
Abstract
p53 tumor-suppressor gene is a master transcription factor which controls cell cycle progression and apoptosis. killin was discovered as one of the p53 target genes implicated in S-phase control coupled to cell death. Due to its extreme proximity to pten tumor-suppressor gene on human chromosome 10, changes in epigenetic modification of killin have also been linked to Cowden syndrome as well as other human cancers. Previous studies revealed that Killin is a high-affinity DNA-binding protein with preference to single-stranded DNA, and it inhibits DNA synthesis in vitro and in vivo. Here, co-localization studies of RFP-Killin with either GFP-PCNA or endogenous single-stranded DNA binding protein RPA during S-phase show that Killin always adopts a mutually exclusive punctuated nuclear expression pattern with the 2 accessory proteins in DNA replication. In contrast, when cells are not in S-phase, RFP-Killin largely congregates in the nucleolus where rRNA transcription normally occurs. Both of these cell cycle specific localization patterns of RFP-Killin are stable under high salt condition, consistent with Killin being tightly associated with nucleic acids within cell nuclei. Together, these cell biological results provide a molecular basis for Killin in competitively inhibiting the formation of DNA replication forks during S-phase, as well as potentially negatively regulate RNA synthesis during other cell cycle phases.
Introduction
p53 is the most frequently mutated, disrupted, and/or allelically lost tumor suppressor gene.Citation1-3 The loss of p53 function in 50–70% of carcinomas and 50% of all human tumors is a key initiating or tumor-promoting event.Citation4,5 In response to genotoxic stress, p53 is stabilized and translocated to the nucleus where p53 transactivates target genes responsible for mediating p53-dependent responses. These genes include those that induce cell cycle arrest, DNA repair, senescence, anti-angiogenesis, and apoptosis.Citation6-9 Activation of cell cycle arrest by p53 results predominantly from the induction of p21WAF1 that mediates G1-S checkpoint arrest and through GADD45 and 14–3–3 proteins for G2-M checkpoint control.Citation10-13
Studies over the last decade have implicated p53-dependent apoptosis as being critical for suppression of tumorigenesis.Citation4 Many p53 target genes play direct roles in p53-dependent apoptosis including the death receptor, Fas; the pro-apoptotic Bcl−2 family members, Bax, Noxa, and Puma; proteins important for p53 hyper-activation, p53AIP1 and p53DINP1; the zinc-finger protein, PAG608; the KH-RNA binding domain containing protein, MCG10; PIG3, Pidd, p53RDL1, mRTVP-1, mtCLIC/CLIC4, PAC1, and NDRG1.Citation8,14-17 In fact majority of these genes were discovered by Differential Display technology that we pioneered, yet the complete network of genes responsible for mediating p53-dependent apoptosis remains unclear.
Using comprehensive Fluorescent Differential Display (FDD) screening strategy, we had found a number of new p53 target genes, including a novel gene that we dubbed killin.Citation14-17 killin is encoded by a single exon located on human chromosome 10 within 140 bp from another major tumor suppressor gene, pTEN.Citation17 The 140 bp intergenic region contains a divergent promoter which drives the p53-dependent expression of killin and largely constitutive expression of pTEN.Citation17 Epigenetic modifications of killin have also been linked to Cowden syndrome as well as other human cancers.Citation18,30-33
Genetic screen and biochemical analysis demonstrate that Killin is a high-affinity DNA-binding protein, which potently inhibits DNA synthesis in vitro and triggers S-phase arrest prior to apoptosis in vivo.Citation17 Based on the preference of Killin to bind single-stranded DNA (ssDNA) and mutually exclusive nuclear localization patterns of RFP-Killin and newly formed replication forks marked by BrdU labeling, we hypothesized that Killin may inhibit DNA synthesis by competitively inhibiting single-stranded DNA binding protein RPA and PCNA loading to the nascent replication forks.Citation17
In this study, we set out to test this hypothesis by co-localization studies of RFP-Killin with either GFP-PCNA or endogenous RPA during S-phase. We show that indeed as expected, Killin always adopts a mutually exclusive punctuated nuclear expression pattern with the 2 important accessory proteins in DNA replication.Citation19–23 Furthermore, we also notice that in non-S-phase cells, RFP-Killin congregates in the nucleolus where rRNA transcription is known to be most active.Citation24 We provide evidence that throughout the cell cycle, RFP-Killin is tightly associated with the nucleic acids. Together, these results provide a molecular basis for Killin in S-phase arrest, as well as potential negative regulation of RNA synthesis during other cell cycle phases of the cell.
Results
Effect of RFP-Killin on RPA binding to ssDNA during DNA replication
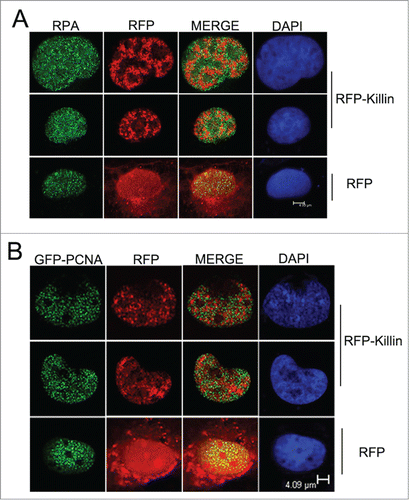
Strong genetic and biochemical evidence presented in our previous studies supports that Killin functions directly as a potent inhibitor of DNA replication, likely through its ability to bind ssDNA.Citation17 The most direct evidence for Killin-mediated S-phase arrest in vivo comes from the visualization of rare cell nuclei with RFP-Killin transiently expressed in CosE5 cells during S-phase, with active DNA replication forks labeled with BrdU.Citation17 CosE5 is a clonally purified COS1 cells with a flatter and more uniformed size opted for cell imaging. The mutually exclusive patterns of DNA replication foci labeled by BrdU with RFP-Killin foci strongly support that Killin inhibits DNA replication during the S-phase. Given the fact that the Kd of Killin to ssDNA template is very similar to that of RPA,Citation17,21 both of which are in the sub μM range, it is possible that Killin could interfere with DNA replication by competitively inhibit RPA binding to ssDNA templates during the onset of DNA replication. To this end, we conducted confocal fluorescent microscopy by looking at the precise RFP-Killin expression pattern in relationship with DNA replication foci marked by immunofluorescent staining of endogenous RPA. RFP-Killin expression vector was transiently transfected into exponentially growing Cos-E5 cells and the expression pattern of RFP-Killin (in Red) in S-phase nuclei marked by RPA antibody (in Green) was visualized 24 hours later by confocal fluorescent microscopy. Consistent with our previous observation with BrdU labeling of DNA replication forks, punctate nuclear signals of RFP-Killin and RPA always showed mutually exclusive patterns ()
Figure 1. RFP-Killin and DNA replication accessory proteins exhibit mutually exclusive nuclear expression pattern during S-phase. (A) S-phase co-localization of RFP-Killin with RPA. The RFP-Killin in-frame fusion protein or RFP control expression vectors were transiently transfected into Cos-E5 cells. Twenty-four hours after the transfection, S phase cells undergoing DNA replication were visualized by punctate staining with anti-RPA70, followed by secondary Alexa Flour488 goat anti-Rabbit IgG (green). Representative images of the co-localization of RPA and RFP-Killin in the nucleus viewed by confocal microscopy. The two proteins showed a mutually exclusive pattern (merge), in contrast to RPA vs RFP control. The scale bar was at 4.93 μm. (B) S-phase co-localization of RFP-Killin with GFP-PCNA. The RFP-Killin or RFP expression vectors were transiently co-transfected with GFP-PCNA into Cos-E5 cells. The S-phase cells undergoing DNA replication as marked by punctate nuclear GFP-PCNA staining were visualized by confocal microscopy. Representative images of the co-localization of GFP-PCNA and RFP-Killin in the nucleus showed a mutually exclusive pattern (merge), in contrast to RFP control. The scale bar was at 4.09 μm.
Effect of RFP-Killin on PCNA loading to DNA replication forks
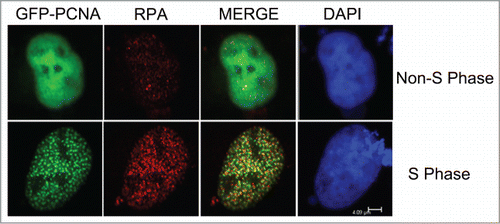
As DNA replication associated proteins, both PCNA and RPA have been shown a high degree of co-location pattern throughout the S-phase.Citation23,25-27 To complement the analysis of the nuclear co-localization of Killin and RPA during S phase described above, expression vectors encoding GFP-PCNA and RFP-Killin were transiently co-transfected into exponentially growing Cos-E5 cells. Twenty-four hours following the transfection, confocal fluorescent microscopy revealed that, like RPA, PCNA also exhibited a mutually exclusive nuclear localization pattern with RFP-Killin (). As key accessory proteins involved in DNA replication, RPA and PCNA bind to replication forks at the onset of S-phase as highly overlapping punctate nuclear foci of DNA replication forks, which was confirmed here by double-labeling with GFP-PCNA (in Green) and endogenous RPA visualized using red fluorescent-labeled secondary antibody (). It should be noted that some discrete faint nuclear RPA signals were also observed in non-S phase cells marked by diffusive nuclear localization pattern of GFP-PCNA (). Some of these loci could be related to DNA repairs reported previously.Citation28
Figure 2. Co-localization of GFP-PCNA with endogenous RPA during S-phase. The GFP-PCNA expression vectors were transiently transfected into Cos-E5 cells. Twenty-four hours after transfection, cells were immune-stained with anti-RPA70 and visualized with Alexa Flour594 Goat Anti-Rabbit IgG (red) and GFP-PCNA (green) by confocal fluorescent microscopy. Co-localization of the 2 proteins (merge) showed largely overlapping signals as yellow colored replication foci. The scale bar was at 4.09 μm.
RFP-Killin is tightly associated with DNA throughout the cell cycle
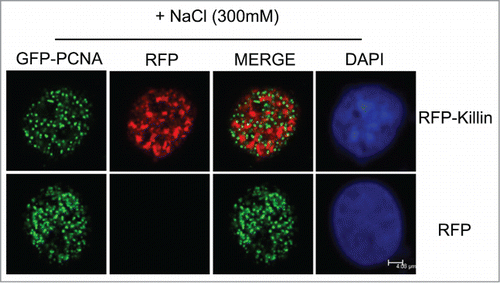
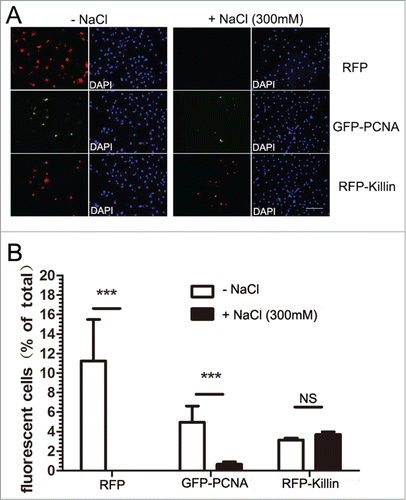
Although RFP-Killin has now been shown to exhibit punctate and mutually exclusive nuclear localization pattern with BrdU, RPA and PCNA, one would argue that RFP-Killin may have nothing to do with DNA synthesis unless RFP-Killin is really associated with DNA in vivo. To this end, we conducted the salt extraction which has been shown to disrupt GFP-PCNA signal in non-S phase nuclei, while GFP-PCNA signal associated with DNA replication forks during S phase remained unperturbed.Citation29 When Cos-E5 cells double transfected with GFP-PCNA and RFP control were treated with 300 mM NaCl, we noticed that none of the S-phase cells marked by punctate nuclear GFP-PCNA foci (replication forks) had any RFP signal compared to untreated cells shown in (). In contrast, when Cos-E5 cells double transfected with GFP-PCNA and RFP-Killin were treated with 300 mM NaCl, we saw mutually exclusive localization pattern of S phase nuclei of both signals (). These results support that unlike RFP protein alone, RFP-Killin is likely associated with DNA, much like GFP-PCNA localized in DNA replication forks during S phase.
Figure 3. GFP-Killin is tightly associated with DNA during S-phase. The RFP-Killin or RFP expression vectors were transiently co-transfected with GFP-PCNA into Cos-E5 cells. Twenty-four hours after transfection, the cells were treated with 300 mM NaCl (in situ salt extractions) to remove proteins that were not bound to DNA. Images were acquired in confocal fluorescent microscopy. GFP-PCNA and RFP-Killin showed a mutually exclusive localization pattern of S phase nuclei (merge) that were salt stable, whereas RFP were completely removed by salt extraction. The scale bar was at 4.08 μm.
RFP-Killin is localized in cell nucleoli in non-S phase cells
Although we have been largely focusing on Killin in S phase and its role as an inhibitor of DNA synthesis, we have also noticed a peculiar localization pattern of RFP-Killin in non-S phase cells. Marked by diffusive GFP-PCNA nuclear localization pattern, we saw RFP-Killin signal congregated in the nucleoli of the non-S phase cells doubly transfected with both expression vectors, while RFP control was randomly distributed through-out the cells (). Upon salt extraction, the diffusive GFP-PCNA signals from these non-S phase cells disappeared, in contrast to that bound to DNA replication forks during S phase (), while RFP-Killin signals in the nucleoli remained intact (). As a negative control, cells transfected with RFP expression vector completely lost the RFP signal upon salt extraction, no matter what cell cycle phases they were in (). Statistical tabulations of multiple fluorescently labeled Cos-E5 cells transfected with RFP, GFP-PCNA, RFP-Killin, respectively before and after salt extraction, indicated that RFP-Killin was always tightly associated with nuclei acids throughout the cell cycle, GFP-PCNA showed S phase-specific DNA binding, while RFP alone was never associated with nucleic acids (). The average ratio of the number of fluorescent cells over that of total cells evaluated for RFP transfected cells was 10% before salt treatment, and none afterwards. In contrast, GFP-PCNA transfected cells had the ratio of 5% and 0.6% without and with salt extraction, respectively; whereas RFP-Killin transfected cells had similar number of fluorescent labeled cells whether they were treated with salt or not.
Figure 4. RFP-Killin is localized in cell nucleoli in non-Sphase cells. (A) The RFP-Killin or RFP expression vectors were transiently co-transfected with GFP-PCNA into Cos-E5 cells for 24 h. Non-S-phase cells marked by diffusive GFP-PCNA nuclear staining were visualized via confocal fluorescent microscopy. Note that RFP-Killin resided in the nucleoli. The scale bar was at 4.09 μm. (B) The RFP-Killin and GFP-PCNA expression vectors were transiently co-transfected into Cos-E5 cells and, treated with 300 mM NaCl (in situ salt extractions) prior to immunostaining. Representative images of RFP-Killin in the nucleoli were acquired by confocal fluorescent microscopy. Note that none of RFP-Killin positive nucleoli showed any GFP-PCNA signals. The scale bar was at 2.16 μm.
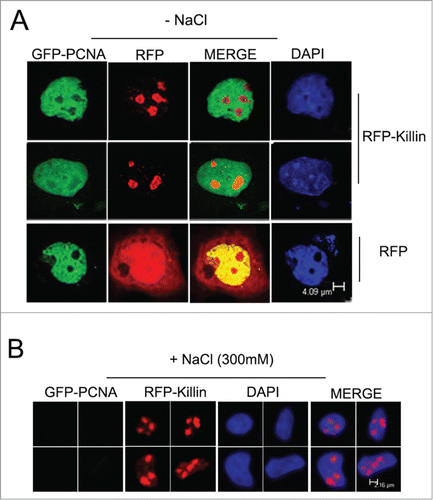
Figure 5. RFP-Killin is tightly associated with DNA throughout the cell cycle. (A) Cos-E5 cells were transfected with RFP or GFP-PCNA and RFP-Killin expression vectors. Changes in the number of fluorescent positive cells without or with 300 mM NaCl extraction were visualized by fluorescence microscopy (X20). The scale bar was at 0.3 cm. (B) Quantification of fluorescence positive cells before and after salt extraction.
Discussion
The elucidation of the entire network of p53 transcriptionally regulated target genes is of great importance for the understanding of the complexities underlying p53-dependent tumor suppression. Through a nonbiased and systematic screen conducted using saturation fluorescent differential display technology, we identified Killin as a p53 target gene involved in p53-mediated S-phase arrest coupled to cell apoptosis. Strong genetic, biochemical and cell biological evidence support that Killin functions directly as a potent inhibitor of DNA replication.Citation17 The duplication of the genome is mediated by a dynamic protein complex called the replisome.Citation34,35 DNA replication starts at the origins of replication where helicase as part of replisome unwinds the DNA duplex, and the resulting single-stranded DNA is stabilized through binding of multiple copies of the heterotrimeric single-strand binding protein RPA. Thereafter, a cascade of events occur, including the loading of the ring-shaped replication factor PCNA that ensure processivity of DNA replication. Both RFP-labeled RPA and GFP-PCNA have been previously co-localized with active replication foci (containing multiple replication forks) visualized by BrdU labeling,Citation22,29 as well as their largely co-localization pattern in S-phase nuclei shown in this study.
To gain more insight into the molecular mechanism by which Killin inhibits DNA replication, we extended our previous finding with BrdU labeling by co-localization studies of RFP-Killin with endogenous RPA and GFP-PCNA. As expected, we show that when exponentially growing Cos-E5 cells where transiently transfected with RFP-Killin, rare S-phase nuclei that exhibit both RFP-Killin (in red fluorescence) and either of the important accessory proteins for DNA replication (in green fluorescence) adopt a mutually exclusive punctuate nuclear expression pattern. It should be noted that although several Killin antibodies have been madeCitation17 and some of them are commercially available, none of them seemed to be able to detect the native Killin protein specifically in vivo (data not shown), thus we have to rely on fluorescent tagged Killin for this study. To further support that Killin may competitively inhibit the initiation of DNA replication fork formation by competitively blocking RPA binding to ssDNA, we show that RFP-Killin not only forms punctate nuclear foci that never overlap with that of RPA or PCNA, these RFP-Killin foci, like S-phase foci marked by GFP-PCNA, are also stable under salt extraction, in contrast to RFP control which is completely removed by 0.3 M NaCl treatment. This important finding indicates that RFP-Killin not only resides in the nucleus, but also is tightly associated with DNA. Conceivably, when RFP-Killin is expressed in the cells that happen to be in S-phase, RFP-Killin would compete against RPA for binding to nascent single-stranded DNA segments, and thus prevent subsequent PCNA loading. Replication forks that are already formed prior to transient expression of RFP-Killin do not appear to be affected, thereby creating a unique mosaic, non-overlapping Red/Green fluorescence pattern. This result is also consistent with our earlier studies showing that the N-terminus of Killin is responsible for DNA binding.Citation17 The tight binding of Killin to nascent DNA replication forks is likely to prevent DNA synthesis machinery from accessing or moving along the template, thus leading to inhibition of DNA synthesis and S phase arrest. The high affinity of Killin to both double- and ssDNA in vitro can now also be reconciled with the beads-on-string distribution pattern of RFP-Killin bound to DNA in S-phase nuclei in vivo. In the contest of Cowden syndrome, a familial cancer syndrome linked to abnormality in both pTEN and killin expression,Citation18,30-33 it is conceivable that the loss of killin expression due to hyper methylation in the promoter region would partially impact DNA damage responses via p53 pathway, thus increasing the susceptibility to cancer. The inability for p53 to induce killin expression would fail to arrest a damage genome entering S-phase due to lack of Killin which would otherwise compete against RPA and loading of PCNA in replication forks. Since pTEN functions as negative regulator of AKT while Killin in p53-mediated S-phase arrest, the extreme close proximity of the 2 genes share by a divergent promoter makes pTEN/killin locus a potent tumor-suppressor dual, and the loss of function in either gene seems to predispose one to increased risk of cancer, as seen in patients with Cowden syndrome.Citation18,30-33
Another interesting finding of our study is the revelation that in non-S-phase cells marked by diffusive expression of GFP-PCNA,Citation22,29 we notice RFP-Killin signals congregate in nucleolus. When these cells were treated with salt extraction prior to fluorescence microscopy, we show that GFP-PCNA signals, in contrast to those being at S-phase described above, are completely lost, whereas RFP-Killin signals remain in the nucleoli of the cells. This surprising finding seems to suggest that when cells are not in S-phase, RFP-Killin may bind to highly abundant rRNA transcription sites in the nucleoli where it may negatively regulate RNA synthesis. Thus Killin may function in multiple cell cycle phases, which is consistent with the static cell cycle profiles following RFP-Killin induction and termination of cell growth previously described.Citation17 Future mechanistic studies will shed light on the role of Killin in RNA synthesis.
Materials and Methods
Cell culture, cell transfection and plasmid
CosE5, derived from clonally purified COS1 cells with a flatter and more uniformed size opted for cell imaging, were obtained from GenHunter (Nashville, TN, USA). CosE5 were maintained in Dulbecco's Modified Eagles Medium (DMEM) with 10% fetal bovine serum (Hyclone) and 1% penicillin-streptomycin (Invitrogen) at 37˚C with 10% CO2. CosE5 cells, plated on glass coverslips, were transiently transfected for 24 hours with GFP-PCNA and either RFP or RFP-Killin using FuGENE-6 (Promega, USA). The GFP-PCNA expression plasmid was a gift from Dr Cristina Cardoso. RFP-Killin expression plasmid was previously described.Citation17 The RFP expression plasmid pDs-Red was obtained from Clonteh.
Antibodies
RPA70 Rabbit polyclonal antibody (Cell Signaling, USA) was used at 1:100 dilution for immune-histochemistry (IHC). Secondary antibodies Alexa Flour488 Goat Anti-Rabbit IgG (H+L) and Alexa Fluor® 594 Goat Anti-Rabbit IgG (H+L) were from Life Technologies (USA) and used at 1:100 dilution for IHC.
Immunofluorescence analysis
Exponentially growing CosE5 cells were transiently transfected on coverslips. After 24 hours, cells were fixed with 4% PFA for 15 min at room temperature and subsequently permeabilized with 0.5% Triton X-100 for 15 min. After blocking with 3% BSA in PBS, cells were incubated with anti-RPA70 overnight at 4°C. After washing with PBS, the secondary antibody Alexa Flour488 Goat Anti-Rabbit IgG (H+L) or Alexa Flour594 Goat Anti-Rabbit IgG (H+L) was added for 1 h at room temperature. Nuclei were stained by DAPI (Bi, Yun Tian, China) for 15 min. After mounting, the slides were visualized by digital images captured with a Leica TCS SP5 II confocal microscope. Selected images were analyzed by Adobe Photoshop CS6.
In situ salt extractions
Salt extractions of non-DNA bound proteins in vivo were performed essentially as previously desribed.Citation29 Briefly, RFP or RFP-Killin were transiently co-transfected and with GFP-PCNA into Cos-E5 cells for 24 h. Cells grown were then permeabilized for about 30 seconds with ice-cold CSK buffer (50 mM NaCl, 250 mM sucrose, 2 mM MgCl2, 2 mM EGTA, 10 mM PIPES, pH 6.8) containing 0.1% Triton-X 100. Thereafter, the cells were further extracted for 1 min with ice-cold phosphate buffer containing 300 mM of NaCl. Then cells were then fixed with 4% PFA for 15 min at room temperature, and subsequently permeabilized with 0.5% Triton X-100 for 15 min. After washing with PBS, cell nuclei were stained with DAPI. Fluorescent microscopy was performed with either a Leica TCS SP5 II confocal microscope or a Leica DM2500 Fluorescent microscope. Selected digital images were analyzed by Adobe Photoshop CS6.
Statistical analysis
The number of fluorescent cells vs total cell numbers viewed (> 100) in at least 2 fields from each experiment were calculated. The values in percentage (fluorescent cells/total cells) were shown as mean values ± SD (shown as error bars). Statistical analysis and comparisons were performed using 2-tailed, unpaired Student t tests.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank instrument support from Center for Cell Growth, Metabolism and Aging (CCGMA) and Kang Han for help with confocal microscopy. We also thank Jiang, Pu for logistic support of this work.
Funding
This work was funded in part by a 973 grant (2012CB910700) from the Chinese Ministry of Education (PL), a grant (81171955) from the Chinese Natural Science Foundation (PL), grants (2011ZX09401–005; 2012AA02A305) from the Chinese Ministry of Science and Technology (PL).
Related Research Data
References
- Levine AJ. p53, the cellular gatekeeper for growth and division. Cell 1997; 88:323-331; PMID:9039259; http://dx.doi.org/10.1016/S0092-8674(00)81871-1
- Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature 2000; 408:307-310; PMID:11099028; http://dx.doi.org/10.1038/35042675
- Vousden K-H, Prives C. p53 and prognosis: New inisights and further complexity. Cell 2005; 120:7-10; PMID:15652475
- Ryan K, Phillips A, Vousden K. Regulation and function of the p53 tumor suppressor protein. Curr Opin Cell Biol 2001; 13:332-7; PMID:11343904; http://dx.doi.org/10.1016/S0955-0674(00)00216-7
- May P, May E. Twenty years of p53 research: Structural and functional aspects of protein. Oncogene 1999; 18:7621-36; PMID:10618702; http://dx.doi.org/10.1038/sj.onc.1203285
- El-Deiry WS. Regulation of p53 downstream genes. Semin Cancer Biol 1998; 8:345-357; PMID:10101800; http://dx.doi.org/10.1006/scbi.1998.0097
- Yu J, Zhang L, Hwang PM, Rago C, Kinzler KW, Vogelstein B. Identification and classification of p53-regulated genes. Proc Natl Acad Sci USA 1999; 96:14517-14522; PMID:10588737; http://dx.doi.org/10.1073/pnas.96.25.14517
- Vousden KH, Lu X. Live or let die: The cell's response to p53. Nat Rev Cancer 2002; 2:594-604; PMID:12154352; http://dx.doi.org/10.1038/nrc864
- Liang P, Pardee AB. Analyzing differential gene expression in cancer. Nat Rev Cancer 2003; 3:869-876; PMID:14668817; http://dx.doi.org/10.1038/nrc1214
- Dulic V, Kaufmann WK, Wilson SJ, Tlsty TD, Lees E, Harper JW, Elledge SJ, Reed SI. p53-dependent inhibition of cyclin-dependent kinase activities in human fibroblasts during radiation-induced G1 arrest. Cell 1994; 76:1013-1023; PMID:8137420; http://dx.doi.org/10.1016/0092-8674(94)90379-4
- El-Deiry WS, Harper J-W, O'Connor P-M, Velculescu VE, Canman C-E, Jackman J, Pietenpol J-A, Burrell M, Hill D-E, Wang Y, et al. WAF1/CIP1 is induced in p53-mediated G1 arrest and apoptosis. Cancer Res 1994; 54:1169-1174; PMID:8118801
- Deng C, Zhang P, Harper JW, Elledge SJ, Leder P. Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell 1995; 82:675-684; PMID:7664346; http://dx.doi.org/10.1016/0092-8674(95)90039-X
- Taylor WR, Stark GR. Regulation of the G2/M transition by p53. Oncogene 2001; 20:1803-1815; PMID:11313928; http://dx.doi.org/10.1038/sj.onc.1204252
- Jackson RS, Cho YJ, Stein S, Liang P. CYFIP2, a direct p53 target, is leptomycin-B sensitive. Cell Cycle 2007; 6(1):95-103; PMID:17245118; http://dx.doi.org/10.4161/cc.6.1.3665
- Jackson RS, Cho YJ, Liang P. TIS11D is a Candidate pro-apoptotic p53 target gene. Cell Cycle 2006; 5(24):2889-93; PMID:17172869; http://dx.doi.org/10.4161/cc.5.24.3539
- Stein S, Thomas EK, Herzog B, Westfall MD, Rocheleau JV, Jackson RS, Wang M, Liang P. NDRG1 is necessary for p53-dependent apoptosis. J Biol Chem 2004; 279(47):48930-40; PMID:15377670; http://dx.doi.org/10.1074/jbc.M400386200
- Cho YJ, Liang P. Killin is a p53-regulated nuclear inhibitor of DNA synthesis. Proc Natl Acad Sci USA 2008; 105(14):5396-401; PMID:18385383; http://dx.doi.org/10.1073/pnas.0705410105
- Kristi L. Bennett , Ph.D, Jessica Mester, MS, CGC, and Charis Eng, MD, Ph.D. Germline epigenetic regulation of KILLIN in cowden and cowden-like syndromes. JAMA 2010; 304(24): 2724-2731; PMID:21177507; http://dx.doi.org/10.1001/jama.2010.1877
- Bravo R. Synthesis of the nuclear protein cyclin (PCNA) and its relationship with DNA replication. Exp Cell Res 1986; 163:287-293; PMID:2869964; http://dx.doi.org/10.1016/0014-4827(86)90059-5
- Moldovan GL, Pfander B, Jentsch S. PCNA, the Maestro of the Replication Fork. Cell 2007; 129(4):665-79; PMID:17512402; http://dx.doi.org/10.1016/j.cell.2007.05.003
- Fanning E, Klimovich V, Nager AR. A dynamic model for replication protein A (RPA) function in DNA processing pathways. Nucleic Acids Res 2006; 34(15):4126-37; PMID:16935876; http://dx.doi.org/10.1093/nar/gkl550
- Leonhardt H, Rahn HP, Weinzierl P, Sporbert A, Cremer T, Zink D, Cardoso MC. Dynamics of DNA replication factories in living cells. J Cell Biol 2000; 149(2):271-80; PMID:10769021; http://dx.doi.org/10.1083/jcb.149.2.271
- Dimitrova DS, Todorov IT, Melendy T, Gilbert DM. Mcm2, but Not RPA, Is a component of the mammalian early G1-phase prereplication complex. J Cell Biol 1999; 146(4):709-22; PMID:10459007; http://dx.doi.org/10.1083/jcb.146.4.709
- Pickard AJ, Bierbach U. The cell's nucleolus: an emerging target for chemotherapeutic intervention. Chem Med Chem 2013; 8(9):1441-9; PMID:23881648; http://dx.doi.org/10.1002/cmdc.201300262
- Bravo R, Macdonald-Bravo H. Existence of two populations of cyclin/proliferating cell nuclear antigen during the cell cycle: association with DNA replication sites. J Cell Biol 1987; 105(4): 1549-54; PMID:2889739; http://dx.doi.org/10.1083/jcb.105.4.1549
- Celis JE, Celis A. Cell cycle-dependent variations in the distribution of the nuclear protein cyclin proliferating cell nuclear antigen in cultured cells: Subdivision of S phase. Proc Natl Acad Sc USA 1985; 82(10):3262-6; PMID:2860667; http://dx.doi.org/10.1073/pnas.82.10.3262
- Cseresnyes Z, Schwarz U, Green CM. Analysis of replication factories in human cells by super-resolution light microscopy. BMC Cell Biol 2009; 10:88; PMID:20015367; http://dx.doi.org/10.1186/1471-2121-10-88
- Diamant N, Hendel A, Vered I, Carell T, Reissner T, de Wind N, Geacinov N, Livneh Z. DNA damage bypass operates in the S and G2 phases of the cell cycle and exhibits differential mutagenicity. Nucleic Acids Res 2012; 40(1):170-80; PMID:21908406; http://dx.doi.org/10.1093/nar/gkr596
- Sporbert A, Gahl A, Ankerhold R, Leonhardt H, Cardoso MC. DNA polymerase clamp shows little turnover at established replication sites but sequential De novo assembly at adjacent origin clusters. Mol Cell 2002; 10(6):1355-65; PMID:12504011; http://dx.doi.org/10.1016/S1097-2765(02)00729-3
- Ng EK, Shin VY, Leung CP, Chan VW, Law FB, Siu MT, Lang BH, Ma ES, Kwong A. Elevation of methylated DNA in KILLIN/PTEN in the plasma of patients with thyroid and/or breast cancer. Onco Targets Ther 2014; 7:2085-92; PMID:25419146
- Bennett KL, Campbell R, Ganapathi S, Zhou M, Rini B, Ganapathi R, Neumann HP, Eng C. Germline and somatic DNA methylation and epigenetic regulation of KILLIN in renal cell carcinoma. Genes Chromosomes Cancer 2011; 50(8):654-61; PMID:21584899; http://dx.doi.org/10.1002/gcc.20887
- Schmidt C. KILLIN gene discovery stirs up research on Cowden syndrome cancers. J Natl Cancer Inst 2011; 103(5):362-4; PMID:21357597; http://dx.doi.org/10.1093/jnci/djr055
- Jelovac D, Park BH. PTEN promoter silencing and Cowden syndrome: the role of epigenetic regulation of KILLIN. JAMA 2010; 304(24):2744-5; PMID:21177512; http://dx.doi.org/10.1001/jama.2010.1863
- Zech J, Dalgaard JZ. Replisome components–post-translational modifications and their effects. Semin Cell Dev Biol 2014; 30:144-53; PMID:24685613; http://dx.doi.org/10.1016/j.semcdb.2014.03.026
- Leman AR, Noguchi E. The replication fork: understanding the eukaryotic replication machinery and the challenges to genome duplication. Genes (Basel) 2013; 4(1):1-32; PMID:23599899; http://dx.doi.org/10.3390/genes4010001