated by drugs that act on the MT cytoskeleton, including taxol or nocodazole,Citation25,26 suggesting the interesting possibility that the SAC may regulate IKK-β.
It is not clear why IKK-β depletionCitation20 and its chemical inhibitionCitation21 have opposite effects on mitotic exit in mammalian cells, with the former causing mitotic arrest while the latter promotes SAC release. These distinct phenotypes may reflect the activity of IKK-β against multiple downstream targets. Within XEEs, a simpler system that does not require gene expression,Citation14 IKK-β depletion and its inhibition by TPCA both caused mitotic exit delays (). It seems likely that p31comet phosphorylation by IKK-β contributes to its capacity to promote mitotic exit in XEE, since phosphomimetic p31comet-EEE was more effective than wild type p31comet in a variety of assays for Mad2 binding and SAC release (). Since structural analysis of the complex between Mad2 with p31comet did not resolve the first 53 residues within p31comet's N-terminal domain,Citation10 it is difficult to speculate mechanistically as to how phosphorylation of this region might enhance the stability of Mad2-p31comet association. The N-terminus of Xenopus p31comet is not closely conserved with mammalian p31comet homologues (Fig. S8). Nevertheless, alignment of their sequences shows that mouse p31comet has residues that may correspond to the S4 and T6 phosphorylation sites, while human p31comet shares the site corresponding to T6. This relationship suggests that p31comet phosphorylation by IKK-β could be important for mitotic control in mammals as well as amphibians, a possibility that we are currently testing.
In summary, our results show that the SAC can modulate anaphase timing in XEE without external perturbation of spindle assembly, and suggest a model in which IKK-β enhances mitotic exit in XEE at least partially through phosphorylation and activation of p31comet as a SAC release factor. While previous reports have indicated a role of IKK-β within mitosis, our findings are among the first mechanistic insights into the nature of this role. These observations point toward an interesting possible convergence of two disparate fields through the action of a classical signaling kinase upon the mitotic machinery.
Materials and Methods
Constructs and protein purification
We performed a blast search using the human p31comet protein sequence against the X. laevis and X. tropicalis mRNA plus EST cluster database (protein query to DNA database) to identify the Xenopus homolog of p31comet. The cDNA sequence that we retrieved (Genbank: CA973841.1) encodes a protein that is 52% identical and 64% similar to the human p31comet protein (Fig. S8).
A cDNA encoding wild type Xenopus p31comet was amplified from a X. laevis cDNA library using forward (ATGGCGCAGAGTGGCACAGATCTACC) and reverse (TCAGTTGTGAAACCCCTTTACTAT) primers. It was subcloned into a pGEX4T1 vector for protein expression in E. Coli, or into pFastBac HT for expression in Baculovirus infected insect cells. p31comet phosphomimetic and phosphodeficient mutants were generated using PCR-based site-directed mutagenesis. Xenopus Mad2 was cloned into pGEX4T1 using forward (ATGTCGTCCATCTTGCCTTTCACCCCGCCAGTAG) and reverse (TTAGGACATGCTTGAGCAGCGGACTGAAGGGGATCCCATCTGTGT) primers. Xenopus Mad2L12A was generated by site direct mutagenesis.
GST-tagged p31comet variants and Mad2 were purified from E. Coli using glutathione Sepharose beads (Amersham). The GST tag of GST-Mad2L12A was subsequently removed by thrombin digestion. His6-tagged p31comet variants were purified from Baculovirus infected insect cell lysates using Ni2+-NTA resin (Qiagen).
Antibodies, Western blotting and kinases
Rabbit anti-Xenopus p31comet, anti-Xenopus Mad2 and anti-Xenopus IKK-β were raised by Pacific Immunology Corporation (Ramona, CA) against p31comet (full length), Mad2 (full length) and IKK-β (aa 100–300) proteins. Anti-peptide antibodies that specifically recognize p31comet phosphorylated on residues S4 and T6 were generated in rabbits by the Pacific Immunology Corporation using peptide MAQ-Sp-G-Tp-DLPLRRAH.
Antibodies were used at the following concentrations: anti-p31comet (1:1,000, rabbit), anti- IKKβ (1:1000, rabbit), anti-Xenopus cyclin B (1:1,000 rabbit, Abcam), anti-CENP-A (1:1,000 rabbit), anti-Mad1 (1:1,000 rabbit) and anti-GST (1:5,000 mouse, Santa Cruz), anti-xMad2 (1:1,000 rabbit), anti-BubR1 (1:1,000 rabbit for western blotting and 1:300 chicken for IF), anti-Cdc20 (1:1,000 rabbit), anti-Spc24 (1:1,000 rabbit), anti-Bub1 (1:1000 rabbit), anti-Aurora B (1:1,000 rabbit), Histone H3 (1:5,000 rabbit, SignalChem), anti-Shugoshin (1:1,000 rabbit), anti-Nup93 (1:1,000 rabbit).
For quantification of Western blots in films were scanned and ImageJ software was used to quantify respective Western blot bands in the resultant images. For quantification of Western blots in and Figure S5A, images were captured with Gel Logic 6000 Pro system (Carestream) and quantified using Carestream Imaging software.
Kinases (Mps1, Aurora B, IKK-β, Cdk1-Cyclin B) were purchased from SignalChem.
Kinase assays
In , 2 µl of XEEs samples were incubated with 1 µCu [γ−32P]ATP and 2.5 µg GST-p31comet in a final volume of 25 µl of reaction buffer (40 mM Hepes pH 7.8, 100 mM NaCl, 2 mM MgCl2, 40 mM β–glycerolphosphate). Reactions were incubated for 1 hr at 23°C. GST-p31comet was precipitated on glutathione sepharose beads, and washed 3 times with buffer A (50 mM Tris-Cl pH 7.5, 150 mM NaCl, 0.05% Tween-20, protease inhibitor (EGTA-free tablet, Roche), 0.1 µM Okadic acid, 40 mM β–glycerolphosphate) for 4 times (5 min each). The samples were subjected to SDS-PAGE and32P labeled GST-p31comet was visualized using a phosphoimager and captured by phospho-scanner. GST-p31comet panel shows the kinase reaction blotted with anti-GST antibodies (third panel).
In , reactions included 500 µM ATP-γ-S, 100 ng/µl GST-p31comet and 4 ng/ul (of each of each purified kinase, as indicated, in a final volume of 25 µl of reaction buffer (10 mM Tris-Cl pH 7.5, 150 mM NaCl, 10 mM MgCl2). The reactions were incubated for 1 hour at 23°C, and terminated though addition of 20 mM EDTA. 2.5 mM p-nitrobenzyl mesylate (PNBM) was added to each sample, followed by an additional hour of incubation at 23°C. The samples were subjected to SDS-PAGE and Western blotting using the thiophosphate ester specific rabbit (Abcam) antibodies to identify phosphorylated proteins.
Preparation and use of XEEs
CSF-XEEs, cycling XEEs and DSN were prepared as described.Citation27,14 Unless otherwise indicated, DSN were added at 10,000 DSN/µl, and CaCl2 was added to a concentration of 0.6 mM to promote mitotic exit of CSF-XEEs. Where indicated, nocodazole was added at 65 µM. Immunodepletions of p31comet, IKKβ and Mad2 were made using antibodies coupled to protein-A Dynal magnetic beads (Life Technologies) through 2 round of depletion. The p31comet variants were added to depleted XEEs when indicated. In , isolated chromatin was prepared and analyzed as previously described.Citation27
To assay MCC disruption in , His6-tagged p31comet-wt, p31comet-AAA or p31comet-EEE were added at the indicated concentrations to CSF-XEE containing DSN and nocodazole, and incubated at 23°C for 30 min. MCC was immunoprecipitated from each sample using anti-Xenopus Cdc20 antibodies coupled to protein-A Dynal magnetic beads. The beads were washed 3 times with buffer A, eluted with SDS sample buffer and subjected to Western blotting.
In vitro binding of recombinant Mad2 and p31comet variants
In , 100 nM Mad2 and 50 nM His6-tagged p31comet-wt, p31comet-AAA or p31comet-EEE were incubated in 1 ml of TBS with 0.05% Tween20 (TBST) buffer at 23°C for 1 hour. Mad2 was Immunoprecipitated using anti-Mad2 antibodies coupled to Protein A-agarose beads. The beads were washed 3 times using Buffer A and the associated proteins were visualized by SDS-PAGE and CBB staining.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
1033590_supplemental_files.zip
Download Zip (3.3 MB)Funding
This study was supported by NICHD project number ZIA HD008816.
Related Research Data
References
- Foley EA, Kapoor TM. Microtubule attachment and spindle assembly checkpoint signalling at the kinetochore. Nat Rev Mol Cell Biol 2013; 14:25-37; PMID:23258294; http://dx.doi.org/10.1038/nrm3494
- Jia L, Kim S, Yu H. Tracking spindle checkpoint signals from kinetochores to APC/C. Trends Biochem Sci 2013; 38:302-11; PMID:23598156; http://dx.doi.org/10.1016/j.tibs.2013.03.004
- Lara-Gonzalez P, Westhorpe FG, Taylor SS. The spindle assembly checkpoint. Curr Biol 2012; 22:R966-80; PMID:23174302; http://dx.doi.org/10.1016/j.cub.2012.10.006
- Scott RJ, Lusk CP, Dilworth DJ, Aitchison JD, Wozniak RW. Interactions between Mad1p and the nuclear transport machinery in the yeast Saccharomyces cerevisiae. Mol Biol Cell 2005; 16:4362-74; PMID:16000377; http://dx.doi.org/10.1091/mbc.E05-01-0011
- Kitagawa R, Rose AM. Components of the spindle-assembly checkpoint are essential in Caenorhabditis elegans. Nat Cell Biol 1999; 1:514-21; PMID:10587648; http://dx.doi.org/10.1038/70309
- Dobles M, Liberal V, Scott ML, Benezra R, Sorger PK. Chromosome missegregation and apoptosis in mice lacking the mitotic checkpoint protein Mad2. Cell 2000; 101:635-45; PMID:10892650; http://dx.doi.org/10.1016/S0092-8674(00)80875-2
- Habu T, Matsumoto T. p31 inactivates the chemically induced Mad2-dependent spindle assembly checkpoint and leads to resistance to anti-mitotic drugs. Springerplus 2013; 2:562; PMID:24255856; http://dx.doi.org/10.1186/2193-1801-2-562
- Hagan RS, Manak MS, Buch HK, Meier MG, Meraldi P, Shah JV, Sorger PK. p31(comet) acts to ensure timely spindle checkpoint silencing subsequent to kinetochore attachment. Mol Biol Cell 2011; 22:4236-46; PMID:21965286; http://dx.doi.org/10.1091/mbc.E11-03-0216
- Ma HT, Chan YY, Chen X, On KF, Poon RY. Depletion of p31comet promotes sensitivity to antimitotic drugs. J Biol Chem 2012; 287:21561-9; PMID:22544748; http://dx.doi.org/10.1074/jbc.M112.364356
- Yang M, Li B, Tomchick DR, Machius M, Rizo J, Yu H, Luo X. p31comet blocks Mad2 activation through structural mimicry. Cell 2007; 131:744-55; PMID:18022368; http://dx.doi.org/10.1016/j.cell.2007.08.048
- Westhorpe FG, Tighe A, Lara-Gonzalez P, Taylor SS. p31comet-mediated extraction of Mad2 from the MCC promotes efficient mitotic exit. J Cell Sci 2011; 124:3905-16; PMID:22100920; http://dx.doi.org/10.1242/jcs.093286
- Teichner A, Eytan E, Sitry-Shevah D, Miniowitz-Shemtov S, Dumin E, Gromis J, Hershko A. p31comet promotes disassembly of the mitotic checkpoint complex in an ATP-dependent process. Proc Natl Acad Sci U S A 2011; 108:3187-92; PMID:21300909; http://dx.doi.org/10.1073/pnas.1100023108
- Fava LL, Kaulich M, Nigg EA, Santamaria A. Probing the in vivo function of Mad1:C-Mad2 in the spindle assembly checkpoint. Embo J 2011; 30:3322-36; PMID:21772247; http://dx.doi.org/10.1038/emboj.2011.239
- Murray AW. Cell cycle extracts. Methods Cell Biol 1991; 36:581-605; PMID:1839804; http://dx.doi.org/10.1016/S0091-679X(08)60298-8
- Xia G, Luo X, Habu T, Rizo J, Matsumoto T, Yu H. Conformation-specific binding of p31(comet) antagonizes the function of Mad2 in the spindle checkpoint. Embo J 2004; 23:3133-43; PMID:15257285; http://dx.doi.org/10.1038/sj.emboj.7600322
- Jia L, Li B, Warrington RT, Hao X, Wang S, Yu H. Defining Pathways of Spindle Checkpoint Silencing: Functional Redundancy between Cdc20 Ubiquitination and p31comet. Mol Biol Cell 2011; 22:4227-35; PMID:21937719; http://dx.doi.org/10.1091/mbc.E11-05-0389
- Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER, 3rd, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini N, Lerenthal Y, et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007; 316:1160-6; PMID:17525332; http://dx.doi.org/10.1126/science.1140321
- Hegemann B, Hutchins JR, Hudecz O, Novatchkova M, Rameseder J, Sykora MM, Liu S, Mazanek M, Lenart P, Heriche JK, et al. Systematic phosphorylation analysis of human mitotic protein complexes. Sci Signal 2011; 4:rs12; PMID:22067460; http://dx.doi.org/10.1126/scisignal.2001993
- Date DA, Burrows AC, Summers MK. Phosphorylation regulates the p31Comet – Mitotic Arrest Deficient 2 (Mad2) interaction to promote Spindle Assembly Checkpoint (SAC) activity. J Biol Chem 2014; 289:11367-73; PMID:24596092; http://dx.doi.org/10.1074/jbc.M113.520841
- Irelan JT, Murphy TJ, DeJesus PD, Teo H, Xu D, Gomez-Ferreria MA, Zhou Y, Miraglia LJ, Rines DR, Verma IM, et al. A role for IkappaB kinase 2 in bipolar spindle assembly. Proc Natl Acad Sci U S A 2007; 104:16940-5; PMID:17939994; http://dx.doi.org/10.1073/pnas.0706493104
- Blazkova H, von Schubert C, Mikule K, Schwab R, Angliker N, Schmuckli-Maurer J, Fernandez PC, Doxsey S, Dobbelaere DA. The IKK inhibitor BMS-345541 affects multiple mitotic cell cycle transitions. Cell Cycle 2007; 6:2531-40; PMID:17704647; http://dx.doi.org/10.4161/cc.6.20.4807
- Landais I, Hiddingh S, McCarroll M, Yang C, Sun A, Turker MS, Snyder JP, Hoatlin ME. Monoketone analogs of curcumin, a new class of Fanconi anemia pathway inhibitors. Mol Cancer 2009; 8:133; PMID:20043851; http://dx.doi.org/10.1186/1476-4598-8-133
- Boyarchuk Y, Salic A, Dasso M, Arnaoutov A. Bub1 is essential for assembly of the functional inner centromere. J Cell Biol 2007; 176:919-28; PMID:17389228; http://dx.doi.org/10.1083/jcb.200609044
- Ledoux AC, Perkins ND. NF-kappaB and the cell cycle. Biochem Soc Trans 2014; 42:76-81; PMID:24450631; http://dx.doi.org/10.1042/BST20130156
- Mistry P, Deacon K, Mistry S, Blank J, Patel R. NF-kappaB promotes survival during mitotic cell cycle arrest. J Biol Chem 2004; 279:1482-90; PMID:14581467; http://dx.doi.org/10.1074/jbc.M310413200
- Rosette C, Karin M. Cytoskeletal control of gene expression: depolymerization of microtubules activates NF-kappa B. J Cell Biol 1995; 128:1111-9; PMID:7896875; http://dx.doi.org/10.1083/jcb.128.6.1111
- Arnaoutov A, Dasso M. The Ran GTPase regulates kinetochore function. Dev Cell 2003; 5:99-111; PMID:12852855; http://dx.doi.org/10.1016/S1534-5807(03)00194-1