
Abstract
Abnormal protein interactions of mutant huntingtin (Htt) triggered by polyglutamine expansion are thought to mediate Huntington's disease (HD) pathogenesis. Here, we explored a functional interaction of Htt with protein arginine methyltransferase 5 (PRMT5), an enzyme mediating symmetrical dimethylation of arginine (sDMA) of key cellular proteins, including histones, and spliceosomal Sm proteins. Gene transcription and RNA splicing are impaired in HD. We demonstrated PRMT5 and Htt interaction and their co-localization in transfected neurons and in HD brain. As a result of this interaction, normal (but to a lesser extend mutant) Htt stimulated PRMT5 activity in vitro. SDMA of histones H2A and H4 was reduced in the presence of mutant Htt in primary cultured neurons and in HD brain, consistent with a demonstrated reduction in R3Me2s occupancy at the transcriptionally repressed promoters in HD brain. SDMA of another PRMT5 substrate, Cajal body marker coilin, was also reduced in the HD mouse model and in human HD brain. Finally, compensation of PRMT5 deficiency by ectopic expression of PRMT5/MEP50 complexes, or by the knock-down of H4R3Me2 demethylase JMJD6, reversed the toxic effects of mutant Htt in primary cortical neurons, suggesting that PRMT5 deficiency may mediate, at least in part, HD pathogenesis. These studies revealed a potential new mechanism for disruption of gene expression and RNA processing in HD, involving a loss of normal function of Htt in facilitation of PRMT5, supporting the idea that epigenetic regulation of gene transcription may be involved in HD and highlighting symmetric dimethylation of arginine as potential new therapeutic target.
Abbreviations
| HD | = | Huntington's disease |
| Htt | = | huntingtin |
| polyQ | = | polyglutamine |
| PRMT5 | = | protein arginine methyltransferase |
| Sm proteins | = | spleceosomal small nuclear ribonucleoproteins |
| sDMA | = | symmetrical arginine dimethylation |
| CB | = | Cajal body |
| SMN | = | survival of motor neurons |
| snRNPs | = | small nuclear ribonucleoprotein particles |
| BDNF | = | brain-derived neurotrophic factor |
| IP | = | immunoprecipitation |
| ChIP | = | the chromatin immunoprecipitation |
| DMEM | = | Dulbecco's modified Eagle's medium |
| FBS | = | fetal bovine serum |
| HEK | = | human embryonic kidney |
| PIC | = | protease inhibitors cocktail |
| IgG | = | immunoglobulin |
Introduction
Huntington's disease (HD) is a progressive autosomal dominant neurodegenerative disorder caused by a polyglutamine (polyQ) expansion in the HD gene product, huntingtin (Htt). While this genetic mutation was found to cause HD,Citation1 mechanisms of HD cellular pathogenesis remain elusive.Citation2-4 Studies using cellular and mouse models of HD have revealed disruptions in many cellular processes and molecular functions, including protein homeostasis, energy expenditure, cellular trafficking, gene transcription and RNA processing. Many of these defects are believed to be mediated by abnormal interactions of mutant Htt with other proteins,Citation5-14 triggered by a conformational change in the mutant Htt protein, but the full range of alterations of protein interactions of mutant Htt is still undefined. Numerous studies, including our recent study of normal and mutant Htt protein interactome in striatal cells, highlighted gene transcription and RNA metabolism as key pathways potentially disrupted in HD.Citation15-26 We have identified many novel Htt interactors within these pathways, including several protein arginine methyltransferases, (PRMTs 1, 3 and 5),Citation18 enzymes that carry out protein methylation resulting in the mono-and di-methylation of arginine either in a symmetrical (sDMA) or asymmetrical (aDMA) manner. Arginine methylation has recently emerged as an important protein modification involved in signaling, RNA processing and regulation of gene transcription.Citation27,28 In particular, PRMT5 is an enzyme mediating symmetrical dimethylation of arginine (sDMA) of histones and spliceosomal Sm proteins. Since both gene transcription and RNA splicing are impaired in HD, we decided to focus on the PRMT5/Htt interaction to investigate a potential involvement of Htt in sDMA protein modification carried out by PRMT5.
PRMT5 is one of the 3 known type II protein arginine methyltransferases, catalyzing ωG-monomethylation and ωG NG-symmetric dimethylation of arginine. PRMT5 has been found in multiple complexes mediating diverse cellular functions including RNA processing and transcriptional regulation. Among the known substrates of PRMT5 are histones H3, H2A and H4.Citation29-33 As a component of the 20S methylosome, PRMT5 methylates SmD1 and SmD3 proteins, and induces their interaction with the survival of motor neuron (SMN) protein, which is essential for cytoplasmic assembly of the small nuclear ribonucleoprotein particles (snRNPs).Citation34,35 PRMT5 has also been co-purified with the members of chromatin remodeling complexes, hSWI/SNF and NURD, and has been implicated in transcriptional repression of genes that control cell cycle and tumor suppression.Citation31-33 PRMT5 was also found to methylate p53, thereby regulating its response to stress signals and affecting the p53 specificity toward its target genes.Citation36
In this study, we demonstrate the Htt/PRMT5 interaction in transfected cells and in HD brain, and show co-localization of these 2 proteins in primary neurons. Our results suggest that a normal function of Htt may include facilitation of PRMT5 activity, while mutant Htt might fail to do it efficiently, possibly due to a conformational shift and its abnormal protein interactions. As a result of PRMT5 impairment, the sDMA of histones H2A and H4 was reduced in neurons expressing mutant Htt, and in HD human brain tissues, compared to controls. Furthermore, using chromatin immunoprecipitation (ChIP), we detected a reduction in R3Me2s occupancy of histones H4 and H2A at the transcriptionally repressed promoters in HD brain. Our findings of the PRMT5/Htt functional interaction raise the possibility that mutant Htt may in part exert its effects on transcription via direct interaction with histone-modifying enzymes, such as PRMT5, and by modulating their activity.
As another evidence of PRMT5 impairment in HD, we showed that sDMA of a Cajal body (CB) marker, coilin, was reduced in HD tissues, which was previously shown to interfere with incorporation of SMN protein in Cajal bodies and pre-mRNA splicing.Citation37,38 This observation is consistent with the defects in RNA processing previously observed in HD models. Thus our studies provide a potential new mechanism for the previously well-documented disruption of gene expression and RNA processing in HD, involving a loss of normal function of Htt in facilitation of PRMT5. Compensation of PRMT5 deficiency by expression of PRMT5/MEP50 complexes improved the survival of primary neurons expressing mutant Htt, suggesting that PRMT5 deficiency may mediate, at least in part, HD pathogenesis. Consistent with that, reducing the levels of H4R3Me2 demethylase JMJD6 reversed the toxic effects of mutant Htt in primary cortical neurons. The broad significance of this work is that it supports the idea that epigenetic regulation of gene transcription may be involved in HD, and highlights, for the first time, symmetric dimethylation of arginine as potential new therapeutic target.
Results
Huntingtin interacts with PRMT5/MEP50 complex and co-localizes with PRMT5
We have previously conducted a comprehensive proteomics analysis of normal and expanded Htt interactome using tandem affinity purification of Htt N586 fragment expressed in striatal cells.Citation18 In this study, we found initial evidence that both normal Htt and expanded Htt could interact with several protein arginine methyltransferases (PRMTs 1, 3 and 5), as well as a few histones and Sm proteins. Since both histones and Sm proteins are known substrates for sDMA by PRMT5, we decided to focus on the PRMT5/Htt interaction to further investigate a potential involvement of Htt in sDMA protein modification carried out by PRMT5.
To validate the PRMT5/Htt interactions, previously identified by mass spectrometry of Htt pull-downs,Citation18 we performed the reverse immunoprecipitation (IP) using FLAG antibody to pull-down FLAG-PRMT5 protein from transfected HEK293 and ST14 cells. As shown on , exogenous Htt-N511–8Q and Htt-N511–52Q proteins were co-precipitated with the exogenous FLAG-PRMT5 in both cell lines.
Figure 1. Htt interacts with PRMT5 in cell lines and human brain. (A) HEK 293 cells were transiently transfected with the indicated plasmids, keeping the amount of Htt and PRMT5 plasmids and total amount of DNA equal between transfections by supplementing with empty vector. Cells were lysed 48 h after transfection, and PRMT5 complexes were immunoprecipitated using the anti-FLAG-agarose beads. Normal and expanded Htt were detected in the immunoprecipitates (IPs) from cells co-transfected with Htt and PRMT5, but not in control samples without the primary antibody or in cells transfected with Htt constructs only (first panel and data not shown). IPs were also analyzed for the presence of PRMT5 using the PRMT5-specific antibody (second panel). The inputs are shown on the right 2 panels. (B) Rat striatal ST14 cells were transiently transfected with indicated plasmids, lysed 48 h after transfection, and PRMT5 complexes were immunoprecipitated using the anti-FLAG-agarose beads. Normal and expanded Htt were detected in the IPs from cells co-transfected with Htt and PRMT5, but not in mock transfection, or in cells transfected with Htt constructs only (left panel). IPs were also analyzed for the presence of PRMT5 using PRMT5-specific antibody (right panel). (C) Endogenous Htt proteins were immunoprecipitated from STHdh Q7/Q7 and Q111/Q111 cells using a specific antibody to Htt (MAB2166). Endogenous PRMT5 monomers and high-molecular weight PRMT5 complexes were detected in the IPs using the PRMT5-specific antibody, but not in control samples without the primary antibody (top panel, left side). PRMT5 inputs are shown on the last 2 lanes. IPs were also analyzed for the presence of Htt using the Htt-specific antibody, and Htt inputs are shown (bottom panel). (D) Htt complexes were immunoprecipitated from total cell homogenates from frozen human front superior cortex of normal controls and HD cases. Endogenous PRMT5 monomers and high-molecular weight PRMT5 complexes were detected in the IPs from HD brain using the PRMT5-specific antibody, but not in normal controls or in control samples where normal IgGs were used for IP (top panel). Htt inputs are shown on the bottom panel.
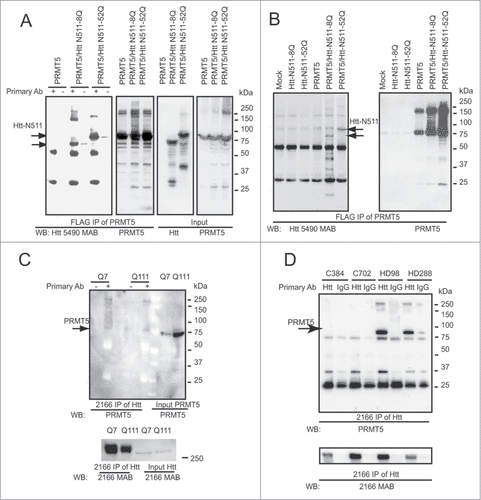
Using MAB2166 antibody to Htt, we further found that endogenous PRMT5 (detected with PRMT5 antibody) was co-precipitated with the endogenous Htt in STHdh Q7/Q7 and Q111/Q111 knock-in cells (). Notably, increased levels of endogenous PRMT5 were observed in Q111/Q111 cells expressing expanded Htt, compared to normal Q7/Q7 cells. We next examined whether the endogenous PRMT5/Htt interaction could be detected in human brain tissues. Htt proteins were precipitated from total cell homogenates of frozen human frontal superior cortex of normal controls and HD cases using MAB2166 (). A robust band was detected with a specific antibody to PRMT5 in the Htt pull-downs of HD brain tissues, but not in normal controls, or negative controls (with normal mouse immunoglobulin, IgG). We have analyzed a total of 12 HD cases and 12 normal controls and obtained similar results (representative gel is shown). Notably, there was no significant change in overall PRMT5 levels between normal and HD brain tissues (data not shown).
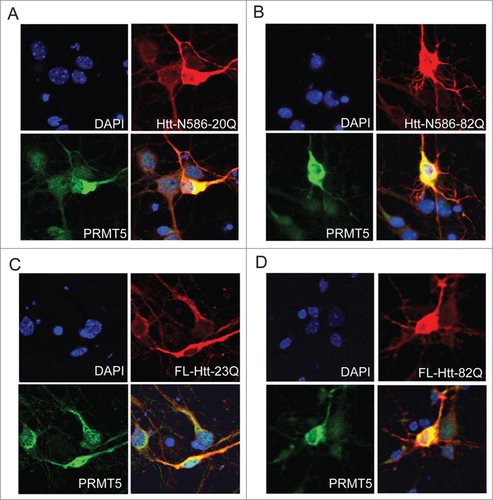
To investigate whether endogenous Htt and PRMT5 co-localize in neuronal cells, primary mouse cortical neurons were co-transfected with normal or polyQ-expanded Htt and PRMT5, and co-labeled with the antibody to Htt (MAB2166) and PRMT5, followed by immunofluorescence confocal microscopy. We detected substantial cytoplasmic co-localization of PRMT5 with both normal and expanded N586 fragment of Htt () and full-length Htt ().
Figure 2. Htt and PRMT5 co-localize in primary neurons. Primary mouse cortical neurons were co-transfected at 5 DIV with the expression vectors for PRMT5 and either normal or polyQ-expanded Htt N586 fragment (A, B) or the full-length Htt constructs (C, D). Confocal immunofluorescence detection of Htt with 2166 monoclonal antibody is shown in red (Alexa Fluor 555); detection of PRMT5 with polyclonal specific antibody is shown in green (Alexa Fluor 488); nuclear staining (DAPI) is shown in blue. Yellow staining in merged images demonstrated partial co-localization. Representative images are shown for each construct.
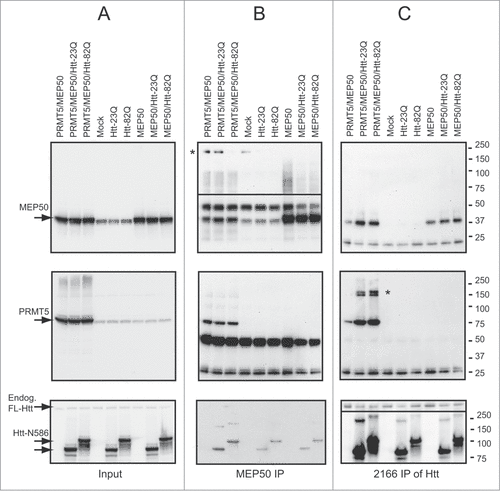
PRMT5 was previously found within multimeric complexes with a few protein partners, regulating its function and specificity.Citation29,31-34,39-42 A core component of these complexes is the WD40 protein MEP50 (methylosome protein 50), which promotes PRMT5 interactions with substrates and stimulates PRMT5 activity.Citation43,44 Since Htt often acts as a scaffolding protein facilitating protein complexes, we investigated whether Htt is involved in PRMT5/MEP50 functional interaction. As expected, PRMT5 protein was detected within the MEP50 IPs, demonstrating PRMT5/MEP50 interaction in HEK293 cells transfected with PRMT5 and MEP50 expression plasmids (, middle panel). Interestingly, transfected Htt-N586 fragments have been also detected within these complexes (, bottom panel), suggesting the interaction of all 3 proteins. Confirming this notion, we have detected both PRMT5 and MEP50 within Htt IPs using 2166 antibody to Htt (, top and middle panels). Notably, more PRMT5 co-precipitated with polyQ-expanded Htt compared to normal Htt, suggesting an increased affinity of expanded Htt toward PRMT5. Consistent with previously reported multimeric nature of PRMT5 and MEP50 complexes, we have detected high-molecular weight complexes (marked by asterisk) using PRMT5 and MEP50 antibodies. Notably, MEP50 high molecular weight complexes were absent in the presence of expanded Htt fragments in transfected HEK293 cells (, top panel), raising a possibility that expanded Htt may affect MEP50 multimerization, and potentially its interaction with PRMT5.
Figure 3. Htt interacts with PRMT5/MEP50 complex. HEK 293 cells were transiently transfected with the indicated plasmids, keeping the amount of Htt-N586, PRMT5 and MEP50 plasmids and total amount of DNA equal between transfections by supplementing with empty vector. Cells were lysed 48 h after transfection, and MEP50 (B) or Htt (C) complexes were immunoprecipitated using either MEP50-or Htt-specific antibodies. All three proteins-MEP50 (top panels), PRMT5 (middle panels) and Htt (bottom panels) were detected in both MEP50 and Htt IPs from cells co-transfected with Htt, PRMT5 and MEP50. High-molecular weight complexes detected with PRMT5 and MEP50 antibodies are marked by asterisk. The inputs are shown in A.
Huntingtin modulates PRMT5 activity toward histones in vitro
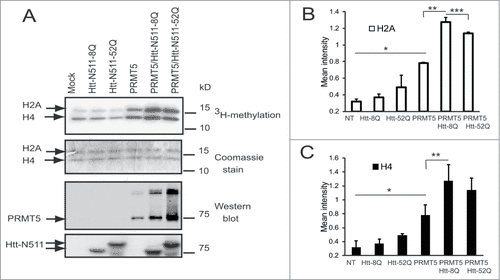
To gain further insights into a potential functional significance of the PRMT5/Htt interaction and co-localization, we investigated whether Htt affected PRMT5 activity in vitro. PRMT5 was shown to methylate histones H2A and H4 at arginine 3.Citation42 A radioactive in vitro methylation assay was conducted using purified histones H2A and H4 as substrates. ST14 rat striatal cells were co-transfected with the recombinant plasmids expressing FLAG-PRMT5 with or without N511 Htt fragments, or with Htt fragments alone. FLAG-agarose beads from IPs were used as an enzyme source. An expected increase in the radiolabeling of both histones was observed in the presence of recombinant PRMT5 compared to non-transfected cells (). In the presence of Htt N511–8Q fragments we detected a further significant increase in the amounts of labeled histones H2A and H4, indicating induction of PRMT5 activity by Htt fragment (). This effect was less evident in the presence of expanded Htt N511–52Q. This difference was significant in case of histone H2A methylation (). Thus, these data demonstrate that normal (but to a lesser extend mutant) Htt stimulates PRMT5 activity in vitro.
Figure 4. Htt modulates PRMT5 activity toward histones. (A) In vitro methyltransferase assay of FLAG IPs from ST14 cells transfected with indicated plasmids, keeping the amount of Htt and PRMT5 plasmids and total amount of DNA equal between transfections by supplementing with empty vector. Autoradiographs (upper panel), Coomassie stained gel (middle panel) are shown. Western blot detections of PRMT5 with PRMT5-specific antibody and of Htt with MAB5490 antibody are shown on bottom panels. (B, C) The graphs show the quantification of autoradiographs from 3 experiments for histones H2A (B) and H4 (C), #n = 3, p = 0.005; ##n = 3, p = 0.02; ###n = 3, p = 0.1.
Symmetric arginine dimethylation of histones is attenuated by mutant Htt in primary neurons and is impaired in HD brain
PRMT5 was previously reported to mediate a specific post-translational modification of histones H2A and H4, sDMA of R3.Citation42 As demonstrated by our methylation assay, normal Htt stimulated PRMT5 activity toward histones in vitro, while this stimulation was decreased in the presence of mutant Htt. Thus we sought to evaluate the appearance of sDMA on H2A/H4 (R3Me2s) in primary neurons transfected with either normal (FL-Htt-22Q) or mutant (FL-Htt-82Q) full-length Htt. 48 h following transfection, neurons were fixed and co-stained with the modification-specific antibody (R3Me2s) and an antibody to Htt (MAB2166) to identify transfected cells (). R3Me2s antibody specifically reacts with the symmetrically dimethylated arginine 3 on both histones H2A and H4 (according to the information provided by supplier, www.abcam.com). Immunofluorescence intensity analysis was performed using the Zen 2012 software. To measure the intensity of the signal in the nucleus, a confocal plane was established at a level passing through the centers of the nuclei of Htt-positive cells. We found that H2A/H4 sDMA was significantly increased in neurons transfected with the normal full-length Htt plasmid compared to cells transfected with an empty vector, and significantly decreased in cells transfected with mutant Htt, with 29% difference between normal and expanded Htt ().\raster="rgFigKCCY_A_1033595_F0005_B"
Figure 5. Symmetric arginine dimethylation of histones is attenuated by mutant Htt in primary neurons and is impaired in HD brain. (A, B) Primary mouse cortical neurons were transfected at 5 DIV with normal (A) or mutant (B) full-length Htt expression constructs. Representative images of confocal immunofluorescence detection of Htt with 2166 monoclonal antibody (in red, Alexa Fluor 555) and of sDMA modification of H2A and H4 with R3Me2s modification-specific antibody (in green, Alexa Fluor 488) are shown. Transfected cells are indicated with white arrows The nuclear staining (DAPI) is shown in blue. (C) Quantification of H4R3me2s staining in transfected cells presented as a ratio of mean intensity of the staining in transfected and non-transfected cells. 100 transfected cells were analyzed for each condition in 3 experiments. (#n = 3, p = 0.004). (D) Total cell homogenates from frozen human front superior cortex of 6 normal controls and 6 HD cases were analyzed by Western blotting using the following antibodies: MW1 for detection of expanded Htt; 2166 MAB for detection of normal and expanded Htt; histone-specific antibodies for detection of histones H2A and H4; modification-specific antibody R3Me2s for detection of sDMA on R3 of histones H2A and H4; β-tubulin as a loading control. Representative blots are shown. (E) Quantification of the levels of R3Me2s modification on histones H2A and H4 presented as a ratio of R3Me2s signal intensity to total levels of histones based on Western blot repeated 3 times (#n = 3, p = 0.045; ## n = 3, p = 0.045).
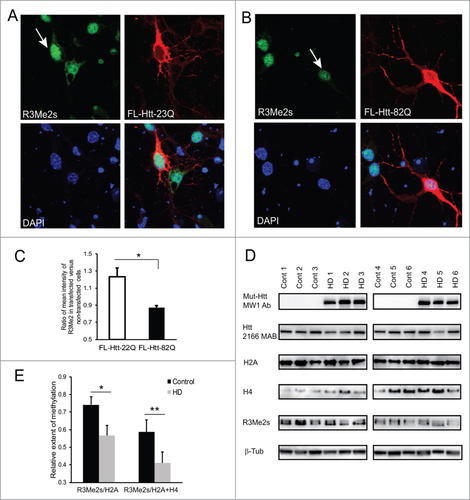
Next, we examined if the total levels of sDMA on H2A/H4 were changed in vivo in human HD brains compared to non-affected controls. Total cell homogenates from frozen human frontal superior cortex of 6 normal controls and 6 HD cases were analyzed by Western blotting using the modification-specific antibody R3Me2s. As expected, expression of expanded Htt was detected with a polyQ-specific MW1 antibody in HD brain, but not in controls (, top panels). The total levels of histones H2A and H4, although somewhat variable, were slightly elevated in HD brains compared to controls, but the difference was not significant. However the levels of R3Me2s modification on histones H2A and H4 (presented as a ratio of R3Me2s signal intensity to total levels of histones and normalized to tubulin as loading control) were significantly decreased (30%) in HD brains compared to normal controls ().
Reduction of H2A/H4R3Me2s occupancy at the γ-globin and BDNF promoters in HD brain
PRMT5 has been shown to mediate gene silencing through the induction of repressive histone marks, such as symmetric dimethylation of H4R3, H2AR3 and H3R8, at the promoters of PRMT5 target genes, including γ-globin,Citation45 cyclin E1,Citation46 tumor suppressor genes ST7 and NM23Citation32 and CUL4.Citation47 As another potential evidence of PRMT5 impairment in HD, we tested whether there is a change in H2A/H4 R3Me2s occupancy at the specific promoters in HD brain. As previously established, PRMT5-mediated methylation of histone H4R3 regulates the silencing of fetal γ-globin gene observed in human and primates after birth.Citation45 Hence, we performed the chromatin immunoprecipitation (ChIP) analysis with the modification-specific antibody to compare R3Me2s enrichment at the human γ-globin promoter P-1 regionCitation45 in human HD and control brains. As more relevant to HD, we have also evaluated occupancy of H2A/H4R3Me2s mark at the BDNF promoter. Transcription from both exon II and exon IV of the BDNF promoter and H4K4 trimethylation at the exon II of the BDNF promoter were previously shown to be reduced in HD mouse and human brains.Citation48,49
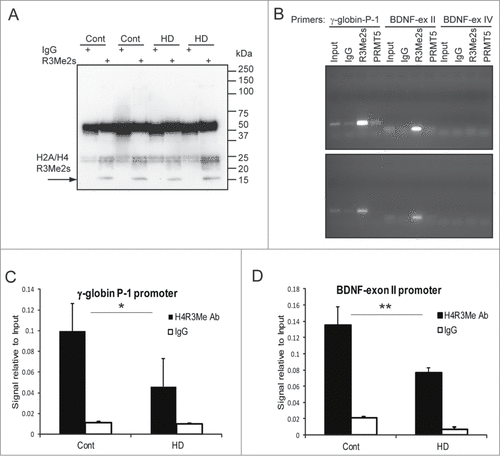
First, the efficiency of the ChIP assay was confirmed by Western blotting, which detected a specific H2A/H4 R3Me2s band following ChIP with R3Me2s antibody, but not with normal rabbit IgG (). Using PCR with promoter-specific primers, we detected binding of R3-methylated histones to the human γ-globin promoter P-1 region and BDNF exon II promoter in both HD and control human brain (). The H2A/H4 R3Me2s mark was not detected at the exon IV of the BDNF promoter, which demonstrated the specificity of the assay. A weak binding of PRMT5 (using available PRMT5-specific antibody) to the human γ-globin promoter P-1 region was observed in normal, but not in HD brain (It should be noted that ChIP-grade antibody for PRMT5 is not currently available). ChIP analysis with R3Me2s antibody was further carried out using 4 HD cases and 4 control human brains, followed by quantitative (q)-PCR with the gene-specific primers. We observed a consistent and significant reduction of H2A/H4R3Me2s occupancy at the γ-globin and BDNF exon II promoters in HD brain compared to control samples ().
Figure 6. Reduction of H2A/H4R3Me2s occupancy at the γ-globin and BDNF promoters in HD brain. (A) Confirmation of ChIP assay by Western blotting with the R3Me2s modification-specific antibody. Representative image for 2 controls and 2 HD samples is shown. (B) Detection of H2A/H4 R3Me2s marks at the γ-globin P-1 and BDNF promoters by ChIP assay. Chromatin fractions from HD (bottom panel) or control (top panel) human brain were immunoprecipitated with the indicated antibodies and amplified with the indicated primer pairs. Normal rabbit IgG used as a negative control. (C and D) R3Me2s enrichment at the γ-globin P-1 (C) and BDNF-ex II promoters (D) were measured by ChIP and q-PCR assays in HD and control human brains (#n = 4, p = 0.1; ##n = 3, p = 0.1).
Coilin symmetric dimethylation is reduced in HD brain
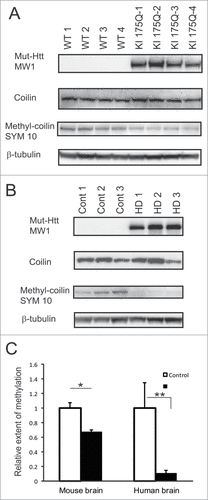
A major group of proteins that contain sDMA includes RNA-binding proteins, particularly those involved in the pre-mRNA splicing. As a part of methylosome, PRMT5 symmetrically methylates Sm proteins, increasing their affinity for SMN protein.Citation34,35,50 Another PRMT5 substrate is a Cajal body marker coilin, and its methylation is required for localization of SMN to Cajal bodies.Citation37,38 sDMA modification of coilin and other proteins can be specifically detected by SYM10 antibody.Citation37 Using this antibody we assessed the levels of coilin methylation in the knock-in HD mouse model (KI175Q homozygous mice obtained from CHDI) and in human HD brain (). Comparison of 4 HD and 4 wild type (WT) littermates at 6 months of age showed that coilin was hypomethylated (34%) in KI175Q HD mouse brain compared to WT brains (). The overall expression of coilin, detected with a coilin-specific antibody, was largely unchanged between HD and WT tissues. The difference in the levels of methylated coilin between HD and control was even more dramatic in human postmortem tissue. The frontal cortex brain homogenates from 5 HD cases and 5 controls were analyzed with SYM10 antibody. As shown on , the extent of methylation of coilin in HD brain was only around 10% of that found in the control tissues. The levels of total coilin were somewhat variable between the cases, however no significant difference between HD and control was observed.
Figure 7. Coilin symmetric dimethylation is reduced in HD mouse and human brain. (A, B) Whole brain homogenates from 4 KI175Q HD and 4 WT mice (A) or homogenates from frozen human frontal superior cortex of 5 normal controls and 5 HD cases (B) were analyzed by Western blotting using the following antibodies: MW1 for detection of expanded Htt; coilin-specific antibody; SYM10 for detection of symmetrically dimethylated coilin and β-tubulin as a loading control. Representative images are shown. (C) Quantification of the relative extent of symmetric dimethylation of coilin presented as a ratio of SYM10 signal intensity to total levels of coilin. (#n = 4, p = 0.007; ##n = 5, p = 0.03).The experiment was repeated 3 times.
Modulation of the levels of arginine methyltransferase PRMT5 and histone arginine demethylase JMJD6 affects the survival of HD neurons
As we found that PRMT5 impairment in HD models resulted in reduced arginine dimethylation of important cellular proteins, we tested whether forced expression of PRMT5 in HD neurons would compensate for such deficit and could rescue primary neurons from mutant Htt-induced toxicity.
Primary cortical neurons were co-transfected with either normal or mutant full-length Htt and PRMT5 expression plasmids, and cell death was measured by nuclear condensation assay (). No benefit for survival was observed in neurons co-transfected with mutant Htt and PRMT5 alone. However co-transfection of PRMT5 with its cofactor MEP50 completely rescued primary neurons from the toxicity of mutant Htt. These results are consistent with the proposed role of MEP50 in stimulation of PRMT5 activity.Citation44\raster="rgFigKCCY_A_1033595_F0008_B"
Figure 8. Modulation of the levels of arginine methyltransferase PRMT5 and histone arginine demethylase JMJD6 affects the survival of HD neurons. (A) Co-transfection of PRMT5 with its cofactor MEP50 rescued primary neurons from the toxicity of mutant Htt. Primary cortical neurons were co-transfected at 5 DIV with either normal or mutant full-length Htt and PRMT5 and MEP50 expression plasmids as indicated, and co-transfected with eGFP to identify transfected cells. Cell death was measured by nuclear condensation assay. # n = 4, P < 0.01; ## (co-transfected with PRMT5 and MEP50 vs with Htt alone) n = 4, P < 0.01. (B–D) JMJD6 knock-down rescued primary neurons from the toxicity of mutant Htt. (B) Primary mouse cortical neurons were co-transfected at 5 DIV with mutant full-length Htt expression constructs, eGFP (to identify transfected cells) and either JMJD6 siRNA (top panels) or with non-targeting control siRNA (bottom panels). Representative images of confocal immunofluorescence detection of JMJD6 (in red, Alexa Fluor 555) in GFP-positive (transfected, indicated with white arrow) and GFP-negative (non-transfected) cells are shown. The nuclear staining (Hoechst) is shown in blue. (C) Primary mouse cortical neurons were co-transfected at 5 DIV with mutant full-length Htt expression constructs and either JMJD6 siRNA (top panels) or with non-targeting control siRNA (bottom panels). Representative images of confocal immunofluorescence detection of Htt with 2166 monoclonal antibody (in red, Alexa Fluor 555) and of sDMA modification of H2A and H4 with R3Me2s modification-specific antibody (in green, Alexa Fluor 488) in transfected (indicated with white arrow) and non-transfected cells are shown. The nuclear staining (Hoechst) is shown in blue. (D) Quantification of H4R3Me2s staining in transfected cells presented as a ratio of mean intensity of the staining in transfected and non-transfected cells. 100 transfected cells were analyzed for each condition in 3 experiments. (#n = 3, P < 0.001). (E) Primary cortical neurons were co-transfected with mutant full-length Htt-82Q and either JMJD6 siRNA or with non-targeting control siRNA as indicated, and co-transfected with eGFP to identify transfected cells. Cell death was measured by nuclear condensation assay. ˜300 cells were analyzed per each condition in each experiment. # n = 3, P < 0.001
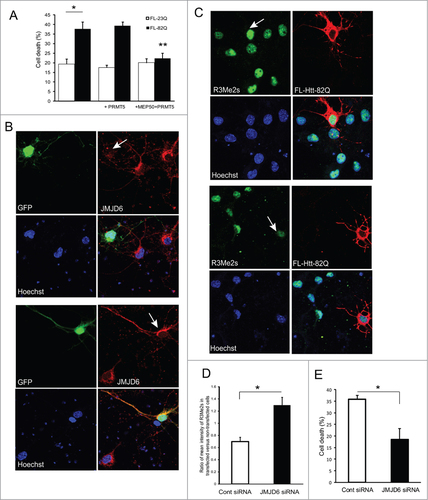
Identification of PRMT5 impairment and the resulting reduction in symmetric arginine dimethylation associated with HD raises the possibility that an intervention designed to compensate for this deregulation may be beneficial in HD. The only enzyme known to erase dimethyl arginine marks from histones is a Jumonji C domain-containing protein 6 (JMJD6), a member of a novel class of iron and 2-oxoglutarate-dependent dioxygenase.Citation51,52 JMJD6 is expressed in brain, and was shown to demethylate H4R3.Citation51,52 We used siRNA to knock-down this enzyme in primary neurons co-transfected with full-length normal or expanded Htt. The effectiveness of JMJD6 siRNA knock-down was first evaluated using immunofluorescence (). JMJD6 contains 5NLS domains for nuclear localization, and an NES domain, suggesting nuclear/cytoplasmic shuttling, and has been previously detected in both nucleus and cytoplasm.Citation52 As shown on , the levels of JMJD6 were barely detectable in cells co-transfected with JMJD6 siRNA, compared to the cells co-transfected with non-targeting control siRNA. As expected, JMJD6 knock-down resulted in an increase of sDMA levels of histones H2A and H4, detected with the modification-specific antibody R3Me2s (). We then assessed the effects of JMJD6 knock-down on the toxicity of mutant Htt as measured by nuclear condensation assay. As shown on , JMJD6 knock-down rescued primary neurons from the toxicity of mutant Htt observed upon co-transfection with expanded Htt and non-targeting control siRNA.
Discussion
Abnormal interactions of polyQ-expanded Htt are frequently observed in HD models, and are thought to play a major role in the disease mechanism.Citation5-14 We focused on Htt/PRMT5 interaction, and demonstrated that an altered interaction of expanded Htt with PRMT5 can affect major cellular functions, such as regulation of gene transcription and RNA splicing, providing a molecular mechanism for the previously observed disruption of these pathways in HD.Citation15-26
Our experiments demonstrate Htt and PRMT5 interaction in cell lines and in human HD brain, and suggest a change in stoichiometry of the PRMT5/Htt complexes in the presence of expanded Htt. In transfected cells we observed an increase in the amounts of expanded Htt (compared to normal Htt) found within PRMT5 complexes (). In a reverse IP using Htt-specific antibody, we detected more PRMT5 co-precipitated with polyQ-expanded Htt compared to normal Htt, also suggesting an increased affinity of expanded Htt toward PRMT5 (). Finally, in human brain, PRMT5/Htt interaction was only detected in HD tissues, but not in normal controls (). This may indicate of altered stoichiometry or of a more transient nature of PRMT5/Htt complexes in normal tissues. Increased levels of endogenous PRMT5 were observed in STHdh cells expressing expanded Htt, compared to normal cells; however there was no significant change in overall PRMT5 levels between normal and HD brain tissues (data not shown). PRMT5/Htt functional interaction was also confirmed by co-localization of the 2 proteins, primarily in the cytoplasm of primary neurons ().
What may be the functional role of PRMT5/Htt interaction? Our in vitro methylation assay () demonstrated that the normal function of Htt may include the stimulation of PRMT5 activity, which is not achieved to the same extent by expanded Htt possibly due to its conformational change and abnormal interactions. A role of Htt as the facilitator of macromolecular complexes has been suggested previously, e.g. Htt was found to increase H3K27 tri-methylation of polycomb repressive complex 2 (PRC2), and the polyQ region augmented this stimulation.Citation53 PRMT5 was also found interacting with several cofactors, including MEP50, pICln,Citation34,39,42-44 COPR5,Citation41 RioK1Citation40 and Blimp1,Citation29 and has been co-purified with the chromatin remodeling complexes, hSWI/SNF and NURD.Citation31-33 Recently the crystal structure of the human and Xenopus laevis PRMT5:MEP50 complex has been determined, revealing a tight hetero-octameric complex between MEP50 and the PRMT5.Citation43,44 In addition, it was demonstrated that MEP50 is required for PRMT5-catalyzed histone H2A and H4 methyltransferase activity and binds substrates independently.Citation44 In this study we found that Htt interacted with both PRMT5 and MEP50 in transfected HEK293 cells (). We have also detected high molecular weight MEP50 complexes in cells expressing normal Htt. These complexes were absent in cells transfected with expanded Htt fragment, suggesting that expanded Htt may affect MEP50/PRMT5 complex formation, providing a potential mechanism for observed PRMT5 impairment in the presence of mutant Htt. Whether Htt is directly affecting PRMT5 activity, its self-association,Citation54,55 or its interaction with other cofactors, is a subject for further investigation. Another intriguing possibility is that Htt may be a substrate for PRMT5 activity itself. Further studies using mass spectrometry combined with advanced enrichment techniques will determine whether Htt protein undergoes symmetric or/and asymmetric arginine dimethylation, potentially carried out by PRMT5 or other protein methyltransferases.
Further evidence of PRMT5 impairment in the presence of mutant Htt comes from our analysis of sDMA modifications of histones H2A and H4 (known substrates of PRMT5Citation28), which can be specifically detected by modification-specific antibody R3Me2s. In two HD models-primary neurons transfected with expanded Htt and in HD brain-we observed a decrease in sDMA on histones H2A and H4 (). This was also reflected in a reduction of H2A/H4R3Me2s occupancy at the γ-globin and BDNF promoters in HD brain (). Methylation of histones is an important step in epigenetic regulation of transcription.Citation56 Transcriptional dysregulation has been widely implicated in HD pathogenesis,Citation15,19,22,23,25,26 however the mechanism of this dysregulation is still elusive. A recent genome-wide analysis of H3K4 trimethylation in mouse and human HD brain found a correlation between decreased H3K4me3 occupancy and decreased gene expression patterns.Citation48 The authors discovered a specific pattern of H3K4 trimethylation at the promoters of down-regulated in HD genes and suggested targeting of this epigenetic signature as a therapeutic strategy for HD. Our findings of the PRMT5/Htt functional interaction raise the possibility that mutant Htt exerts its well-documented effects on transcription via direct interaction with histone-modifying enzymes, such as PRMT5, and by modulating their activity.
Using ChIP assay we found that H2A/H4R3Me2s occupancy at the BDNF exon II promoter was decreased in HD brain compared to control. Transcription from the exon II BDNF promoter has been previously shown to be reduced in HD mouse and human brain.Citation49 So far PRMT5 has been mostly implicated in transcriptional repression of target genes.Citation31-33 However, the effects of histone methylation are thought to be context-dependent, and the same modifications can be associated with both transcriptional activation and repression possibly due to different effector proteins within the chromatin remodeling complexes at specific promoters.Citation56 Furthermore, a recent analysis of genome-wide pattern of H4R3Me2s revealed that up to 87% of both active and inactive promoters in ES cells were enriched in this mark.Citation57 Thus the impairment of PRMT5 may lead to a widespread dysregulation of both transcriptional activation and repression.
RNA processing and splicing abnormalities has emerged as a molecular mechanism associated with HD based on several expression profiling studies in cells, mouse, yeast and fly models of HD.Citation15-26 Our recent analysis of Htt interactome in striatal cells also demonstrated that expanded Htt complexes were enriched in the proteins related to RNA processing and regulation of translation, and identified several novel Htt interactors within these pathways.Citation18 The more recent collaborative protein profiling study in HD iPS cells also highlighted RNA metabolism as one of the major pathways altered in HDCitation58 (and unpublished data). Arginine methylation is found in many RNA-binding proteins important for mRNP biogenesis and RNA splicing.Citation59 An important group of proteins known to be methylated by PRMT5 are the components of spliceosome, Sm proteins. The sDMA modification on Sm proteins is recognized by Tudor domain protein SMN and is required for their interaction and cytoplasmic assembly of the spliceosome.Citation34,35 Methylation of another PRMT5 substrate, Cajal body marker coilin, is required for localization of SMN to Cajal bodies and pre-mRNA splicing.Citation37,38 In this study, we showed that the symmetric dimethylation of coilin was reduced in HD mouse and human brain, likely due to PRMT5 impairment (). The absence of PRMT5 in the NPCs from PRMT5 knock-out mice has been shown to result in a reduced methylation of Sm proteins and aberrant constitutive and alternative RNA splicing, suggesting that PRMT5 is a master regulator of RNA splicing.Citation60 Further studies are needed to show whether Sm proteins methylation and snRNPs assembly is disrupted in HD models due to PRMT5 deficiency and may underlie the observed splicing abnormalities in HD.
We found that total levels of sDMA on histones and coilin and R3Me2s occupancy at 2 transcriptionally repressed promoters were reduced in HD brain likely due to PRMT5 impairment. Furthermore, increasing the levels of PRMT5/MEP50 complexes by ectopic expression improved the survival of primary neurons expressing mutant Htt (), suggesting that PRMT5 deficiency may mediate, at least in part, mutant Htt toxicity and thus restoring the levels of sDMA may be beneficial for neuronal survival in HD.
The promoter occupancy of another histone methyl-modification, H3K4 trimethylation, was previously found to be reduced as well in HD mouse and human brain.Citation48 This reduction was reversed in primary neurons by a knock-down of an enzyme with proven H3K4Me3 demethylase activity, Jarid1c. The knock-down of Jarid1c also rescued HD phenotypes in primary neurons of BACHD mice and in fly HD model.Citation48 In our study we employed a similar approach to restore the balance of sDMA disrupted in the tested HD systems due to the impairment of PRMT5. We used siRNA knock-down to reduce the levels of JMJD6, the only enzyme known to reverse arginine dimethylation.Citation51,52 Although JMJD6 has no lysine demethylase activity, it is a dual action enzyme: In addition to its arginine demethylase activity, it acts as lysyl-hydroxylase that catalyzes 5-hydroxylation on specific lysine residues of target proteins, such as U2AF65Citation61 and p53.Citation62 We found that the JMJD6 knock-down in primary neurons resulted in an increase of sDMA levels of histones H2A and H4, and reversed the toxic effects of exogenous mutant Htt on transfected neurons (). These data support the notion of the dynamic nature of protein post-translational modifications, and suggest that a delicate balance of protein methylation/demethylation is essential for neuronal survival in HD. Therapeutic strategies restoring this balance may be an effective approach to ameliorate mutant Htt toxicity.
Materials and Methods
Plasmid construction and mutagenesis
Full-length Htt constructs (HD-FL-23/82Q) were described previously.Citation63 Truncated Htt expression constructs N511–8Q/82Q were generated as previously describedCitation64 from the full-length Htt constructs by an introduction of a stop codon after amino acid 511 of Htt. The stop codons were introduced by site-directed mutagenesis using the QuikChange II XL kit (Agilent Technologies) according to the manufacturer's protocol. Htt-N511–8Q/52Q constructs were produced from N511–82Q by random contractions of polyQ repeat in bacterial cells. Htt-N586–20Q and Htt-N586–82Q plasmids were described previously.Citation18 Human PRMT5 cDNA subcloned into pcDNA expression vector was obtained from Origene. The N-terminal FLAG tag was introduced using QuikChange II XL kit (Agilent Technologies). Human MEP50 cDNA subcloned into pcDNA expression vector was obtained from Origene.
Cell culture and transfection
Human embryonic kidney (HEK) 293FT cells were obtained from Invitrogen. Rat striatal ST14 cells were originally derived from rat embryonic striatum.Citation65 Mouse striatal STHdh neuronal progenitor lines were generated in the MacDonald lab from E14 striatal primordia of wild type (Q7) or HdhQ111 knock-in mouse embryos.Citation66 HEK293 and striatal cells were grown in DMEM (with 4.5 g/L D-Glucose, Life Technologies) supplemented with 10% FBS, 100 μg/ml Geneticin, 100 units/ml penicillin and 100 units/ml streptomycin. Primary mouse cortical neurons were prepared as described previously.Citation67 Cortices of CD1 mice at embryonic day 15.5 were dissected out, treated with trypsin (0.05% with EDTA, Life Technologies) and mechanically dissociated. Neurons were resuspended in Neurobasal medium supplemented with B27 (Life Technologies) and plated at 1 × 106 cell/cm2 on poly-D-lysine coated 24 well plates (Corning). Cells were 1 kept in Neurobasal-2 medium supplemented with B27 and 2mM GlutaMAX (Life Technologies) in 5% CO2 at 37°C until 3 the day of the experiment. All cells were transfected using Lipofectamine 2000 reagent (Life Technologies) according to the manufacturer's protocol.
Western blotting and immunoprecipitation
For Western blotting analysis, cells were lysed 48 h after transfection in Triton lysis buffer (50 mM Tris, pH 7.0, 150 mM NaCl, 5 mM EDTA, 50 mM MgCl2, 0.5% Triton X100) supplemented with protease inhibitors (PIC, Protease Inhibitor Cocktail III, Calbiochem). Total cell homogenates from frozen human front superior cortex of normal controls and HD cases were prepared by the Dounce homogenization in Triton lysis buffer, supplemented with 0.5% Na deoxycholate and PIC), followed by centrifugation at 13,000 g. The whole brain homogenates from HD KI 175Q 6 months-old mice were prepared as above. Protein concentrations were estimated using BCA method (BioRad). Lysates were fractionated on NuPAGE 4–12% Bis-Tris polyacrylamide gels (Life Technologies), transferred to nitrocellulose membranes, and probed with primary antibodies for 1 h at room temperature. Immunoblots were developed with peroxidase-conjugated secondary antibodies (GE Healthcare), and enhanced chemi-luminescence (ECL-Plus detection reagent, GE Healthcare). Protein bands were visualized using Molecular Imager Gel Doc XR System (BioRad) and quantified using Image J software. For IP, cells were lysed 48 h after plating or after transfection in Triton lysis buffer, supplemented with PIC, followed by centrifugation at 13,000g. Lysates were pre-cleared by incubating with Protein G-Sepharose beads (GE Healthcare) for 1 h at 4°C, followed by the incubation (ON at 4°C) with primary antibodies and then were incubated with Protein G-Sepharose for 1 h at 4°C. FLAG IPs were carried out using anti-FLAG M2 Affinity Gel (Sigma) according to the manufacturer's protocol. The IPs were washed 3 times with the lysis buffer, and protein complexes were eluted from the beads with 2xSDS Laemmli sample buffer (Bio-Rad), and fractionated on SDS-PAGE as described above.
Antibodies
Htt MAB5490 (against residues 115–129 of Htt) and Htt MAB2166 antibody (against residues 181–810 of Htt) were from EMD Millipore; MW1 monoclonal antibody to expanded polyQ was a gift from Paul Patterson;Citation68 PRMT5, SYM10 and JMJD6 antibodies were from EMD Millipore; FLAG M2 antibody was from Sigma; β-tubulin and SMN antibodies were from Santa Cruz Biothechology; MEP50 antibody, antibodies to histones H4 and H2A and coilin were from Cell Signaling Technology; Anti-Histone H4/H2A (symmetric di-methyl R3, R3Me2s) ChIP Grade antibody was from Abcam.
Immunofluorescence
Primary mouse cortical neurons were prepared and transfected as described above, fixed (24 h after transfection) with 4% paraformaldehyde for 15 min, permealized with 0.1% Triton X-100 (Sigma) for 10 min, blocked in 10% normal donkey serum (Sigma) for 30 min, and incubated (1 h, room temperature or overnight at 4°C) with the following antibodies: for Htt and PRMT5 co-localization study-with the mouse monoclonal 2166 antibody to Htt and rabbit polyclonal antibody to PRMT5, followed by donkey anti-mouse Alexa Flour-555 (Life Technologies) and donkey anti-rabbit Alexa Flour-488 (Life Technologies); for the detection of JMJD6-with the rabbit polyclonal antibody to JMJD6, followed by donkey anti-rabbit Alexa Flour −555; for the detection of H2A/H4 R3Me2s symmetric dimethylation-with the mouse monoclonal 2166 antibody to Htt and rabbit H4R3me2s antibody, followed by donkey anti-mouse Alexa Flour-555 (Life Technologies) and donkey anti-rabbit Alexa Flour-488 (Life Technologies). Quantification was performed using ZEN 2012 software. 100 transfected cells were quantitated for each condition.
In vitro methylation assay
FLAG-agarose beads following the IPs from ST14 cells transfected with FLAG-PRMT5 alone, or in combination with Htt N511 constructs, were used as an enzyme source in the in vitro methylation assays. Washed beads were incubated with 1 μg of purified histone H2A and H4 (Roche) and 2 μCi of
S-adenosyl-L-methyl-3H-methionine (3H-SAM, GE Healthcare) as the methyl donor in a mixture of 30 μl of the reaction buffer (25 mM NaCl, 25 mM Tris, 10 Mm MgCl2, 0.25 Mm DTT and PIC, pH 8.8) for 2 h at 37°C. Proteins were resolved on a 12% (w/v) SDS-PAGE gel, stained with Coomassie blue and the gel was treated with Amplify (GE Healthcare) to enhance the radioactive signal prior to autoradiography.
ChIP-qPCR analysis
150 mg of human frontal cortex tissue were finely chopped and fixed with 1.5% formaldehyde. Chromatin isolation and IP were carried out using SimpleChIP Plus Enzymatic Chromatin immunoprecipitation Kit (Cell Signaling) according to the manufacturer's protocol, except the protocol was scaled-up 5 times. Enzymatic chromatin digestion resulted in the fragments of about 150–800 base pairs, as assessed by resolving DNA on 1% agarose gel. We used ChIP-grade antibody to symmetrically dimethylated arginine H4R3Me2s (Abcam) for IP. Normal rabbit immunoglobulin (IgG) served as a control. 2% of each sample was saved as an input before IP. After elution with 150 μl of ChIP buffer, 100 μl were saved for the confirmation of ChIP by Western blotting, and the rest of the sample was processed for reversal of cross-link and DNA isolation. DNA was dissolved in 50 μl of deionized water and 2 μl of Input and ChIP samples were used for PCR. For ChIP quantitation we used the previously published primers to amplify the following gene-specific promoters: the P-1 region of the human γ-globin promoter;Citation45 the human BDNF exon 2 and exon 4 promoters.Citation48 Q-PCR was carried out on the QuantStudio™ 12K Flex Real-Time PCR system (Life Technologies) using SYBR Green method with SsoAdvanced Universal SYBR Green Supermix (Bio-Rad). The differences in threshold cycle (ΔCt) between IP and Input samples were calculated and the results were expressed as a percent of the total Input chromatin using the formula: Percent Input = 2% × 2ΔCt
Nuclear condensation cell death assay
To measure the survival of transfected primary neurons in culture, we used an analysis based on the nuclear condensation observed during cell death.Citation18,67,69 Neurons were co-transfected at DIV5 with either normal (FL-Htt-22Q) or mutant (FL-Htt-82Q) full-length Htt, siRNA to JMJD6 or non-targeting control (at 20 nM, ON-TARGET siRNA, Dharmacon) and eGFP (10:1 ratio) using Lipofectamine 2000 (Life Technologies). After 48 h of expression, cells were fixed with 4% paraformaldehyde for 30 min and nuclei were stained with Hoechst 3325 (bis-benzimide, Sigma-Aldritch). Image acquisition was done using the Axiovision imaging software on an Axiovert 100 inverted microscope (Carl Zeiss) using the automated Mozaix function to cover the integral surface of the wells. Analysis and quantification were performed using the Volocity software (Perkin-Elmer). Transfected cells were identified based on GFP fluorescence and the average intensity of the nuclear staining was measured. The percentage of dead cells was calculated as the percentage of cells for which the average intensity of nuclear staining exceeded 200% of the average intensity of healthy untransfected nuclei. Results are shown as a percentage of cell death of transfected cells, and each independent experiment represent the average of 4 wells per condition.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank David Borchelt (University of Florida, Gainesville, FL) for providing us with the Htt N586–82Q expression construct, Michael Hayden (University of British Columbia, Vancouver) for the Htt-N586–20Q expression constructs, Paul Patterson (California Institute of Technology, Pasadena, CA) for MW1 monoclonal antibody and Marcy MacDonald (Massachusetts General Hospital, Boston, MA) for STHdh striatal cell lines.
Related Research Data
References
- A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. The Huntington's Disease Collaborative Research Group. Cell 1993; 72:971-83; PMID:8458085; http://dx.doi.org/10.1016/0092-8674(93)90585-E
- Ross CA, Aylward EH, Wild EJ, Langbehn DR, Long JD, Warner JH, Scahill RI, Leavitt BR, Stout JC, Paulsen JS, et al. Huntington disease: natural history, biomarkers and prospects for therapeutics. Nat Rev Neurol 2014; 10:204-16; PMID:24614516; http://dx.doi.org/10.1038/nrneurol.2014.24
- Ross CA, Tabrizi SJ. Huntington's disease: from molecular pathogenesis to clinical treatment. Lancet Neurol 2011; 10:83-98; PMID:21163446; http://dx.doi.org/10.1016/S1474-4422(10)70245-3
- Walker FO. Huntington's disease. Lancet 2007; 369:218-28; PMID:17240289; http://dx.doi.org/10.1016/S0140-6736(07)60111-1
- Boutell JM, Thomas P, Neal JW, Weston VJ, Duce J, Harper PS, Jones AL. Aberrant interactions of transcriptional repressor proteins with the Huntington's disease gene product, huntingtin. Hum Mol Genet 1999; 8:1647-55; PMID:10441327; http://dx.doi.org/10.1093/hmg/8.9.1647
- Harjes P, Wanker EE. The hunt for huntingtin function: interaction partners tell many different stories. Trends Biochem Sci 2003; 28:425-33; PMID:12932731; http://dx.doi.org/10.1016/S0968-0004(03)00168-3
- Li SH, Cheng AL, Zhou H, Lam S, Rao M, Li H, Li XJ. Interaction of Huntington disease protein with transcriptional activator Sp1. Mol Cell Biol 2002; 22:1277-87; PMID:11839795; http://dx.doi.org/10.1128/MCB.22.5.1277-1287.2002
- Li SH, Gutekunst CA, Hersch SM, Li XJ. Interaction of huntingtin-associated protein with dynactin P150Glued. J Neurosci 1998; 18:1261-9; PMID:9454836
- Li SH, Li XJ. Huntingtin-protein interactions and the pathogenesis of Huntington's disease. Trends Genet 2004; 20:146-54; PMID:15036808; http://dx.doi.org/10.1016/j.tig.2004.01.008
- Lim J, Crespo-Barreto J, Jafar-Nejad P, Bowman AB, Richman R, Hill DE, Orr HT, Zoghbi HY. Opposing effects of polyglutamine expansion on native protein complexes contribute to SCA1. Nature 2008; 452:713-8; PMID:18337722; http://dx.doi.org/10.1038/nature06731
- Nucifora FC, Jr., Sasaki M, Peters MF, Huang H, Cooper JK, Yamada M, Takahashi H, Tsuji S, Troncoso J, Dawson VL, et al. Interference by huntingtin and atrophin-1 with cbp-mediated transcription leading to cellular toxicity. Science 2001; 291:2423-8; PMID:11264541; http://dx.doi.org/10.1126/science.1056784
- Steffan JS, Kazantsev A, Spasic-Boskovic O, Greenwald M, Zhu YZ, Gohler H, Wanker EE, Bates GP, Housman DE, Thompson LM. The Huntington's disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc Natl Acad Sci U S A 2000; 97:6763-8; PMID:10823891; http://dx.doi.org/10.1073/pnas.100110097
- Subramaniam S, Sixt KM, Barrow R, Snyder SH. Rhes, a striatal specific protein, mediates mutant-huntingtin cytotoxicity. Science 2009; 324:1327-30; PMID:19498170; http://dx.doi.org/10.1126/science.1172871
- Zoghbi HY, Orr HT. Pathogenic mechanisms of a polyglutamine-mediated neurodegenerative disease, spinocerebellar ataxia type 1. J Biol Chem 2009; 284:7425-9; PMID:18957430; http://dx.doi.org/10.1074/jbc.R800041200
- Hodges A, Strand AD, Aragaki AK, Kuhn A, Sengstag T, Hughes G, Elliston LA, Hartog C, Goldstein DR, Thu D, et al. Regional and cellular gene expression changes in human Huntington's disease brain. Hum Mol Genet 2006; 15:965-77; PMID:16467349; http://dx.doi.org/10.1093/hmg/ddl013
- Lee J, Hwang YJ, Kim KY, Kowall NW, Ryu H. Epigenetic mechanisms of neurodegeneration in Huntington's disease. Neurotherapeutics 2013; 10:664-76; PMID:24006238; http://dx.doi.org/10.1007/s13311-013-0206-5
- Ma B, Culver BP, Baj G, Tongiorgi E, Chao MV, Tanese N. Localization of BDNF mRNA with the Huntington's disease protein in rat brain. Mol Neurodegener 2010; 5:22; PMID:20507609; http://dx.doi.org/10.1186/1750-1326-5-22
- Ratovitski T, Chighladze E, Arbez N, Boronina T, Herbrich S, Cole RN, Ross CA. Huntingtin protein interactions altered by polyglutamine expansion as determined by quantitative proteomic analysis. Cell Cycle 2012; 11:2006-21; PMID:22580459; http://dx.doi.org/10.4161/cc.20423
- Runne H, Regulier E, Kuhn A, Zala D, Gokce O, Perrin V, Sick B, Aebischer P, Deglon N, Luthi-Carter R. Dysregulation of gene expression in primary neuron models of Huntington's disease shows that polyglutamine-related effects on the striatal transcriptome may not be dependent on brain circuitry. J Neurosci 2008; 28:9723-31; PMID:18815258; http://dx.doi.org/10.1523/JNEUROSCI.3044-08.2008
- Savas JN, Ma B, Deinhardt K, Culver BP, Restituito S, Wu L, Belasco JG, Chao MV, Tanese N. A role for huntington disease protein in dendritic RNA granules. J Biol Chem 2010; 285:13142-53; PMID:20185826; http://dx.doi.org/10.1074/jbc.M110.114561
- Savas JN, Makusky A, Ottosen S, Baillat D, Then F, Krainc D, Shiekhattar R, Markey SP, Tanese N. Huntington's disease protein contributes to RNA-mediated gene silencing through association with Argonaute and P bodies. Proc Natl Acad Sci U S A 2008; 105:10820-5; PMID:18669659; http://dx.doi.org/10.1073/pnas.0800658105
- Sipione S, Rigamonti D, Valenza M, Zuccato C, Conti L, Pritchard J, Kooperberg C, Olson JM, Cattaneo E. Early transcriptional profiles in huntingtin-inducible striatal cells by microarray analyses. Hum Mol Genet 2002; 11:1953-65; PMID:12165557; http://dx.doi.org/10.1093/hmg/11.17.1953
- Tauber E, Miller-Fleming L, Mason RP, Kwan W, Clapp J, Butler NJ, Outeiro TF, Muchowski PJ, Giorgini F. Functional gene expression profiling in yeast implicates translational dysfunction in mutant huntingtin toxicity. J Biol Chem 2010; 286:410-9; PMID:21044956; http://dx.doi.org/10.1074/jbc.M110.101527
- Valor LM. Transcription, Epigenetics and Ameliorative Strategies in Huntington's Disease: a Genome-Wide Perspective. Mol Neurobiol 2015; 51:406-23; PMID:24788684
- Wyttenbach A, Swartz J, Kita H, Thykjaer T, Carmichael J, Bradley J, Brown R, Maxwell M, Schapira A, Orntoft TF, Kato K, Rubinsztein DC. Polyglutamine expansions cause decreased CRE-mediated transcription and early gene expression changes prior to cell death in an inducible cell model of Huntington's disease. Hum Mol Genet 2001; 10:1829-45; PMID:11532992; http://dx.doi.org/10.1093/hmg/10.17.1829
- Zucker B, Luthi-Carter R, Kama JA, Dunah AW, Stern EA, Fox JH, Standaert DG, Young AB, Augood SJ. Transcriptional dysregulation in striatal projection-and interneurons in a mouse model of Huntington's disease: neuronal selectivity and potential neuroprotective role of HAP1. Hum Mol Genet 2005; 14:179-89; PMID:15548548; http://dx.doi.org/10.1093/hmg/ddi014
- Blackwell E, Ceman S. Arginine methylation of RNA-binding proteins regulates cell function and differentiation. Mol Reprod Dev 2012; 79:163-75; PMID:22345066; http://dx.doi.org/10.1002/mrd.22024
- Wolf SS. The protein arginine methyltransferase family: an update about function, new perspectives and the physiological role in humans. Cell Mol Life Sci 2009; 66:2109-21; PMID:19300908; http://dx.doi.org/10.1007/s00018-009-0010-x
- Ancelin K, Lange UC, Hajkova P, Schneider R, Bannister AJ, Kouzarides T, Surani MA. Blimp1 associates with Prmt5 and directs histone arginine methylation in mouse germ cells. Nat Cell Biol 2006; 8:623-30; PMID:16699504; http://dx.doi.org/10.1038/ncb1413
- Branscombe TL, Frankel A, Lee JH, Cook JR, Yang Z, Pestka S, Clarke S. PRMT5 (Janus kinase-binding protein 1) catalyzes the formation of symmetric dimethylarginine residues in proteins. J Biol Chem 2001; 276:32971-6; PMID:11413150; http://dx.doi.org/10.1074/jbc.M105412200
- Le Guezennec X, Vermeulen M, Brinkman AB, Hoeijmakers WA, Cohen A, Lasonder E, Stunnenberg HG. MBD2/NuRD and MBD3/NuRD, two distinct complexes with different biochemical and functional properties. Mol Cell Biol 2006; 26:843-51; PMID:16428440; http://dx.doi.org/10.1128/MCB.26.3.843-851.2006
- Pal S, Vishwanath SN, Erdjument-Bromage H, Tempst P, Sif S. Human SWI/SNF-associated PRMT5 methylates histone H3 arginine 8 and negatively regulates expression of ST7 and NM23 tumor suppressor genes. Mol Cell Biol 2004; 24:9630-45; PMID:15485929; http://dx.doi.org/10.1128/MCB.24.21.9630-9645.2004
- Pal S, Yun R, Datta A, Lacomis L, Erdjument-Bromage H, Kumar J, Tempst P, Sif S. mSin3A/histone deacetylase 2-and PRMT5-containing Brg1 complex is involved in transcriptional repression of the Myc target gene cad. Mol Cell Biol 2003; 23:7475-87; PMID:14559996; http://dx.doi.org/10.1128/MCB.23.21.7475-7487.2003
- Friesen WJ, Paushkin S, Wyce A, Massenet S, Pesiridis GS, Van Duyne G, Rappsilber J, Mann M, Dreyfuss G. The methylosome, a 20S complex containing JBP1 and pICln, produces dimethylarginine-modified Sm proteins. Mol Cell Biol 2001; 21:8289-300; PMID:11713266; http://dx.doi.org/10.1128/MCB.21.24.8289-8300.2001
- Meister G, Eggert C, Buhler D, Brahms H, Kambach C, Fischer U. Methylation of Sm proteins by a complex containing PRMT5 and the putative U snRNP assembly factor pICln. Curr Biol 2001; 11:1990-4; PMID:11747828; http://dx.doi.org/10.1016/S0960-9822(01)00592-9
- Jansson M, Durant ST, Cho EC, Sheahan S, Edelmann M, Kessler B, La Thangue NB. Arginine methylation regulates the p53 response. Nat Cell Biol 2008; 10:1431-9; PMID:19011621; http://dx.doi.org/10.1038/ncb1802
- Boisvert FM, Cote J, Boulanger MC, Cleroux P, Bachand F, Autexier C, Richard S. Symmetrical dimethylarginine methylation is required for the localization of SMN in Cajal bodies and pre-mRNA splicing. J Cell Biol 2002; 159:957-69; PMID:12486110; http://dx.doi.org/10.1083/jcb.200207028
- Hebert MD, Shpargel KB, Ospina JK, Tucker KE, Matera AG. Coilin methylation regulates nuclear body formation. Dev Cell 2002; 3:329-37; PMID:12361597; http://dx.doi.org/10.1016/S1534-5807(02)00222-8
- Friesen WJ, Wyce A, Paushkin S, Abel L, Rappsilber J, Mann M, Dreyfuss G. A novel WD repeat protein component of the methylosome binds Sm proteins. J Biol Chem 2002; 277:8243-7; PMID:11756452; http://dx.doi.org/10.1074/jbc.M109984200
- Guderian G, Peter C, Wiesner J, Sickmann A, Schulze-Osthoff K, Fischer U, Grimmler M. RioK1, a new interactor of protein arginine methyltransferase 5 (PRMT5), competes with pICln for binding and modulates PRMT5 complex composition and substrate specificity. J Biol Chem 2010; 286:1976-86; PMID:21081503; http://dx.doi.org/10.1074/jbc.M110.148486
- Lacroix M, El Messaoudi S, Rodier G, Le Cam A, Sardet C, Fabbrizio E. The histone-binding protein COPR5 is required for nuclear functions of the protein arginine methyltransferase PRMT5. EMBO Rep 2008; 9:452-8; PMID:18404153; http://dx.doi.org/10.1038/embor.2008.45
- Wilczek C, Chitta R, Woo E, Shabanowitz J, Chait BT, Hunt DF, Shechter D. Protein arginine methyltransferase Prmt5-Mep50 methylates histones H2A and H4 and the histone chaperone nucleoplasmin in Xenopus laevis eggs. J Biol Chem 2011; 286:42221-31; PMID:22009756; http://dx.doi.org/10.1074/jbc.M111.303677
- Antonysamy S, Bonday Z, Campbell RM, Doyle B, Druzina Z, Gheyi T, Han B, Jungheim LN, Qian Y, Rauch C, et al. Crystal structure of the human PRMT5:MEP50 complex. Proc Natl Acad Sci U S A 2012; 109:17960-5; PMID:23071334; http://dx.doi.org/10.1073/pnas.1209814109
- Ho MC, Wilczek C, Bonanno JB, Xing L, Seznec J, Matsui T, Carter LG, Onikubo T, Kumar PR, Chan MK, et al. Structure of the arginine methyltransferase PRMT5-MEP50 reveals a mechanism for substrate specificity. PLoS One 2013; 8:e57008; PMID:23451136; http://dx.doi.org/10.1371/journal.pone.0057008
- Zhao Q, Rank G, Tan YT, Li H, Moritz RL, Simpson RJ, Cerruti L, Curtis DJ, Patel DJ, Allis CD, et al. PRMT5-mediated methylation of histone H4R3 recruits DNMT3A, coupling histone and DNA methylation in gene silencing. Nat Struct Mol Biol 2009; 16:304-11; PMID:19234465; http://dx.doi.org/10.1038/nsmb.1568
- Fabbrizio E, El Messaoudi S, Polanowska J, Paul C, Cook JR, Lee JH, Negre V, Rousset M, Pestka S, Le Cam A, et al. Negative regulation of transcription by the type II arginine methyltransferase PRMT5. EMBO Rep 2002; 3:641-5; PMID:12101096; http://dx.doi.org/10.1093/embo-reports/kvf136
- Aggarwal P, Vaites LP, Kim JK, Mellert H, Gurung B, Nakagawa H, Herlyn M, Hua X, Rustgi AK, McMahon SB, et al. Nuclear cyclin D1/CDK4 kinase regulates CUL4 expression and triggers neoplastic growth via activation of the PRMT5 methyltransferase. Cancer Cell 2010; 18:329-40; PMID:20951943; http://dx.doi.org/10.1016/j.ccr.2010.08.012
- Vashishtha M, Ng CW, Yildirim F, Gipson TA, Kratter IH, Bodai L, Song W, Lau A, Labadorf A, Vogel-Ciernia A, et al. Targeting H3K4 trimethylation in Huntington disease. Proc Natl Acad Sci U S A 2013; 110:E3027-36; PMID:23872847; http://dx.doi.org/10.1073/pnas.1311323110
- Zuccato C, Marullo M, Conforti P, MacDonald ME, Tartari M, Cattaneo E. Systematic assessment of BDNF and its receptor levels in human cortices affected by Huntington's disease. Brain Pathol 2008; 18:225-38; PMID:18093249; http://dx.doi.org/10.1111/j.1750-3639.2007.00111.x
- Brahms H, Meheus L, de Brabandere V, Fischer U, Luhrmann R. Symmetrical dimethylation of arginine residues in spliceosomal Sm protein B/B' and the Sm-like protein LSm4, and their interaction with the SMN protein. RNA 2001; 7:1531-42; PMID:11720283; http://dx.doi.org/10.1017/S135583820101442X
- Chang B, Chen Y, Zhao Y, Bruick RK. JMJD6 is a histone arginine demethylase. Science 2007; 318:444-7; PMID:17947579; http://dx.doi.org/10.1126/science.1145801
- Hahn P, Bose J, Edler S, Lengeling A. Genomic structure and expression of Jmjd6 and evolutionary analysis in the context of related JmjC domain containing proteins. BMC Genomics 2008; 9:293; PMID:18564434; http://dx.doi.org/10.1186/1471-2164-9-293
- Seong IS, Woda JM, Song JJ, Lloret A, Abeyrathne PD, Woo CJ, Gregory G, Lee JM, Wheeler VC, Walz T, et al. Huntingtin facilitates polycomb repressive complex 2. Hum Mol Genet 2009; 19:573-83; PMID:19933700; http://dx.doi.org/10.1093/hmg/ddp524
- Krause CD, Yang ZH, Kim YS, Lee JH, Cook JR, Pestka S. Protein arginine methyltransferases: evolution and assessment of their pharmacological and therapeutic potential. Pharmacol Ther 2007; 113:50-87; PMID:17005254; http://dx.doi.org/10.1016/j.pharmthera.2006.06.007
- Rho J, Choi S, Seong YR, Cho WK, Kim SH, Im DS. Prmt5, which forms distinct homo-oligomers, is a member of the protein-arginine methyltransferase family. J Biol Chem 2001; 276:11393-401; PMID:11152681; http://dx.doi.org/10.1074/jbc.M008660200
- Greer EL, Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet 2012; 13:343-57; PMID:22473383; http://dx.doi.org/10.1038/nrg3173
- Girardot M, Hirasawa R, Kacem S, Fritsch L, Pontis J, Kota SK, Filipponi D, Fabbrizio E, Sardet C, Lohmann F, et al. PRMT5-mediated histone H4 arginine-3 symmetrical dimethylation marks chromatin at G + C-rich regions of the mouse genome. Nucleic Acids Res 2013; 42:235-48; PMID:24097435; http://dx.doi.org/10.1093/nar/gkt884
- Induced pluripotent stem cells from patients with Huntington's disease show CAG-repeat-expansion-associated phenotypes. Cell Stem Cell 2012; 11:264-78; PMID:22748968; http://dx.doi.org/10.1016/j.stem.2012.04.027
- Yu MC. The Role of Protein Arginine Methylation in mRNP Dynamics. Mol Biol Int 2011; 2011:163827; PMID:22091396; http://dx.doi.org/10.4061/2011/163827
- Bezzi M, Teo SX, Muller J, Mok WC, Sahu SK, Vardy LA, Bonday ZQ, Guccione E. Regulation of constitutive and alternative splicing by PRMT5 reveals a role for Mdm4 pre-mRNA in sensing defects in the spliceosomal machinery. Genes Dev 2013; 27:1903-16; PMID:24013503; http://dx.doi.org/10.1101/gad.219899.113
- Webby CJ, Wolf A, Gromak N, Dreger M, Kramer H, Kessler B, Nielsen ML, Schmitz C, Butler DS, Yates JR, 3rd, et al. Jmjd6 catalyses lysyl-hydroxylation of U2AF65, a protein associated with RNA splicing. Science 2009; 325:90-3; PMID:19574390; http://dx.doi.org/10.1126/science.1175865
- Wang F, He L, Huangyang P, Liang J, Si W, Yan R, Han X, Liu S, Gui B, Li W, et al. JMJD6 promotes colon carcinogenesis through negative regulation of p53 by hydroxylation. PLoS Biol 2014; 12:e1001819; PMID:24667498; http://dx.doi.org/10.1371/journal.pbio.1001819
- Cooper JK, Schilling G, Peters MF, Herring WJ, Sharp AH, Kaminsky Z, Masone J, Khan FA, Delanoy M, Borchelt DR, et al. Truncated N-terminal fragments of huntingtin with expanded glutamine repeats form nuclear and cytoplasmic aggregates in cell culture. Hum Mol Genet 1998; 7:783-90; PMID:9536081; http://dx.doi.org/10.1093/hmg/7.5.783
- Ratovitski T, Gucek M, Jiang H, Chighladze E, Waldron E, D'Ambola J, Hou Z, Liang Y, Poirier MA, Hirschhorn RR, et al. Mutant huntingtin N-terminal fragments of specific size mediate aggregation and toxicity in neuronal cells. J Biol Chem 2009; 284:10855-67; PMID:19204007; http://dx.doi.org/10.1074/jbc.M804813200
- Cattaneo E, Conti L. Generation and characterization of embryonic striatal conditionally immortalized ST14A cells. J Neurosci Res 1998; 53:223-34; PMID:9671979; http://dx.doi.org/10.1002/(SICI)1097-4547(19980715)53:2%3c223::AID-JNR11%3e3.0.CO;2-7
- Trettel F, Rigamonti D, Hilditch-Maguire P, Wheeler VC, Sharp AH, Persichetti F, Cattaneo E, MacDonald ME. Dominant phenotypes produced by the HD mutation in STHdh(Q111) striatal cells. Hum Mol Genet 2000; 9:2799-809; PMID:11092756; http://dx.doi.org/10.1093/hmg/9.19.2799
- Nucifora LG, Burke KA, Feng X, Arbez N, Zhu S, Miller J, Yang G, Ratovitski T, Delannoy M, Muchowski PJ, et al. Identification of novel potentially toxic oligomers formed in vitro from mammalian-derived expanded huntingtin exon-1 protein. J Biol Chem 2012; 287:16017-28; PMID:22433867; http://dx.doi.org/10.1074/jbc.M111.252577
- Ko J, Ou S, Patterson PH. New anti-huntingtin monoclonal antibodies: implications for huntingtin conformation and its binding proteins. Brain Res Bull 2001; 56:319-29; PMID:11719267; http://dx.doi.org/10.1016/S0361-9230(01)00599-8
- Watkin EE, Arbez N, Waldron-Roby E, O'Meally R, Ratovitski T, Cole RN, Ross CA. Phosphorylation of mutant huntingtin at serine 116 modulates neuronal toxicity. PLoS One 2014; 9:e88284; PMID:24505464; http://dx.doi.org/10.1371/journal.pone.0088284