
Abstract
Accumulating evidence suggests that obesity and enhanced inflammatory reactions are predisposing conditions for developing colon cancer. Obesity is associated with high levels of circulating leptin. Leptin is an adipocytokine that is secreted by adipose tissue and modulates immune response and inflammation. Lipid droplets (LD) are organelles involved in lipid metabolism and production of inflammatory mediators, and increased numbers of LD were observed in human colon cancer. Leptin induces the formation of LD in macrophages in a PI3K/mTOR pathway-dependent manner. Moreover, the mTOR is a serine/threonine kinase that plays a key role in cellular growth and is frequently altered in tumors. We therefore investigated the role of leptin in the modulation of mTOR pathway and regulation of lipid metabolism and inflammatory phenotype in intestinal epithelial cells (IEC-6 cells). We show that leptin promotes a dose- and time-dependent enhancement of LD formation. The biogenesis of LD was accompanied by enhanced CXCL1/CINC-1, CCL2/MCP-1 and TGF-β production and increased COX-2 expression in these cells. We demonstrated that leptin-induced increased phosphorylation of STAT3 and AKT and a dose and time-dependent mTORC activation with enhanced phosphorilation of the downstream protein P70S6K protein. Pre-treatment with rapamycin significantly inhibited leptin effects in LD formation, COX-2 and TGF-β production in IEC-6 cells. Moreover, leptin was able to stimulate the proliferation of epithelial cells on a mTOR-dependent manner. We conclude that leptin regulates lipid metabolism, cytokine production and proliferation of intestinal cells through a mechanism largely dependent on activation of the mTOR pathway, thus suggesting that leptin-induced mTOR activation may contribute to the obesity-related enhanced susceptibility to colon carcinoma.
Keywords:
Abbreviations
| mTOR | = | mammalian target of rapamycin |
| Rapa | = | rapamycin |
| PI3K | = | phosphoinositide 3-kinase |
| IEC-6 | = | rat intestinal epithelial cells |
| LD | = | lipid droplets |
Introduction
The incidence of obesity and its associated disorders is increasing markedly worldwide. Numerous results from epidemiological studies have concluded that people with obesity are at increased risk of developing several types of cancer, including adenocarcinoma of the esophagus, colon cancer, endometrial and breast cancer.Citation1 Colon cancer is complex, multifactorial disease that involves the interaction of genetic and environmental factors. Obesity and high-fat diet intake may predispose to the development of colorectal cancer in humans,Citation1,2 a phenomenon also shown in animal models reinforcing the existence of a link between obesity and colorectal cancer.Citation3,4 Different players and interconnected mechanisms are believed to contribute to obesity-driven cancer, including altered levels of adipokines, increased release of hormones, growth-factors and free fatty acids, as well as microbiotal dysbiosis.Citation1,5,6 However, the molecular mechanisms involved in obesity-related tumors are not fully understood.
Adipose tissue is no longer considered to be an inert tissue functioning solely for energy storage, but is emerging as an important player in the regulation of many pathological processes.Citation7 It is also important in the regulation of energy balance and lipid metabolism through the release of adipokines such as leptin, adiponectin, resistin and tumor necrosis factor-α (TNFα).Citation7,8 Leptin is an adipocyte-derived cytokine that links nutritional status with neuroendocrine and immune functions.Citation9 Leptin was the first adipocyte-derived hormone/cytokine described, and the plasma leptin concentration is directly proportional to the amount of adipose tissue.Citation7,10 Studies in cellular and animal models indicate that leptin can potentiate the growth of cancer cells (breast, esophageal, gastric, pancreatic, colorectal, prostate, ovarian and lung carcinoma cell lines).Citation11 Brandon and cols demonstrated that leptin may accelerate tumor growth while leptin deficiency in the absence of obesity attenuates tumor growth.Citation12 Furthermore, the leptin receptor is important for the progression of aberrant crypt foci to colonic tumors, mediated by activation of STAT3 signaling.Citation13
Our group has established that mTOR activity is an important intracellular player in the activation and regulation of lipid metabolism of macrophages induced by leptin.Citation14,15 The mTOR is a serine/threonine kinase that regulates cellular growth by modulating many processes, including protein synthesis, ribosome biogenesis and autophagy.Citation16 mTORC1 pathway, that is inhibited by rapamycin, regulates growth through downstream effectors, the regulators of translation 4EBP1 (eukaryotic translation initiation factor 4E binding protein 1) and S6K1 (ribosomal S6 kinase 1).Citation17 Rapamycin forms a complex with FK506-binding protein (FKBP)12 and mTOR, resulting in potent inhibition of mTOR signaling.Citation18,19 Recently was demonstrated that the mTORC1 complex induce the genes important for glycolysis, the pentose phosphate pathway, and sterol and lipid biosynthesis.Citation20
It is now well established that upregulated lipogenesis is a common phenotype to numerous human carcinomas and has been associated to poor prognosis in breast, prostate and colon cancer.Citation21-23 In different cell systems, including adipocytes and macrophages, intracellular lipids are stored and metabolized in hydrophobic organelles called lipid bodies/lipid droplets (LD). Of note, increased lipid droplet accumulation in liver, macrophage and skeletal muscle is a common feature in obesity and alterations in the regulation of lipid droplet physiology and metabolism has been associated with the pathophysiology of metabolic disease.Citation24 The mechanisms that regulate lipid droplet formation and their functional significance to the cellular biology of inflammation and tumorigenesis are under investigation.Citation25 Recent studies have described increased numbers of LD in several neoplastic processes including adenocarcinoma of colon,Citation26 invasive squamous cervical carcinoma,Citation27 human brain tumorCitation28 and hepatocarcinoma.Citation29 Human colon adenocarcinoma cell lines and colon cancer biopsies from patients have been shown to exhibit an increase in adipose differentiation-related protein (ADRP), that is a major structural protein associated with LD. The increase in ADRP in those cells is accompained by COX-2-enriched LD. Furthermore, inhibition of LD formation inhibits PGE2 production and correlates with diminished cancer cell proliferation in vitro.Citation26
Nevertheless, the effects and signaling pathways triggered by leptin in intestinal epithelial cell lipid metabolism and proliferation has not been fully investigated. Here we evaluate the ability of leptin to trigger LD biogenesis and the inflammatory microenvironment in epithelial cells. Leptin effects on LD formation, COX-2 expression and cell proliferation are strictly dependent on the mTOR pathway. Our results demonstrate new activities of leptin in inflammation and lipid metabolism in epithelial cells.
Results
Leptin triggers lipid droplets biogenesis within rat epithelial cells
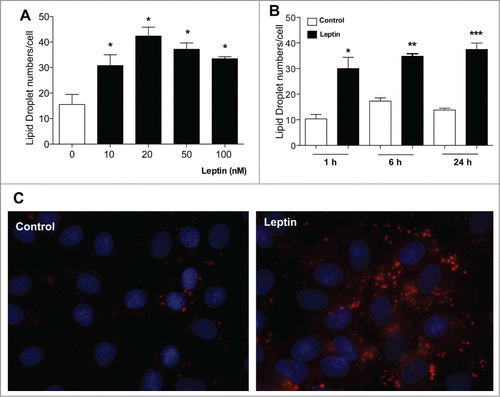
Lipid droplets are emerging as dynamic organelles involved in lipid metabolism and inflammation. Our group demonstrated that leptin directly activates macrophages to form adipose differentiation-related protein (ADRP)-enriched lipid droplets.Citation14 Increased lipid droplet numbers have been described in colon cancer cells where it was shown to be centrally involved in PGE2 synthesis. Inhibition of lipid droplet formation by fatty acid synthase inhibitors diminishes cancer cell proliferation, suggesting that lipid droplets might potentially have implications to the pathogenesis of colon adenocarcinoma.Citation25,26 We hypothesized a role for leptin in regulating epithelial cell lipid droplet biogenesis. As shown in , leptin induced a time and concentration-dependent lipid droplet biogenesis in epithelial cells as quantified by counting the lipid droplets in osmium (OsO4) stained cells. Fig. 1C shows the fluorescence microscopy of lipid droplets staining by oil red O (ORO). The increase in lipid droplets was significant at 1 h and maintained for 24 h after leptin stimulation (). In addition, we analyzed the lipid droplet biogenesis after 6 hours of stimulation with 10, 20, 50 and 100 nM of leptin. Leptin induced an increase in the number of lipid droplets in intestinal epithelial cells at all concentrations used, with an optimal dose of 20 nM. This induction was already significant at the 10 nM dose. Taken together, our results suggest that leptin induced lipid accumulation in epithelial cells and have a role in regulation of lipid metabolism in these cells.
Figure 1. Effects of Leptin on IEC cells lipid droplet formation. IEC-6 cells were treated with different leptin concentrations for 6 h (A) or with 20 nM leptin for different times (B). After incubations the cells were stained with Osmiun tetroxide and the lipid droplets counted. (C) IEC-6 cells were stimulated with 20 nM of leptin in 6 h and the lipid droplets were labeled with Oil Red O and wide field fluorescence images were taken. The results are representative of at least 3 experiments. The symbols (*, ** ;***) represent significant differences from controls according to the Student's T test when p<0.05.
Leptin-induced production of inflammatory mediators
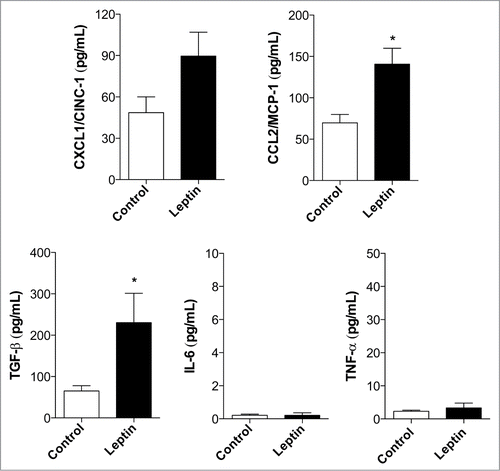
Inflammation is a critical player in the development of both colitis-associated and sporadic colon cancers. Even in cases of sporadic colon cancer, inflammatory mediators have been associated with tumor promotion within the tumor microenvironment.Citation22,30 We investigated the role of leptin in regulating chemokine/cytokine production by epithelial cells. As observed in , leptin (20 nM) was able to trigger the release of CXCL1/CINC-1 and CCL2/MCP-1, chemokines involved in neutrophil and monocyte recruitment, respectively. Leptin also lead to increased production of TGFβ, but not TNF, IL-6 or CCL5/RANTES, by intestinal epithelial cells within 6 h ( and not shown).
Figure 2. Leptin induces inflammatory mediator production. Measurents of CXCL1/CINC-1, CCL2/MCP-1, TGFβ, IL-6 and TNF after 6 hours of incubation with leptin (20 nM) in the culture supernatants. (*) represent significant differences from controls; according to the Student's T test when p < 0.05.
Intracellular Signaling triggered by leptin in epithelial cells
The signaling pathways activated by leptin in intestinal epithelial cells were investigated. As show in , leptin (20 nM, 20 min) induced increased phosphorylation of STAT3 and AKT. The role of mTOR in leptin-induced effects on epithelial cell function has never been addressed. mTOR is a serine/threonine kinase that control translation of specific transcripts by regulating key translational mechanisms and is activated downstream of the PI 3-kinase pathway.Citation31 Several stimuli, mainly signals from the cell environment and nutrient availability contribute to the activation of mTOR. Insulin and other growth factors can activate mTOR by activation of AKT downstream PI3K with subsequent phosphorylation and inhibition of TSC1 and TSC2 (tuberous sclerosis complex 1 and 2), which is an inhibitor of mTOR.Citation22 We have demonstrated that leptin regulates macrophage lipid metabolism and inflammation in a PI3K/mTOR pathway dependent manner.Citation14 Of note, mTOR is a kinase that plays a key role in cellular growth and is frequently overexpressed and activated in tumors.Citation18 As shown in , leptin induced a time- and concentration-dependent phosphorylation of p70S6K in epithelial cells in vitro. p70S6K and 4EBP1 are the main downstream protein targets of mTORC1 phosphorylation and activate the translation initiation by activation of the S6 protein and release of the eIF4E, respectively.Citation32 Maximum effects of leptin on p70S6K phosphorylation was observed at 20nM of leptin for 20 min. We employed rapamycin and everolimus, specific inhibitors of mTOR activity. FK-506, that binds to FKBP12 but does not inhibit mTOR activity, was used as a negative control. Rapamycin and everolimus were able to inhibit the phosphorilation of the p70S6K induced by leptin ( and data not shown).
Figure 3. The leptin effects on IEC cells are dependent on mTOR pathway. (A) IEC cells were incubated with leptin (20nM) for 20 minutes and the westerns blots for the p-STAT3, total STAT3, p-AKT, total AKT and of the cell extracts are shown. (B) IEC cells were incubated with 10, 20, 40 e 100 nM of leptin for 20 minutes (upper panel) and with leptin (20nM) for 0, 5, 10, 20 and 40 minutes (lower panel). After incubations the cells were harvested and Western blottings for phosphorilated P70S6K (p-P70S6K) and total P70S6K were performed. In (C), it is shown the respective densitometries of blots in B.
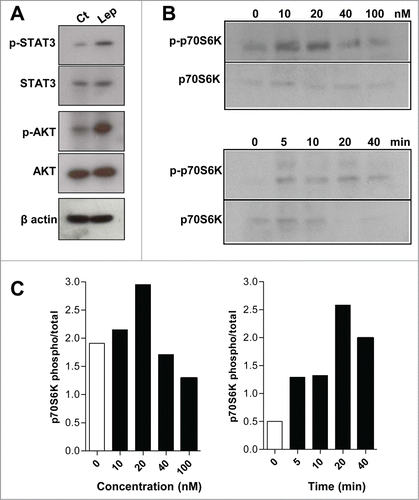
Figure 4. Leptin-induced epithelial cell lipid droplet formation is requisitely dependent on mTOR activity. The IEC cells were incubated with leptin (20 nM, 20 min) in the absence or presence of mTOR inhibitor, rapamycin (20nM). (A) After incubations the cells were harvested and Western blottings for phosphorilated P70S6K (pP70S6K) and total P70S6K were performed. (B) Lipid droplet counting in IEC-6 cells after in vitro incubation for 6h with leptin (20 nM) alone or in the presence of rapamycin (20 nM). (C) The fluorescent area of ORO-labeled lipid droplets were measured with FiJi/ImageJ after incubation for 6h with leptin (20 nM) alone or in the presence of rapamycin (20 nM).*, statistically significant differences between leptin and control; +, statistically significant difference between rapamycin-treated and leptin stimulated groups. (Ct: control; Rapa: Rapamycin; Lep: Leptin; Lep+Rap: Leptin plus rapamycin)
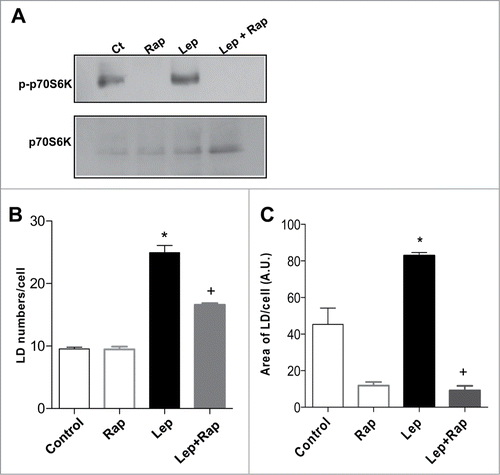
To investigate the functional role of mTOR activation in leptin-induced epithelial cell activation we employed the pretreatment with rapamycin. Rapamycin significantly inhibited lipid droplet formation in epithelial cell stimulated by leptin in vitro, as shown in . The fluorescent area of ORO-labeled lipid droplets were determined, confirming the mTOR dependent effect of leptin in lipid droplet biogenesis (). Collectively, these results indicate that leptin-induced alterations in lipid metabolism were largely dependent on mTOR activation, providing novel links between nutrient regulatory pathways and epithelial cell responses and lipid metabolism.
Leptin modulates COX-2 expression and inflammatory mediator production in mTOR-dependent and independent fashion in IEC-6 cells
Next, we investigated the effects of leptin in COX-2 expression. Different products of arachidonic acid (AA) metabolism are also implicated in carcinogenesis. As much as 80% to 90% of colon carcinomas shown enhanced COX-2 (prostaglandin H synthase 2) expression compared with normal intestinal mucosa.Citation33-35 Several evidences suggest that the inflammatory microenvironment and COX-2 overexpression have a central role in colorectal carcinogenesis and accumulating evidence have established a role for leptin in prostaglandin and leukotriene synthesis by leukocytes and suggest physiological activities for leptin in inflammation.Citation36,37 We demonstrated that leptin induces increased COX-2 expression (). Rapamycin was able to inhibit COX-2 expression induced by leptin in epithelial cells, as can be observed by COX-2 immunofluorescence (). The effect of leptin on TGF-β, but not CCL2/MCP-1, production was also significantly inhibited by rapamycin ().
Figure 5. Leptin stimulates COX-2 overexpression and TGF-β production in a mTOR dependent fashion. (A) IEC-6 cells were incubated with leptin (20nM) for 6 h and the westers blot for COX-2 was performed and quantified. (B) Cells were incubated with leptin (20 nM) in the absence or presence of rapamycin (20 nM). After incubations, cells were processed for Imunofluorescence detection. The cells were labeled with anti-COX-2 antibody and a secondary dylight 488 labeled antibody. (C) The supernatants from incubations with leptin (20nM) treated or not with rapamycin (20 nM) were obtained after 6 h. TGF-β and CCL2/MCP-1 were measured in cell-free supernatants by ELISA. The results are representative of at least 3 experiments; (*) represent significant differences from controls; (+) or (#) represent signifficant differences from leptin treated samples. (Ct: control; Rapa: Rapamycin; Lep: Leptin; Lep+Rap: Leptin plus rapamycin)
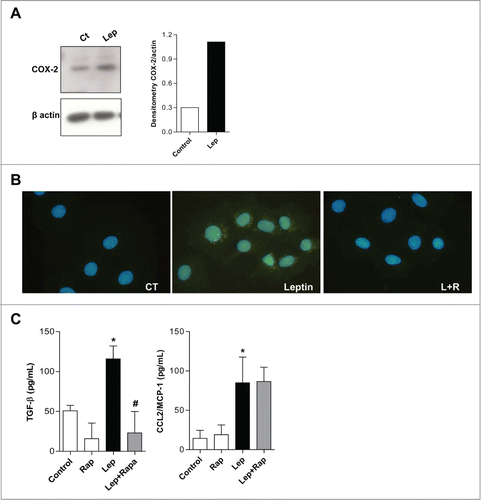
We can, therefore, conclude that leptin may participate in tumor microenviroment inflammatory modulation through COX-2 overexpression and inflammatory cytokine release in a mTOR-dependent and independent fashion, and as such may contribute to the altered tumor-proned microenvironment observed in obesity.
In vitro leptin-induced epithelial cell proliferation depends on mTOR downstream signaling
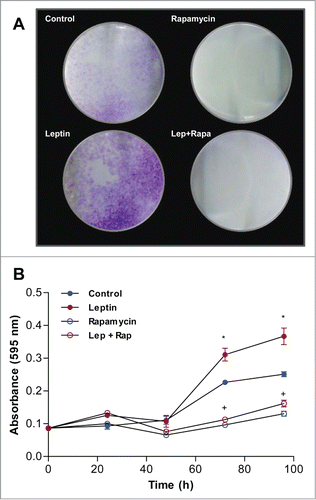
We next examined the effect of leptin on epithelial cell proliferation by staining treated cells with crystal violet. For these experiments, epithelial cells were serum-starved for 16 h followed by treatment with 20 nM of recombinant murine leptin for different time intervals (0, 24, 48, 72 and 96 h or 120 h). In , images from clonogenic assays after 120 h of incubation is shown. Leptin induced increased colony progression compared to control, whereas rapamycin lead to reduction in the area of cell colonies, which suggest impaired growth. Leptin treatment stimulated the proliferation of epithelial cells in a time dependent manner when compared to non treated IEC-6 cells. Significant stimulation was observed 72h and 96h after treatment of cells with leptin (). Treatment with rapamycin, significantly decreased IEC-6 cell proliferation (). To gain insight into the cell-transforming potential of leptin, IEC-6 cells were plated in semisolid agarose medium, an assay where there is no solid substratum for cell adhesion to occur. Neither control nor leptin treated IEC6 cells were able to establish colonies under this condition (not shown). Together, this data suggest that although leptin lead to increased cell proliferation, it is not sufficient to trigger mechanism of cell transformation in epithelial intestinal cells.
Figure 6. Leptin stimulates IEC-6 cell proliferation in a mTOR dependent manner. Cells were incubated with leptin (20 nM) in the absence or presence of rapamycin (20nM). After incubations the cells were incubated with Crystal Violet. (A) Image of the plate wells of cells treated for 120 h. (B) Quantification of the Crystal Violet stain absorbance after extracting it from the culture plates in different time points. (*) represent significant differences between leptin treated and control; and (+) represent significant differences between leptin treated and leptin plus rapamycin groups; according to the Student's T test when p < 0.05.
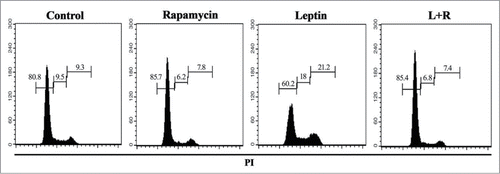
Cellular expansion depends on a balance between cell death and proliferation. Since we observed an important increase in total cell number upon treatment of IEC-6 with leptin (), we decided to evaluate cell cycle progression. IEC-6 cells were cultured as described in the materials and methods and the distribution of cells along the phases of the cell cycle assayed by propidium iodide (PI) staining and FACS analysis. We detected an accumulation of cells at the S phase upon treatment with leptin () and the treatment with rapamycin leads to an important G0/G1 cell cycle arrest. We also evaluate if rapamycin induces apoptosis of leptin-treated IEC-6 cells. To test if the treatment could induce apoptosis cells were stained with PI and analyzed by FACS. We did not observe significant cell death induced by any of the treatments in the time points tested (not shown). These results indicate that leptin-induced activation of the mTOR pathway play a role in the increase of colon epithelial cell proliferation, and therefore may contribute to the promotion of obesity-related colon cancer.
Figure 7. Leptin stimulates cell cycle entrance and progression of IEC cells in a mTOR dependent manner. Cells were incubated with leptin (20 nM) in the absence or presence of rapamycin (20nM) for 48 hours. After incubation, cell cycle progression was evaluated by propidium iodide staining and flow cytometry analysis. The percentages of cells in each stage of the cell cycle (G0/G1, S, and G2/M) are indicated.
Discussion
Obesity is an important risk factor for several chronic diseases, including hypertension, dyslipidemia, insulin resistance, diabetes mellitus and many cancers.Citation1,38-40 Leptin is a hormone/cytokine secreted from adipocytes that is involved with appetite control and energy metabolism through its effects on the hypothalamus. In the non-obese state, higher leptin levels result in decreased appetite through neuroendocrine changes, but during obesity there is the establishment of chronic high leptin plasma levels with hypothalamic resistance.Citation41 The characterization of leptin function in regulating lipid metabolism, inflammatory mediator production and lipid droplet biogenesis in epithelial cells is of importance for understanding its role in the pathogenesis of colon adenocarcinoma and other obesity-related tumors. Here we show that leptin directly activates epithelial cells to form lipid droplets and enhance inflammatory mediator production, in a mechanism dependent on activation of the mTOR pathway. This is the first demonstration that leptin directly regulates the accumulation of lipids within lipid droplets in epithelial cells, thus suggesting that leptin has roles in regulating the lipid metabolism in these cells. We also demonstrated that, in IEC-6 cells, leptin treatment leads to enhanced CCL2/MCP-1,CXCL1/CINC-1 production, as well as COX-2 expression and TGF-β production, that may contribute to the recruitment of inflammatory cells and establishment of an inflammatory tumor microenvironment.
mTOR's major functions include activation by phosphorylation of S6Kinase and activation of eIF4E by inhibition of 4E-BP1 (4E binding protein). These effects result in protein translation, metabolic regulation and cell proliferation.Citation18 It has been described that mutations that enhance the PI3K/mTOR pathway, the signaling pathway activated upon nutrient sensing, contribute to tumorigenesis of epithelial cells, which include colon carcinoma.Citation42 Leptin signals through the PI3K/Akt in different cell types, including macrophages. Our group demonstrated that leptin activates the PI3K/mTOR pathway in macrophages, leading to lipid droplet formation and LTB4 generation that are strictly dependent on PI3K and mTOR.Citation14 Moreover, leptin-induced hypothalamic effect on food intake is dependent on the mTOR pathway.Citation43 Leptin has also been shown to activate Src kinase and PI3K in the colon cancer cells LS174T and HM7Citation44 and promotes invasiveness of the epithelial cells via PI3K-Rho-Rac.Citation45 Recently, Wang et al. showed that leptin receptor is present in colon carcinoma biopsies and correlates with tumor severity.Citation46 During obesity leptin levels are enhanced long before the tumor development, therefore, we hypothesized that leptin might modulate also the non tumor epithelial cell responses through the mTOR pathway. Indeed, in IEC-6 cells, leptin induced a time and concentration-dependent phosphorylation of p70S6K in a rapamycin-sensitive mechanism. Moreover, we show, for the first time, that the mTORC1 inhibitor rapamycin drastically inhibited the lipid droplet formation induced by leptin in epithelial cells, suggesting that mTOR activity, downstream of PI3K, is crucial for leptin effects on the epithelial lipid metabolism. In addition, the sterol-regulatory element-binding proteins, which induce the expression of a large number of genes involved in de novo lipid and sterol biosynthesis, have emerged as major effectors of mTORC1 signaling, and mTORC1 activation stimulates lipogenesis.Citation47,48 Nevertheless, we can not exclude a participation of mTORC2 complex in the lipid droplet accumulation under leptin stimuli, since it has been demonstrated that mTORC2 regulates hepatic lipogenesis and participates on early adipocyte differentiation in different settingsCitation48 and prolonged rapamycin inhibition may also inhibit the mTORC2 complex.Citation49 Further studies will be necessary to determine the contributions of mTORC1 and mTORC2 in leptin-induced effects in epithelial cells. The altered cholesterol phenotype was induced by loss of tumor suppressor PTEN, upregulation of PI3K/AKT/mTOR pathway, and consequent activation of sterol regulatory element-binding protein (SREBP) and low-density lipoprotein receptor (LDLr) in prostate cancer. Inhibition of cholesterol esterification significantly suppressed cancer proliferation, migration, invasion, and tumor growth in vivo.Citation12 Cellular proliferation, a common feature of all cancers, requires fatty acids for synthesis of membranes and signaling molecules and most cells store fatty acids in triacilglycerois in the cytosolic lipid droplets. Thus, fatty acids are essential for cancer cell proliferation and limiting their availability could be a new therapeutic strategy.Citation50 Recently, our group demonstrated that lipid droplets in colon cancer cells are dynamic and functional organelles centrally involved in eicosanoid synthesis. In addition, lipid droplets have been described as sites to where enzymes involved in prostaglandin synthesis are localized, including cPLA2 and COX-2.Citation26,51 Proteins with well-established roles in oncogenic cell transformation, tumorigenesis, and metastasis, including PI3K, extracellular signal-regulated kinase 1 (ERK1), ERK2, p38 and PKC, were shown to localize to lipid droplets in a variety of cell types.Citation52-54 Moreover, the increase in lipid droplet density stimulates proliferation of colon cancer cells, mainly by loss of FOXO3.Citation55 Together, emerging evidence suggest roles of lipid droplets in cell signaling and metabolism and as such induction of these organelles may contribute to the proliferation-proned phenotype observed in epithelial cells stimulated by leptin.
Cellular transformation and cancer progression depends on dysregulations not only in cell's physiology but also on the interactions with the tissue microenvironment. Accordingly, obesity leads to a state of low grade chronic inflammation that is believed to contribute largely to the mechanisms associated with cancer.Citation1,6,56 Our data show that leptin was able to induce COX-2 increased expression in IEC-6 cells. This data is in agreement with previous data that leptin increases arachidonic acid availability, increases phospholipase A2 protein expression and activity, and induces COX-2 expression in macrophages and other cell types.Citation10,37 Overexpression of COX-2 is observed in different epithelial human tumors, and both genetic and pharmacological inactivation of COX-2 has been shown to protect from the development of colon cancer.Citation57 Our data also indicate that leptin may regulate tumor microenvironment through the induced expression of chemokines and cytokines by epithelial cells. The adipokines that are secreted by adipose tissue may activate epithelial cells, macrophages and other inflammatory cells. It has been demonstrated that tumor-associated macrophages and neutrophils (TAM and TAN) have key roles in cancer development and behavior, including in colon cancer.Citation58 TAMs are believed to contribute to tissue invasion,Citation59 angiogenesisCitation60 and metastasis.Citation61 Thus, it is possible that the recruitment and polarization of macrophages or other inflammatory cells in the setting of obesity may contribute to increased tumorigenesis. Leptin triggered the increased production of both the neutrophil (CXCL1/CINC1) and monocyte/macrophage (CCL2/MCP-1) chemoatractants by the epithelial cells. Increased CCL2/MCP1 and CXCL1 has been proposed as predictors of early relapse, poor prognosis and metastasis in human colon and breast.Citation61,62,63 Leptin also induced increased expression of TGF-β that has been associated with differantiation of macrophages into a M2-like phenotype.Citation63 The pretreatment with the mTOR inhibitor rapamycin blocked TGF-β but not CCL2/MCP-1 production, suggesting that leptin modulates tissue environment through mTOR dependent and independent pathways.
Accumulating evidence indicate a mitogenic effect of leptin on gastric,Citation64 breast,Citation65 ovarian,Citation66 prostate,Citation67 and endometrialCitation68 cancer cells. However, the mecanisms involved in leptin induced cell proliferation are not completely understood. Ogunwobi and Beales reported that leptin stimulates proliferation through activation of the Epidermal Growth Factor Receptor (EGFR) system.Citation69 Moreover, Leptin promotes hepatocellular carcinoma growth, invasiveness, and migration and implicate the JAK/STAT pathway as a critical mediator of leptin action.Citation70 Here, we demonstrated that leptin stimulates the proliferation of nontransformed epithelial intestinal cells through mechanisms largelly dependent on mTOR signaling. In agreement to our findings of a role of leptin in obesity-associated colon cancer development, recent findings demonstrate that obese Lepob mice did not exhibit increased risk of colonic cancer.Citation71
In conclusion, our results add to the current knowledge that mTOR is critically involved in the leptin signaling to activation of epithelial cells, leading to intracellular lipid accumulation, inflammatory mediator production and cell proliferation. Such alterations in lipid metabolism and inflammatory mediator production induced by leptin may provide a favorable microenvironment for increased cell proliferation. Taken together, our results suggest that leptin-induced mTOR activation may contribute to the obesity-related enhanced susceptibility to colon carcinoma by altering the intracellular lipid metabolism and inflammatory environment.
Materials and Methods
Cell culture and in vitro stimulation
IEC-6 cells were also grown in DMEM containing 10% FBS supplemented with 0.1 U/mL of insulin at 37°C in a humidified 5% CO2 incubator. Rat intestinal epithelial cells, IEC-6, were stimulated with leptin (20 nM) for 20 min or 6 h in DMEM medium with 2% fetal bovine serum. In designated groups, cells were pretreated with everolimus (10 nM), FK-506 (10 nM) or rapamycin (20 nM) for 30 min before the addition of the stimuli. Cell viability was always greater than 90%, as determined by trypan blue dye exclusion at the end of each experiment.
Lipid droplet staining and enumeration
Cells (5×105 per well) were stimulated with leptin (20 nM) and untreated or treated with rapamycin (20 nM), everolimus (10 nM) or FK-506 (10 nM) for 6 h. Lipid droplets were stained as previously described in each triplicate well. In brief, cells (5×105 cells/mL) were fixed in 3.7% formaldehyde in calcium/magnesium-free HBSS (pH 7.4), rinsed in 0.1 mol/L cacodylate buffer, stained in 1.5% OsO4 (30 min), rinsed in distilled water (dH2O), immersed in 1.0% thiocarbohydrazide (5 min), rinsed in 0.1 mol/L cacodylate buffer, restained in 1.5% OsO4 (3 min), rinsed, and then dried and mounted. The morphology of fixed cells was observed, and lipid droplets were enumerated by light microscopy with 100x objective lens in 50 consecutively scanned cells. Alternatively cells were stained with Oil Red O (lipid fluorescent stain) and Dapi for the analyses of epithelial cell lipid droplet formation in vitro performed 6 h after leptin (20 nM) stimuli and treatment with rapamycin (20 nM). The area of fluorescence was analyzed with the use of FiJi/ImageJ public domain software.
Western Blotting
Cell lysates were prepared in reducing and denaturing conditions and subjected to SDS-PAGE. Samples were submitted to electrophoresis on polyacrylamide gel. After transfer onto nitrocellulose membranes, nonspecific binding sites were blocked with 5% bovine serum albumin in Tris-buffered saline-Tween (TBST; 50 mM Tris- HCl, pH 7.4, 150 mM NaCl, 0.05% Tween 20). Membranes were probed with anti-phospho-p70S6K, anti-p70S6K monoclonal antibody, anti-phospho-STAT3 and anti-STAT3 (Cell Signaling, Danvers, MA), or anti-COX2, anti-AKT and anti-phospho-AKT (Santa Cruz Biotechnology, Dallas, TX) in TBST;. Proteins of interest were then identified by incubating the membrane with horseradish peroxidase conjugated specific secondary antibody in TBST, followed by detection of antigen-antibody complexes by Supersignal chemiluminescence (Pierce).
Immunolocalization
IEC-6 cells (1×105) were stimulated with leptin (24 h) and treated with rapamycin and fixed in 3% formaldehyde at room temperature for 10 min. After permeabilization in 0.2% Triton X-100 for 20 min at room temperature, slides were washed in PBS. The unspecific sites were blocked with 1% normal donkey serum for 10 min. The cells were incubated overnight with goat polyclonal antibody anti-COX-2 (C-20; Santa Cruz Biotechnology). Then, cells were blocked with 1% donkey serum for 10 min before simultaneous incubation with Dylight 488 donkey anti-goat (Jackson ImmunoResearch Laboratories) for 1 h. Then, cells were washed several times with PBS, mounted with an aqueous mounting medium with DAPI (Prolong Gold antifade reagent; Life technologies). Slides were viewed by both phase-contrast and fluorescence microscopy, and digital images were obtained using a Hoper Scientific digital camera with the Image-Express software.
Cell proliferation assay
Cells (5×104 per well) were stimulated with leptin (20 nM) and untreated or treated with rapamycin (20 nM) for 24, 48, 72, and 96 h. After treatment, cell proliferation was assayed in each triplicate well. Briefly, cells were fixed with ethanol for 10 min, stained with 0.05% violet crystal in 20% ethanol for 10 min, the crystal was extracted with methanol, and the absorbance was quantified in a spectrometer at 595 nm.
Cell cycle analysis
IEC-6 cells were stimulated with leptin (20 nM) and untreated or treated with rapamycin (20 nM) for 48 hours. After treatment, cells were washed and stained with propidium iodide staining solution (4 mM Tris-HCl pH 7.6, 10 nM NaCl, 0.1% NP-40, 700 U/L Ribonuclease A, 0.075 mM propidium iodide). DNA content analysis was performed by flow cytometry in a FACScalibur equipment (BD Biosciences).
Statistical analysis
Statistical analysis of values from control and treated groups was determined using an unpaired Student's t test for single comparison or an ANOVA followed by the Student-Newman-Keuls for multiple comparisons. P < 0.05 was considered to be statistically significant.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors are in debt to Dr. Leonardo K Teixeira and Dr. Alan C Brito for assistance and helpful suggestions, Dr. Jens Rietdorf (CDTS) for assistance with imaging processing and to Edson F. Assis for technical assistance.
Funding
This work was supported by grants from by FAPERJ, CNPq, CAPES, INCT-Cancer.
References
- Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of US adults. N Engl J Med 2003; 348:1625–38; PMID:12711737
- Giovannucci E, Goldin B. The role of fat, fatty acids, and total energy intake in the etiology of human colon cancer. Am J Clin Nutr 1997; 66:1564S–71S.
- Reddy BS. Dietary fat and colon cancer: animal model studies. Lipids 1992; 27:807–13; PMID:1435100; http://dx.doi.org/10.1007/BF02535855
- Padidar S, Farquharson AJ, Williams LM, Kearney R, Arthur JR, Drew JE. High-fat diet alters gene expression in the liver and colon: links to increased development of aberrant crypt foci. Dig Dis Sci 2012; 57:1866–74; PMID:22373862; http://dx.doi.org/10.1007/s10620-012-2092-9
- Rogers CJ, Prabhu KS, Vijay-Kumar M. The microbiome and obesity-an established risk for certain types of cancer. Cancer J 2014; 20:176–80; PMID:24855004; http://dx.doi.org/10.1097/PPO.0000000000000049
- Velloso LA, Folli F, Saad MJ. TLR4 at the crossroads of nutrients, gut microbiota and metabolic inflammation. Endocr Rev 2015:er20141100; PMID:25811237
- Flier JS. Obesity wars: molecular progress confronts an expanding epidemic. Cell 2004; 116:337–50; PMID:14744442; http://dx.doi.org/10.1016/S0092-8674(03)01081-X
- Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature 1994; 372:425–32; PMID:7984236; http://dx.doi.org/10.1038/372425a0
- Lam QL, Lu L. Role of leptin in immunity. Cell Mol Immunol 2007; 4:1–13; PMID:17349207
- La Cava A, Matarese G. The weight of leptin in immunity. Nat Rev Immunol 2004; 4:371–9; PMID:15122202; http://dx.doi.org/10.1038/nri1350
- Garofalo CS, E. Leptin and cancer. J Cell Physiol 2005; 207:12–22 ; http://dx.doi.org/10.1002/jcp.20472
- Yue S, Li J, Lee SY, Lee HJ, Shao T, Song B, Cheng L, Masterson TA, Liu X, Ratliff TL, et al. Cholesteryl ester accumulation induced by PTEN loss and PI3K/AKT activation underlies human prostate cancer aggressiveness. Cell Metab 2014; 19:393–406; PMID:24606897; http://dx.doi.org/10.1016/j.cmet.2014.01.019
- Higurashi T, Endo H, Uchiyama T, Uchiyama S, Yamada E, Ohkubo H, Sakai E, Takahashi H, Maeda S, Wada K, et al. Conditional knockout of the leptin receptor in the colonic epithelium revealed the local effects of leptin receptor signaling in the progression of colonic tumors in mice. Carcinogenesis 2014; 35:2134–41; PMID:24958593; http://dx.doi.org/10.1093/carcin/bgu135
- Maya-Monteiro CM, Almeida PE, D'Avila H, Martins AS, Rezende AP, Castro-Faria-Neto H, Bozza PT. Leptin induces macrophage lipid body formation by a phosphatidylinositol 3-kinase- and mammalian target of rapamycin-dependent mechanism. J Biol Chem 2008; 283:2203–10; PMID:18039669; http://dx.doi.org/10.1074/jbc.M706706200
- Maya-Monteiro CM, Bozza PT. Leptin and mTOR: partners in metabolism and inflammation. Cell Cycle 2008; 7:1713–7; PMID:18583936; http://dx.doi.org/10.4161/cc.7.12.6157
- Sarbassov DD, Ali SM, Sabatini DM. Growing roles for the mTOR pathway. Curr Opin Cell Biol 2005; 17:596–603; PMID:16226444; http://dx.doi.org/10.1016/j.ceb.2005.09.009
- Sabatini DM. mTOR and cancer: insights into a complex relationship. Nat Rev Cancer 2006; 6:729–34; PMID:16915295; http://dx.doi.org/10.1038/nrc1974
- Easton JB, Houghton PJ. mTOR and cancer therapy. Oncogene 2006; 25:6436–46; PMID:17041628; http://dx.doi.org/10.1038/sj.onc.1209886
- Li J, Kim SG, Blenis J. Rapamycin: one drug, many effects. Cell Metab 2014; 19:373–9; PMID:24508508; http://dx.doi.org/10.1016/j.cmet.2014.01.001
- Duvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, Triantafellow E, Ma Q, Gorski R, Cleaver S, et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell 2010; 39:171–83; PMID:20670887; http://dx.doi.org/10.1016/j.molcel.2010.06.022
- Kuhajda FP. Fatty acid synthase and cancer: new application of an old pathway. Cancer Res 2006; 66:5977–80; PMID:16778164; http://dx.doi.org/10.1158/0008-5472.CAN-05-4673
- Schetter AJ, Nguyen GH, Bowman ED, Mathe EA, Yuen ST, Hawkes JE, Croce CM, Leung SY, Harris CC. Association of inflammation-related and microRNA gene expression with cancer-specific mortality of colon adenocarcinoma. Clin Cancer Res 2009; 15:5878–87; PMID:19737943; http://dx.doi.org/10.1158/1078-0432.CCR-09-0627
- Swinnen JV, Brusselmans K, Verhoeven G. Increased lipogenesis in cancer cells: new players, novel targets. Curr Opin Clin Nutr Metab Care 2006; 9:358–65; PMID:16778563; http://dx.doi.org/10.1097/01.mco.0000232894.28674.30
- Greenberg AS, Coleman RA, Kraemer FB, McManaman JL, Obin MS, Puri V, Yan QW, Miyoshi H, Mashek DG. The role of lipid droplets in metabolic disease in rodents and humans. J Clin Invest 2011; 121:2102–10; PMID:21633178; http://dx.doi.org/10.1172/JCI46069
- Bozza PT, Viola JP. Lipid droplets in inflammation and cancer. Prostaglandins Leukot Essent Fatty Acids 2010; 82:243–50; PMID:20206487; http://dx.doi.org/10.1016/j.plefa.2010.02.005
- Accioly MT, Pacheco P, Maya-Monteiro CM, Carrossini N, Robbs BK, Oliveira SS, Kaufmann C, Morgado-Diaz JA, Bozza PT, Viola JP. Lipid bodies are reservoirs of cyclooxygenase-2 and sites of prostaglandin-E2 synthesis in colon cancer cells. Cancer Res 2008; 68:1732–40; PMID:18339853; http://dx.doi.org/10.1158/0008-5472.CAN-07-1999
- Than NG, Sumegi B, Bellyei S, Berki T, Szekeres G, Janaky T, Szigeti A, Bohn H, Than GN. Lipid droplet and milk lipid globule membrane associated placental protein 17b (PP17b) is involved in apoptotic and differentiation processes of human epithelial cervical carcinoma cells. Eur J Biochem 2003; 270:1176–88; PMID:12631276; http://dx.doi.org/10.1046/j.1432-1033.2003.03475.x
- Opstad KS, Bell BA, Griffiths JR, Howe FA. An investigation of human brain tumour lipids by high-resolution magic angle spinning 1H MRS and histological analysis. NMR Biomed 2008; 21:677–85; PMID:18186027; http://dx.doi.org/10.1002/nbm.1239
- Dvorak AM, Weller PF, Harvey VS, Morgan ES, Dvorak HF. Ultrastructural localization of prostaglandin endoperoxide synthase (cyclooxygenase) to isolated, purified fractions of guinea pig peritoneal macrophage and line 10 hepatocarcinoma cell lipid bodies. Int Arch Allergy Immunol 1993; 101:136–42; PMID:8508051; http://dx.doi.org/10.1159/000236511
- Terzic J, Grivennikov S, Karin E, Karin M. Inflammation and colon cancer. Gastroenterology 2010; 138:2101-14 e5; PMID:20420949; http://dx.doi.org/10.1053/j.gastro.2010.01.058
- Brunn GJ, Fadden P, Haystead TA, Lawrence JC, Jr. The mammalian target of rapamycin phosphorylates sites having a (Ser/Thr)-Pro motif and is activated by antibodies to a region near its COOH terminus. J Biol Chem 1997; 272:32547–50; PMID:9405468; http://dx.doi.org/10.1074/jbc.272.51.32547
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005; 307:1098–101; PMID:15718470; http://dx.doi.org/10.1126/science.1106148
- Eberhart CE, Coffey RJ, Radhika A, Giardiello FM, Ferrenbach S, DuBois RN. Up-regulation of cyclooxygenase 2 gene expression in human colorectal adenomas and adenocarcinomas. Gastroenterology 1994; 107:1183–8; PMID:7926468
- Kargman SL, O'Neill GP, Vickers PJ, Evans JF, Mancini JA, Jothy S. Expression of prostaglandin G/H synthase-1 and −2 protein in human colon cancer. Cancer Res 1995; 55:2556–9; PMID:7780968
- Sano H, Kawahito Y, Wilder RL, Hashiramoto A, Mukai S, Asai K, Kimura S, Kato H, Kondo M, Hla T. Expression of cyclooxygenase-1 and −2 in human colorectal cancer. Cancer Res 1995; 55:3785–9; PMID:7641194
- Mancuso P, Canetti C, Gottschalk A, Tithof PK, Peters-Golden M. Leptin augments alveolar macrophage leukotriene synthesis by increasing phospholipase activity and enhancing group IVC iPLA2 (cPLA2gamma) protein expression. Am J Physiol Lung Cell Mol Physiol 2004; 287:L497–502; PMID:15145787; http://dx.doi.org/10.1152/ajplung.00010.2004
- Raso GM, Pacilio M, Esposito E, Coppola A, Di Carlo R, Meli R. Leptin potentiates IFN-gamma-induced expression of nitric oxide synthase and cyclo-oxygenase-2 in murine macrophage J774A.1. Br J Pharmacol 2002; 137:799–804; PMID:12411410; http://dx.doi.org/10.1038/sj.bjp.0704903
- Kahn BB, Flier JS. Obesity and insulin resistance. J Clin Invest 2000; 106:473–81; PMID:10953022; http://dx.doi.org/10.1172/JCI10842
- Rahmouni K, Correia ML, Haynes WG, Mark AL. Obesity-associated hypertension: new insights into mechanisms. Hypertension 2005; 45:9–14; PMID:15583075; http://dx.doi.org/10.1161/01.HYP.0000151325.83008.b4
- Ravussin E, Smith SR. Increased fat intake, impaired fat oxidation, and failure of fat cell proliferation result in ectopic fat storage, insulin resistance, and type 2 diabetes mellitus. Ann N Y Acad Sci 2002; 967:363–78; PMID:12079864; http://dx.doi.org/10.1111/j.1749-6632.2002.tb04292.x
- Hursting SD, Smith SM, Lashinger LM, Harvey AE, Perkins SN. Calories and carcinogenesis: lessons learned from 30 years of calorie restriction research. Carcinogenesis 2010; 31:83–9; PMID:19969554; http://dx.doi.org/10.1093/carcin/bgp280
- Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov 2009; 8:627–44; PMID:19644473; http://dx.doi.org/10.1038/nrd2926
- Cota D, Proulx K, Smith KA, Kozma SC, Thomas G, Woods SC, Seeley RJ. Hypothalamic mTOR signaling regulates food intake. Science 2006; 312:927–30; PMID:16690869; http://dx.doi.org/10.1126/science.1124147
- Jaffe T, Schwartz B. Leptin promotes motility and invasiveness in human colon cancer cells by activating multiple signal-transduction pathways. Int J Cancer 2008; 123:2543–56; PMID:18767036; http://dx.doi.org/10.1002/ijc.23821
- Attoub S, Noe V, Pirola L, Bruyneel E, Chastre E, Mareel M, Wymann MP, Gespach C. Leptin promotes invasiveness of kidney and colonic epithelial cells via phosphoinositide 3-kinase-, rho-, and rac-dependent signaling pathways. FASEB J 2000; 14:2329–38; PMID:11053255; http://dx.doi.org/10.1096/fj.00-0162
- Wang D, Chen J, Chen H, Duan Z, Xu Q, Wei M, Wang L, Zhong M. Leptin regulates proliferation and apoptosis of colorectal carcinoma through PI3K/Akt/mTOR signalling pathway. J Biosci 2012; 37:91–101; PMID:22357207; http://dx.doi.org/10.1007/s12038-011-9172-4
- Laplante M, Sabatini DM. An emerging role of mTOR in lipid biosynthesis. Curr Biol 2009; 19:R1046–52; PMID:19948145; http://dx.doi.org/10.1016/j.cub.2009.09.058
- Lamming DW, Sabatini DM. A Central role for mTOR in lipid homeostasis. Cell Metab 2013; 18:465–9; PMID:23973332; http://dx.doi.org/10.1016/j.cmet.2013.08.002
- Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, Markhard AL, Sabatini DM. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell 2006; 22:159–68; PMID:16603397; http://dx.doi.org/10.1016/j.molcel.2006.03.029
- Currie E, Schulze A, Zechner R, Walther TC, Farese RV, Jr. Cellular fatty acid metabolism and cancer. Cell Metab 2013; 18:153–61; PMID:23791484; http://dx.doi.org/10.1016/j.cmet.2013.05.017
- Moreira LS, Piva B, Gentile LB, Mesquita-Santos FP, D'Avila H, Maya-Monteiro CM, Bozza PT, Bandeira-Melo C, Diaz BL. Cytosolic phospholipase A2-driven PGE2 synthesis within unsaturated fatty acids-induced lipid bodies of epithelial cells. Biochim Biophys Acta 2009; 1791:156–65; PMID:19367763; http://dx.doi.org/10.1016/j.bbalip.2009.01.003
- Chen JS, Greenberg AS, Wang SM. Oleic acid-induced PKC isozyme translocation in RAW 264.7 macrophages. J Cell Biochem 2002; 86:784–91; PMID:12210744; http://dx.doi.org/10.1002/jcb.10266
- Yu W, Bozza PT, Tzizik DM, Gray JP, Cassara J, Dvorak AM, Weller PF. Co-compartmentalization of MAP kinases and cytosolic phospholipase A2 at cytoplasmic arachidonate-rich lipid bodies. Am J Pathol 1998; 152:759–69; PMID:9502418
- Yu W, Cassara J, Weller PF. Phosphatidylinositide 3-kinase localizes to cytoplasmic lipid bodies in human polymorphonuclear leukocytes and other myeloid-derived cells. Blood 2000; 95:1078–85; PMID:10648425
- Qi W, Fitchev PS, Cornwell ML, Greenberg J, Cabe M, Weber CR, Roy HK, Crawford SE, Savkovic SD. FOXO3 growth inhibition of colonic cells is dependent on intraepithelial lipid droplet density. J Biol Chem 2013; 288:16274–81; PMID:23603907; http://dx.doi.org/10.1074/jbc.M113.470617
- Lumeng CN, Saltiel AR. Inflammatory links between obesity and metabolic disease. J Clin Invest 2011; 121:2111–7; PMID:21633179; http://dx.doi.org/10.1172/JCI57132
- Wang D, Dubois RN. Eicosanoids and cancer. Nat Rev Cancer 2010; 10:181–93; PMID:20168319; http://dx.doi.org/10.1038/nrc2809
- Galdiero MR, Garlanda C, Jaillon S, Marone G, Mantovani A. Tumor associated macrophages and neutrophils in tumor progression. J Cell Physiol 2013; 228:1404–12; PMID:23065796; http://dx.doi.org/10.1002/jcp.24260
- Coussens LM, Tinkle CL, Hanahan D, Werb Z. MMP-9 supplied by bone marrow-derived cells contributes to skin carcinogenesis. Cell 2000; 103:481–90; PMID:11081634; http://dx.doi.org/10.1016/S0092-8674(00)00139-2
- Murdoch C, Muthana M, Coffelt SB, Lewis CE. The role of myeloid cells in the promotion of tumour angiogenesis. Nat Rev Cancer 2008; 8:618–31; PMID:18633355; http://dx.doi.org/10.1038/nrc2444
- Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, Kaiser EA, Snyder LA, Pollard JW. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 2011; 475:222–5; PMID:21654748; http://dx.doi.org/10.1038/nature10138
- Ogata H, Sekikawa A, Yamagishi H, Ichikawa K, Tomita S, Imura J, Ito Y, Fujita M, Tsubaki M, Kato H, et al. GROalpha promotes invasion of colorectal cancer cells. Oncol Rep 2010; 24:1479–86; PMID:21042742
- Erreni M, Mantovani A, Allavena P. Tumor-associated Macrophages (TAM) and Inflammation in Colorectal Cancer. Cancer Microenvir 2011; 4:141–54; PMID:21909876; http://dx.doi.org/10.1007/s12307-010-0052-5
- Pai R, Lin C, Tran T, Tarnawski A. Leptin activates STAT and ERK2 pathways and induces gastric cancer cell proliferation. Biochem Biophys Res Commun 2005; 331:984–92; PMID:15882975; http://dx.doi.org/10.1016/j.bbrc.2005.03.236
- Rose DP, Komninou D, Stephenson GD. Obesity, adipocytokines, and insulin resistance in breast cancer. Obes Rev 2004; 5:153–65; PMID:15245384; http://dx.doi.org/10.1111/j.1467-789X.2004.00142.x
- Choi JH, Park SH, Leung PC, Choi KC. Expression of leptin receptors and potential effects of leptin on the cell growth and activation of mitogen-activated protein kinases in ovarian cancer cells. J Clin Endocrinol Metab 2005; 90:207–10; PMID:15522945; http://dx.doi.org/10.1210/jc.2004-0297
- Somasundar P, Frankenberry KA, Skinner H, Vedula G, McFadden DW, Riggs D, Jackson B, Vangilder R, Hileman SM, Vona-Davis LC. Prostate cancer cell proliferation is influenced by leptin. J Surg Res 2004; 118:71–82; PMID:15093720; http://dx.doi.org/10.1016/j.jss.2004.01.017
- Sharma D, Saxena NK, Vertino PM, Anania FA. Leptin promotes the proliferative response and invasiveness in human endometrial cancer cells by activating multiple signal-transduction pathways. Endocr Relat Cancer 2006; 13:629–40; PMID:16728588; http://dx.doi.org/10.1677/erc.1.01169
- Ogunwobi OO, Beales IL. Leptin stimulates the proliferation of human oesophageal adenocarcinoma cells via HB-EGF and Tgfalpha mediated transactivation of the epidermal growth factor receptor. Br J Biomed Sci 2008; 65:121–7; PMID:18986098
- Saxena NK, Sharma D, Ding X, Lin S, Marra F, Merlin D, Anania FA. Concomitant activation of the JAK/STAT, PI3K/AKT, and ERK signaling is involved in leptin-mediated promotion of invasion and migration of hepatocellular carcinoma cells. Cancer Res 2007; 67:2497–507; PMID:17363567; http://dx.doi.org/10.1158/0008-5472.CAN-06-3075
- Sikalidis AK, Fitch MD, Fleming SE. Risk of colonic cancer is not higher in the obese Lep(ob) mouse model compared to lean littermates. Pathol Oncol Res 2013; 19:867–74; PMID:23813464; http://dx.doi.org/10.1007/s12253-013-9656-7