
Abstract
Progranulin (PGRN) has recently emerged as an important regulator for glucose metabolism and insulin sensitivity. However, the direct effects of PGRN in vivo and the underlying mechanisms between PGRN and impaired insulin sensitivity are not fully understood. In this study, mice treated with PGRN for 21 d exhibited the impaired glucose tolerance and insulin sensitivity, remarkable ER stress as well as attenuated insulin signaling in liver and adipose tissue but not in skeletal muscle. Furthermore, treatment of mice with phenyl butyric acid (PBA), a chemical chaperone alleviating ER stress, resulted in a significant restoration of systemic insulin sensitivity and recovery of insulin signaling induced by PGRN. Consistent with these findings in vivo, we also observed that PGRN treatment induced ER stress, impaired insulin signaling in cultured hepatocytes and adipocytes, with such effects being partially nullified by blockade of PERK. Whereas PGRN-deficient hepatocytes and adipocytes were more refractory to palmitate-induced insulin resistance, indicating the causative role of the PERK-eIF2α axis of the ER stress response in action of PGRN. Collectively, our findings supported the notion that PGRN is a key regulator of insulin resistance and that PGRN may mediate its effects, at least in part, by inducing ER stress via the PERK-eIF2α dependent pathway.
Introduction
Progranulin (PGRN), also known as proepithelin, granulin/epithelin precursor or PC cell-derived growth factor, is identified to have multiple functions, and plays a critical role in physiological and disease processes such as wound healing, tumorigenesis, degenerative diseases and inflammation.Citation1-4 Recently, PGRN has emerges as a novel regulator associated with proinflammatory properties and insulin resistance.Citation5-10 Specifically, PGRN-deficient mice exhibited resistant to diet-induced obesity and insulin insensitivity through the modulation of inflammation, whereas adipocytes exposed to PGRN have increased susceptibility to be insulin-resistant.Citation7 Moreover, circulating PGRN is significantly higher in clinical subjects with type 2 diabetes and positively correlated with macrophage infiltration in omental adipose tissue.Citation9,11,12 In particular, PGRN is more highly expressed in visceral adipose tissue of the insulin-resistant patients with morbid obesity than in their age-, sex- and BMI-matched insulin-sensitive counterparts.Citation13-15 Thus, PGRN may be an important modulator in a variety of glucose and energy regulation with specific effect on target tissues.
Although the role of PGRN in energy metabolism has just recently been identified, the direct mechanism underlying this pivotal effect has not been established. However, previous evidences suggested that endoplasmic reticulum (ER) stress may be involved in PGRN action in some other pathological processes. For instance, PGRN activates ERK1/2 signaling pathway,Citation16 whereas XBP1S, a key transcription factor in ER stress, inhibits ER stress-mediated apoptosis through the Erk1/2 signaling pathway.Citation17 In addition, tumor necrosis factor-α (TNFα), a putative inducer of inflammation, ER stress and insulin resistance,Citation18–22 significantly augments PGRN expression in 3T3-L1 adipocytes.Citation7 PGRN has also been introduced as a cytoprotective stress-response factor in fibroblasts subjected to hypoxia and acidosis, while mutant seipin in HeLa cells leads not only to ER stress, but also to the impairment secretion of PGRN.Citation23 Moreover, pgrn-1 mutants have been reported to be refractory to ER stress-induced apoptosis measured by resistance to tunicamycin, an inhibitor of N-linked glycosylation.Citation24 Collectively, these findings indicate the possibility that ER stress may contribute to the regulatory role of PGRN on insulin resistance and metabolic dysfunction.
The fact that high fat diet (HFD)-induced insulin resistance could be considerably improved in PGRN−/− mice suggests that chronically elevated PGRN in obese mice could potentially contribute to the progression of insulin resistance and metabolic dysfunction.Citation7 The next critical question to answer is whether PGRN has a direct effect on energy metabolism in wild-type mice in vivo, which would firmly establish the notion that it is a physiologically important factor in the pathogenesis of metabolic disorders. In this study, we embarked on a systematic analysis of the effects of PGRN in vivo and aimed to evaluate the potential interaction of impaired insulin sensitivity and PGRN action involving ER stress. Our results demonstrated that PGRN triggers ER stress and insulin resistance via the PERK-eIF2α dependent pathway, supporting the therapeutic potential of this novel player in the regulation of glucose metabolism and metabolic diseases.
Results
The effects of PGRN on glucose metabolism, insulin sensitivity, and lipid metabolism in vivo
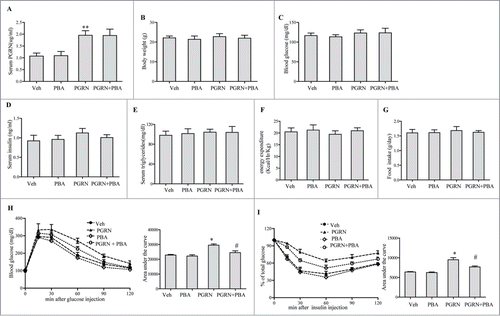
To test whether direct intraperitoneal administration of PGRN is capable of regulating peripheral glucose metabolism, we administrated rmPGRN ranging from 100-fold lower than its physiological concentration (≈1 μg/ml in WT adult mice) to fold4- higher in C57BL/6J mice for 21 d under standard diet (SD) condition. After ip administration, ITT revealed that mice receiving 10, 20 and 40 μg/day of PGRN exhibited a significant and dose-dependent decrease in insulin sensitivity, whereas no changes were observed in mice treated with PGRN<1 μg/day, compared with vehicle (data not shown). Given previous reports that PGRN is biologically active at an effective dose of 20 ug/day, our results also suggest that 20 μg/day of PGRN were optimal to assess the effect of PGRN in mice. After a 3-week study period, we found that serum PGRN level was remarkably increased about 1.5-2.5-fold in PGRN-treated littermates (). Compared with vehicle treatment, administration of PGRN had essentially no effect on body weight, nor did it alter fasting glucose, energy expenditure, food intake, and serum triglycerides levels. Although fasting plasma insulin levels tended to be increased in PGRN-treated mice, this effect did not achieve statistical significance (). When challenged with glucose (GTT), PGRN administration mildly induced glucose intolerance (). Additionally, mice receiving 20 μg/day PGRN exhibited reduced insulin sensitivity than mice receiving vehicle under a normal diet condition as shown in .
Figure 1. Effect of PGRN on glucose metabolism and insulin sensitivity, and its abrogation by PBA in vivo. All analyses compared age- and sex-matched mice fed a normal diet. Mice were distributed in 4 groups (12–15/per group): (1) vehicle (2) PGRN (i.p. 20 μg/day); (3) 4-PBA (orally, 1 g/Kg of body weight); (4) PGRN + 4-PBA. 12–15 mice per group were analyzed. (A) Effect of 3 weeks administration of rmPGRN and PBA on serum PGRN levels. (B) Body weight. (C) Blood glucose. (D) Serum insulin. (E) Serum triglyceride. (F) Energy expenditure. (G) Food intake. (H) GTT. (I) ITT. Data are means ± SD in each bar graph. *P < 0.05, **P < 0.01 (vs. vehicle). #P < 0.05 (vs. PGRN).
To gain further insight into the mechanism underlying the insulin-desensitizing effects of PGRN, we then performed hyperinsulinemic-euglycemic clamp studies. During the clamp procedure, glucose infusion rate (GIR) to maintain euglycemia (120–140 mg/dl) was reduced by 25% in PGRN-treated mice compared with vehicle (Fig. S1A). This was accompanied by an effect of PGRN to impair slightly the action of insulin to suppress hepatic glucose production both at baseline and during the clamp studies (Fig. S1B). In agreement, treatment did not affect glucose uptake by muscle (Fig. S1C). Moreover, PGRN-treated mice also showed impaired pyruvate tolerance relative to vehicle (Fig. S1D). Correspondingly, hepatic expression of both glucose-6-phosphatase (G6pase) and phosphosphoenolpyruvate carboxykinase 1 (Pck1) mRNA, and protein levels were slightly increased in mice receiving PGRN (Fig. S1E, F), indicating that PGRN inhibited insulin-mediated suppression of hepatic gluconeogenesis in mice.
Our observation of glucose metabolism tendency to change despite no alterations in total body in mice receiving PGRN led us to assess lipid metabolism in those mice. When we evaluated in vivo β-oxidation by measuring release of 14CO2 after administration of [1−14C] oleic acid, intraperitoneal administration of PGRN did not result in any detectable decreases in β-oxidation (). Although there was a little bit of difference in expression of genes related to β-oxidation, such as Ppara, in liver and adipose tissue of PGRN-treated mice (), no changes could be observed in lipid accumulation in the liver and skeletal muscle sections stained with Oil Red O in PGRN-treated animals (), suggesting that the impact of PGRN on lipid metabolism could not be readily assessed. However, histomorphometric quantification demonstrated that both the number and the area of mitochondria were mildly decreased after PGRN injections ().
Figure 2. Effect of PGRN on lipid metabolism and liver steatosis in vivo. Mice were distributed as in . (A) β-oxidation of infused [1–14C] oleic acid in vivo. (B) Relative mRNA levels from genes associated with fatty acid and triacylglycerol (TG) synthesis, or β-oxidation in the liver. (C) Relative expression of genes related to β-oxidation in adipose tissue. (D) Oil Red O staining of liver sections (magnification 100×), scale bars: 20μm. (E) Mitochondria number and mitochondria area in liver. Data are means ± SEM in each bar graph. *P < 0.05, #P < 0.05 (vs. PGRN).
![Figure 2. Effect of PGRN on lipid metabolism and liver steatosis in vivo. Mice were distributed as in Figure 1. (A) β-oxidation of infused [1–14C] oleic acid in vivo. (B) Relative mRNA levels from genes associated with fatty acid and triacylglycerol (TG) synthesis, or β-oxidation in the liver. (C) Relative expression of genes related to β-oxidation in adipose tissue. (D) Oil Red O staining of liver sections (magnification 100×), scale bars: 20μm. (E) Mitochondria number and mitochondria area in liver. Data are means ± SEM in each bar graph. *P < 0.05, #P < 0.05 (vs. PGRN).](/cms/asset/cf1c0bc4-6714-4213-a625-1053431bb8d5/kccy_a_1041686_f0002_oc.gif)
Administration of PGRN triggers impaired insulin signaling and ER stress in vivo
Considering the effects of PGRN on glucose intolerance and insulin insensitivity in mice, we began to evaluate the potential impact of PGRN on insulin signaling in insulin-target tissues by western blotting and immunoprecipitation. Remarkably, insulin signaling was inhibited in the liver, and adipose tissue of mice upon PGRN treatment, evidenced by reduction of insulin mediated phosphorylation of IRS-1 and Akt in PGRN-treated mice compared to their controls (). However, PGRN almost has no significant effect on the ability of insulin to phosphorylate Akt and IRS1 in skeletal muscle ().
Figure 3. Effects of PGRN on ER stress and insulin signaling in liver, adipose tissue and skeletal muscle. All analyses compared age- and sex-matched mice injected daily with saline solution or PGRN for 21 d. For insulin signaling, they were injected with 2 IU/kg of insulin (Ins). The relative quantity of proteins was analyzed using the Image J software. (A–C) Phosphorylation of IRS-1 tyrosine and Akt Ser-473 in liver, adipose tissue and skeletal muscle. (D–F) The phosphorylation of PERK and eIF2α in the liver, adipose tissue and skeletal muscle of mice co-treated with insulin and PGRN. The data expressed as means ± SEM in each bar graph. *P < 0.05, **P < 0.01 (vs. vehicle). #P < 0.05 (vs. Ins).
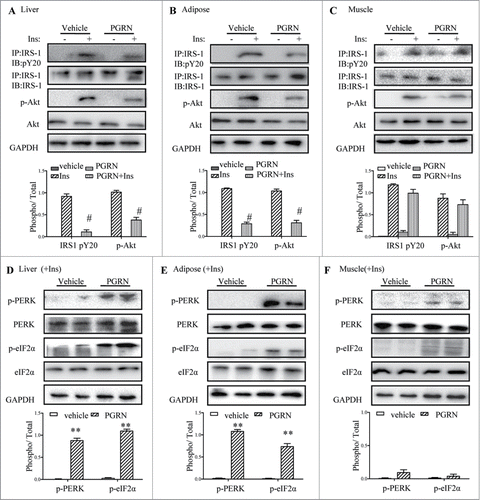
Because IRS-1 is a substrate for insulin receptor tyrosine kinase, and insulin-stimulated tyrosine phosphorylation of IRS-1, particularly mediated by ER stress, it is possible the change in insulin sensitivity involves ER stress. Thus, we determined whether intraperitoneal administration of PGRN was capable of triggering ER stress in liver, adipose tissue and skeletal muscle in the presence of insulin. The pancreatic ER kinase or PKR-like kinase (PERK) is an ER transmembrane protein kinase that phosphorylates α subunit of translation initiation factor 2 (eIF2α) in response to ER stress. The phosphorylation status of PERK and eIF2α is therefore a key indicator of the presence of ER stress. As expected, PGRN markedly elevated PERK activation and eIF2α phosphorylation in liver, and adipose tissue of PGRN-treated mice compared with those of untreated controls (). In contrast, the phosphorylation of PERK and eIF2α in muscle were almost not affected by PGRN (). In addition, the mRNA levels of ATF6 and the spliced form of X-box binding protein 1 (sXBP-1), which are known to be responsive to ER stress, were dramatically up-regulated in liver and adipose tissue in mice treated with PGRN (Fig. S2). Overall, the effects of PGRN injection appear mainly to involve impaired insulin signaling and ER stress in liver and adipose tissue.
Reduction of ER stress via a chemical chaperone, PBA, rescues insulin sensitivity in mice subjected to PGRN administration
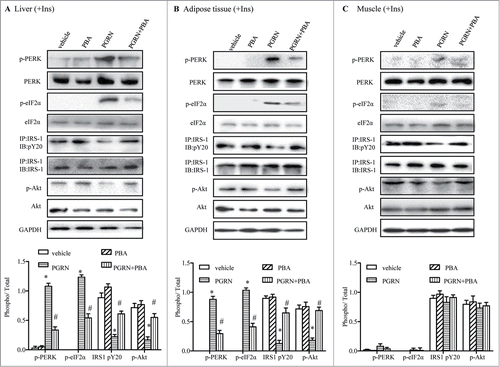
Next, we determined whether the induction of ER stress after intraperitoneal PGRN treatment was functionally related to the ability of PGRN to inhibit insulin sensitivity. PBA, a small chemical chaperone, has been shown to improve ER folding capacity, and restore glucose homeostasis in obese mice. Consistent with its effect to reduce ER stress in obese mice,Citation25 mice co-treated with PBA and PGRN exhibited considerable attenuation of PERK and eIF2α phosphorylations in liver and adipose tissue, with grossly alleviated ER stress (). Notably, oral administration of PBA to PGRN-treated mice abrogated PGRN′s ability to alter insulin sensitivity, as evidence by an increase in the GIR required to attain euglycemia and a reduction in HGP (Fig. S1A–B), as well as GTT and ITT (). Likewise, PBA augmented insulin-mediated inhibition of hepatic G6pase and Pck1 expression (Fig. S1E, F). Furthermore, there were no substantial changes in serum levels of glucose, insulin, or lipids, as well as other measured basal values in mice treated with PBA (). We next examined whether these biochemical alterations enhanced the signaling capacity of the insulin receptor in liver and adipose tissues of PGRN-treated mice. The failure of PGRN to modulate insulin sensitivity in the presence of PBA was also manifested by a lack of change in insulin-induced phosphorylation of IRS-1 tyrosine and Akt Ser-473 in the liver and adipose tissue compared with mice receiving PGRN (), indicating that amelioration of ER stress via PBA treatment partially protected against PGRN-induced impaired insulin signaling.
Figure 4. Effect of PBA treatment on markers of ER stress in the liver, adipose tissue, and muscle tissue. All analyses compare mice distributed in 4 groups (12–15/per group): (1) vehicle (2) PGRN (i.p. 20 μg/day); (3) 4-PBA (orally, 1 g/Kg of body weight); (4) PGRN + 4-PBA. For insulin signaling, they were injected with 2I U/Kg for insulin (Ins). (A–C) The indicators of ER stress and insulin receptor signaling were measured in liver, adipose and skeletal muscle at protein level. The data expressed as means ± SEM in each bar graph. *P < 0.05 (vs. vehicle). #P < 0.05 (vs. PGRN).
ER stress regulates the ability of PGRN to inhibit insulin sensitivity in hepatocytes, 3T3-L1 adipocytes, and L6 muscle cells
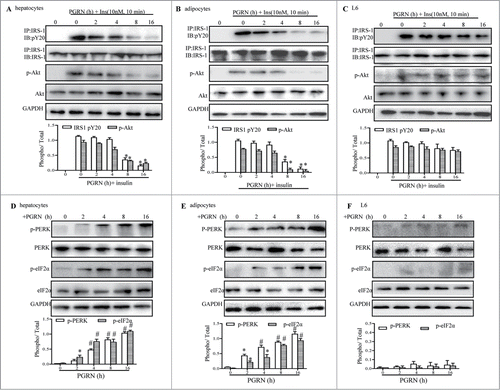
To gain further insight into the role of PGRN on insulin signaling in vitro, we pretreated fully differentiated 3T3-L1 adipocytes, hepatocytes, and L6 muscle cells with 100ng/ml PGRN at different time points, and then stimulated with insulin for 10 min, and total membranes were probed by Western blotting with the appropriate anti-body. As shown in , insulin-stimulated phosphorylation of both IRS-1 and Akt was diminished slightly in hepatocytes upon exposure to PGRN for 8 hr, and a further reduction was observed after treatment with PGRN for 16 hr, indicating that insulin receptor signaling was time-dependently blocked. Similarly, insulin signaling as in hepatocytes was also impaired in adipocytes after exposure to PGRN for 8 hr, and was more dramatically down-regulated at longer periods of time (). Phosphorylation of both IRS-1 and Akt was slightly decreased by PGRN in L6 cells even in the 16 hrs (). Notably, hepatocytes, and 3T3-L1 adipocytes exposed to PGRN exhibited a marked increase in phosphorylation of PERK and eIF2α proteins expression at 2 hr compared to the controls, and their expression levels tended to increase in a time-dependent manner (). In contrast, we observed no significant changes in insulin-promoted phosphorylation of IRS-1 and Akt until 8 hr, indicating a potential causal involvement of ER stress in the induction of insulin insensitivity by PGRN. Meanwhile, ER stress was barely induced in L6 muscle cells with exposure to PGRN (). Consistent with what we found in the animal models, when hepatocytes, adipocytes, and muscle cells were treated with PBA in the presence of PGRN for 16 hr, the phosphorylation of PERK and eIF2α were almost completely reversed (Fig. S3A–C). Of note, there was a pronounced recovery of insulin signaling capacity in PGRN-treated cells exposed to PBA treatment, as evidence by the increased phosphorylation of IRS-1 and Akt (Fig. S3A–C), raising the possibility that the activation of ER stress by PGRN might contribute to promote the suppression of insulin receptor signaling.
Figure 5. Time course of the trigger of ER stress and the inhibition of insulin signaling in vitro. Cells were treated with or without 100 ng/ml PGRN at indicated times. The indicators of ER stress and insulin receptor signaling were measured at protein level. The relative quantity of proteins was analyzed using the Image J software. (A–C) The phosphorylation of IRS-1 and Akt in hepatocytes, adipocytes and L6 cells. (D–F) The phosphorylation of PERK and eIF2α in hepatocytes, adipocytes and L6 cells. A representative blot is shown and data expressed as means ± SEM in each bar graph represent the average of 3 independent experiments. *P < 0.05, #P < 0.01 (vs. cells at time point of 0 minutes).
PGRN deficiency partly prevents palmitate-induced ER stress and repressive insulin signaling in hepatocytes and 3T3-L1 adipocytes
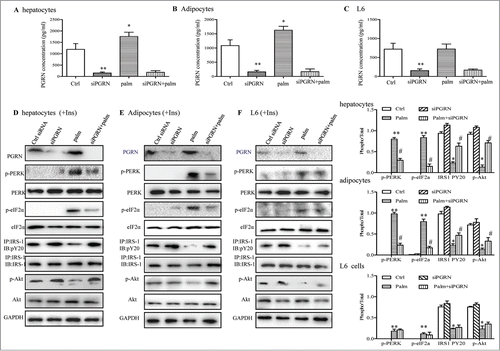
The predominant saturated nonesterified fatty acids palmitate has been shown in numerous studies to induce insulin resistance in pancreatic β cells, hepatocytes, myocytes, and adipocytes.Citation26-28 Consistent with previous experimental observations, palmitate (500 μM) was sufficient to significantly block the effects of insulin and trigger ER stress, evidenced by the decreased phosphorylation of Akt and IRS-1, and elevated phosphorylation of PERK and eIF2α (). To address whether these toxic effects of palmitate were partly due to PGRN, we first measured the PGRN secretion in culture media of cells with or without 500 μM palmitate treatment for 16 h. PGRN levels were significantly increased in hepatocytes, and 3T3-L1 adipocytes by palmitate but not in L6 muscle cells (). This prompted us to investigate the possibility that PGRN knockdown could protect against insulin resistance and ER stress from the inhibitory effects of palmitate. The efficiency of PGRN siRNA was validated by protein gel blotting (). As is evident, PGRN deficiency was capable to block the inhibitory effect of 500 µM palmitate in the regulation of insulin-stimulated Akt and IRS-1 phosphorylation in hepatocytes and adipocytes (). In agreement, these cells also showed pronounced suppression of palmitate-induced PERK and eIF2α phosphorylation, indicating the amelioration of ER stress (). Nevertheless, there were no substantial changes in insulin resistance and ER stress markers in palmitate-treated L6 cells (). Collectively, these data suggest that the loss of PGRN expression in hepatocytes, and 3T3-L1 adipocytes results in enhanced susceptibility to insulin action.
Figure 6. Involvements of PGRN in palmitate (palm)-induced IR in vitro. Control cells and PGRN-/- cells were pre-incubated with 0.5 mM palmitate for 16 h prior to incubation with 10 nM insulin (Ins) for 10 min. (A–C) PGRN content in culture media of cells with or without palmitate treatment. (D) The phosphorylation of IRS-1, Akt, PERK, and eIF2α in hepatocytes. (E) The phosphorylation of IRS-1, Akt, PERK, and eIF2α in adipocytes. (F) The phosphorylation of IRS-1, Akt, PERK, and eIF2α in L6 cells. The right is the quantification of proteins phosphorylation with normalization to total protein levels for each molecule under this condition. A representative blot is shown and all graphs show means ± SEM from at least 3 independent experiments. *P < 0.05, **P < 0.01 vs. the control groups. #P < 0.05 vs. the palmitate-treated groups.
Knockdown of the PERK pathway blocks insulin resistance induced by PGRN in vitro
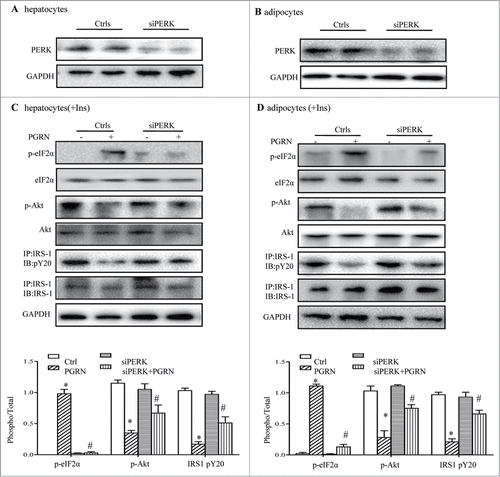
To shed light on the mechanism by which PGRN impacts on insulin signaling, we then first addressed the contribution of PERK activation to PGRN-induced insulin insensitivity by blocking PERK expression with siRNA. PERK siRNA was validated by measurement of reduced PGRN protein expression by western blot in PERK siRNA-transfected cells (). As anticipated, PERK knockdown completely suppressed protein levels of PERK and its downstream target, p-eIF2α (). Importantly, when these cells were exposed to PGRN, phosphorylation of IRS-1 and Akt was partly augmented, and the downstream ER stress-related proteins (p-eIF2α) were diminished in PGRN-treated PERK−/− cells compared to PGRN-treated WT cells (). As in hepatocytes, improved insulin signaling and attenuated ER stress markers were all evident in adipocytes pretreated with PGRN (), suggesting that PERK-eIF2α pathway plays an essential role in insulin resistance induced by PGRN and that activation levels of PERK-eIF2α pathway correlate with insulin resistance during ER stress.
Figure 7. Knockdown of the PERK-eIF2α pathway blocks insulin resistance induced by PGRN in vitro. (A) Expression of PERK in hepatocytes. (B) Expression of PERK in adipocytes. Posphorylated eIF2α, total eIF2α, phosphorylated IRS-1 and total IRS-1 of PERK knockdown hepatocytes (C) and adipocytes (D) treated with PGRN (100 ng/ml) for 16 hr. The bottom is the quantification of proteins phosphorylation with normalization to total protein levels for each molecule under this condition. A representative blot is shown and all the data expressed as means ± SEM in each bar graph represent the average of at least 3 independent experiments. *P < 0.05 vs. the control siRNA groups. #P < 0.05 vs. the control siRNA + PGRN groups.
Tumor necrosis factor receptor (TNFR) 1 is required for PGRN-induced ER stress and insulin resistance in vitro
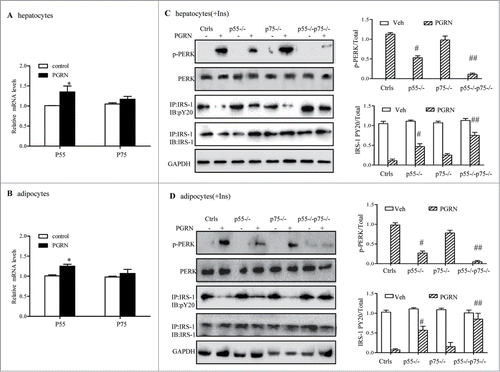
Recent findings suggested that PGRN bound to TNFR in chondrocytes and that the administration of PGRN could prevent TNF-α-induced inflammatory arthritis.Citation29 To clarify whether the effects of PGRN on ER stress and insulin resistance was dependent on TNFR pathway, we first verified that mRNA encoding both receptors was present and upregulated by PGRN exposure in hepatocytes and 3T3-L1 adipocytes (). The interaction between PGRN and TNFR in hepatocytes and adipocytes was also demonstrated by co-immunoprecipitation (Co-IP) (Fig. S4A, B). Next, specific siRNA pool to TNFR1 (p55−/−) and TNFR2 (p75−/−) were used to reduce the endogenous level of TNFR1 and TNFR2, and the efficiency was validated by protein gel blotting (data not shown). As observed in , PGRN induced impaired insulin sensitivity as well as ER stress, while p55−/− hepatocytes displayed an improved response to insulin, as evident by the phosphorylation of IRS-1, and less effect was observed in p75−/− hepatocytes compared with their PGRN-treated controls. Likewise, PGRN-induced ER stress was partially alleviated in p55−/− cells, and knockdown of p75 resulted in a similar trend in the alleviation of ER stress though these changes did not reach statistical significance. While double knockdown of TNFR p55 and p75 exhibited a higher effect than either single knockdown in the presence of PGRN, indicating a complimentary effect of both subunits and a potential role of TNFR mediating PGRN function (). Similar results were also observed in adipocytes (), suggesting that TNFR1 may function as a member receptor of PGRN in the induction of defective insulin signaling.
Figure 8. Improvement of PGRN-induced ER stress and insulin-stimulated IRS-1 phosphorylattion by blockade of TNFR in vitro. WT and TNFR1/2−/- cells were treated with vehicle or PGRN (100 ng/ml) for 16 hr. For insulin signaling, cells were stimulated with 10 nM of insulin (Ins) for 10 minutes. Indicators of ER stress and insulin signaling were measured at protein levels. The relative quantity of proteins was analyzed with Quantity One software. (A, B) Relative expression of TNFR1 (p55) receptor and TNFR2 (p75) in hepatocytes and adipocytes treated with PGRN (100 ng/ml) for 16 hr normalized to β-actin (real-time PCR). (C, D) phosphorylated PERK, total PERK, phosphorylated IRS-1 and total IRS-1 of WT, p55−/−, p75−/−, and p55−/− p75−/− hepatocytes and adipocytes. The right is the quantification of proteins phosphorylation with normalization to total protein levels for each molecule under this condition. The data expressed as means ± SEM in each bar graph represent the average of 3 independent experiments. *P < 0.05 vs. vehicle. #P < 0.05, #P < 0.01 vs. PGRN. IB, immunoblotting; IP, immunoprecipitation.
Discussion
Growing evidences have shown that PGRN is over-expressed in adipose tissues of dietary models of obesity and implicated in the pathogenesis of insulin resistance and glucose intolerance.Citation7,14,30-32 However, much remains to be elucidated regarding the mechanism of PGRN action before its putative potential as therapeutic target can be realized, since obesity is always characterized with chronic inflammation and enhanced intracellular lipid accumulation.Citation33 In the present study, mice developed signs of insulin resistance and ER stress selectively in the liver and adipose tissue upon long-term treatment with PGRN and these effects were ameliorated when PBA was administered simultaneously. Consistent with these findings in vivo, we observed the causative role of PGRN in terms of impaired insulin signaling and ER stress partially through TNFR1 via the PERK-eIF2α signaling in cultured hepatocytes and adipocytes.
PGRN is an extracellular glycoprotein, containing 7.5 repeats of a cysteine-rich motif.Citation3 Proteolytic cleavage of this precursor protein by extracellular proteases gives rise to smaller peptide fragments termed granulins (GRNs) or epithelins.Citation5,6,34 Consistent with previous evidences implicating the aggressive inflammation and insulin resistance,Citation9,11,14 we demonstrated that PGRN treatment led to the attenuated insulin signaling and ER stress, which, in turn, contributes to insulin resistance and relates with inflammation.Citation35,36 However, the normal function of PGRN is complex, with intact PGRN having anti-inflammatory properties, whereas GRNs have been shown to promote inflammation.Citation37,38 It is therefore unclear at this point whether GRNs derived by proteolysis of PGRN is involved in ER stress and the inhibition of insulin sensitivity. Due to GRNs range in size from 6 to 25 kDa, so far it is difficult to measure serum GRNs or to investigate the function of GRNs in vivo.Citation39 Besides, the function of PGRN might quite be diverse in different tissues. In line with our results, previous studies demonstrated that ablation of PGRN prevented mice from HFD-induced insulin resistance and blocked elevation of an inflammatory cytokine, IL-6.Citation7,39
PGRN has most recently been introduced as a novel adipokine inducing insulin resistance and circulating PGRN levels are associated with such as impaired glucose tolerance, which may implicate that promising therapeutic approach through the modulation of PGRN secretion/action and consequent amelioration of insulin resistance in subjects with the metabolic disor-ders.Citation7,11-14 Indeed, Matsubara et al. identified PGRN as an adipokine induced by TNF-α and dexamethasone by differential proteome analysis in cellular models of insulin resistance,Citation7 however, the mechanisms by which PGRN inhibits insulin signaling have not been addressed. In the present study, the initiation of ER stress was dramatically evident at earlier stage, and tended to increase in a time-dependent manner, while the signals for the impaired insulin action were not obvious until 16 hours in hepatocytes and adipocytes, indicating the causal involvement of ER stress in the induction of insulin resistance by PGRN. Evidence for this concept can also be seen from previous work on adipocytes, which showed a critical role of ER stress in the development of insulin resistance by triggering JNK activity via IRE1.Citation36,40 Furthermore, our data demonstrated that relieving ER stress via PBA prevents the further worsening of insulin resistance and ER stress induced by PGRN. During the early stages of obesity, ER stress may contribute to the dampening of glucose homeostasis and the initiation of autophagic machinery,Citation41-43 which would further the organelle dysfunction, and disrupt metabolic homeostasis, thus suggesting a critical role of ER stress in the induction of insulin resistance by PGRN. One critical difference, however, is highlighted by the fact that ER stress is completely dispensable for the role of PGRN in L6 cells or the muscle tissues. This observation suggests that PGRN may have unexplored functions related to insulin action in muscle cells that do not simply involve ER stress. Additional work will be needed to fully determine whether other mechanisms are also involved in integrating the role of PGRN to insulin action, and the exact mechanisms underlying ER stress upon PGRN treatment in the development of insulin resistance.
ER stress activates a set of signaling pathways collectively termed as the unfolded protein response (UPR), and plays a key role as a chronic stimulus in triggering insulin resistance and generally associates with chronic inflammation in obesity.Citation36,44 PERK signaling activates NF-κB, a transcriptional regulator that plays a central role in mediating the responses to inflammatory signaling, while translation of IκBα, the main negative regulator of NF-κB, is known to be inhibited by phosphorylation of eIF2α.Citation45 Likewise, the 2 other canonical branches of the UPR, IRE1 and ATF6, have been reported to up-regulate the expression of inflammatory cytokines.Citation19,46 In addition, inflammatory cytokines can disrupt insulin signaling by interfering with IRS-1–insulin receptor binding and promoting IRS-1 degradation.Citation47 In the present study, we showed that knockdown of PERK partially up-regulated the insulin sensitivity in cells treated with PGRN. Additionally, PGRN ablation resulted in decreased levels of IL-6 in serum and adipose tissue,Citation7 and IL-6 itself is a key player of insulin resistance.Citation48 Thus, a state of inflammation could be the part of reason for our observations. Moreover, a recent report showed that the IRE1–JNK signaling pathway directly inhibits cytoplasmic insulin signaling in ob/ob mice.Citation36 Our findings showed that PGRN caused impaired insulin sensitivity and alleviation of ER stress improved insulin signaling via TNFR pathway although it is unclear whether the mechanisms involves the IRE1–JNK signaling. In brief, the effect of PGRN-induced ER stress on insulin signaling requires further comprehensive study to elucidate the detailed mechanisms underlying the development of insulin resistance.
Although PGRN plays crucial roles in multiple physiological and pathological conditions, efforts to exploit the actions of PGRN and understand the mechanisms involved have been hampered by the inability to identify its binding receptor(s). Some report that PGRN action is not mediated through TNFR,Citation49,50 while more studies suggest that PGRN is a TNFR antagonist or a co-factor for TNFα action.Citation51 Recently, it has been shown that PGRN binds to TNFR, interfering with the interaction between TNFα and TNFR.Citation29 However, the possibility that TNFR could be a specific receptor for PGRN-mediated effects in insulin resistance has never been tested, and it is still hard to clearly define the early stages of PGRN-mediated signaling from the plasma membrane. TNFR1 and TNFR2 do not share homology in the cytoplasmic domains but exhibit a low degree of similarity, which suggests that they are capable of inducing distinct cellular responses.Citation52 In our analysis, PGRN caused impaired insulin signaling and ER stress mainly through TNFR1 rather than TNFR2 in vitro. Indeed, a growing number of evidences have demonstrated that TNFR1 is the predominant receptor mediating insulin resistance, whereas TNFR2 deficiency alone almost did not affect insulin sensitivity.Citation20,22 Several other groups also independently reproduced the binding of PGRN to TNFR1 and TNFR2, and inhibitory effect of this binding on TNFα-induced effects.Citation53-56 Nevertheless, one recent study implied that the disturbance of the interaction between PGRN and TNFR2 abolished PGRN-mediated activation of Erk1/2 pathway.Citation53 These discrepancies with our results might be possibly because there is a different distribution between TNFR1 and TNFR2 in different cell types, and the role of PGRN in energy metabolism might be more complicated than expected. Our findings support the notion that PGRN is a key regulator of hepatic and adipocytic insulin resistance and that PGRN may mediate its effects, at least in part, by binding to TNF receptors. Whether this mechanism accounts for all of the effects we observed remains to be further delineated.
When compared with TNFα PGRN exhibited a higher affinity to both TNFR1 and TNFR2, and PGRN has approximately 600-fold higher binding affinity to TNFR2 than TNFα.Citation38,57-59 Similar to PGRN, Atsttrin, an engineered protein made of 3 PGRN fragments, inhibited the interaction between TNF and TNFR, and in turn, the downstream events of TNF/TNFR signaling. In contrast to TNFα, Atsttrin exhibited a higher binding affinity for TNFR2, but a lower affinity for TNFR1.Citation29 In addition, it was also observed that TNF family ligands bind to the extracellular regions of TNFR1 and TNFR2 in which each receptor subunit contacts 2 adjacent ligand subunits typically via CRD2 and CRD3(cysteine-rich repeat domains, CRDs).Citation51,60 Deletion mutants of TNFR1 and TNFR2 used to map the binding of PGRN revealed that CRD2 and CRD3 of TNFR are essential for the interaction with PGRN, similar to the binding to TNFα.Citation55,56,61,62
In conclusion, our present study revealed that administration of PGRN attenuated insulin signaling and triggered ER stress in vivo and in vitro studies, with such effects being drastically reversed by PBA, suggesting a causative role of ER stress in PGRN-induced impaired insulin sensitivity and implicated that decreasing PGRN levels by influencing its turnover or production is consequently a promising therapeutic approach applied to metabolic disorders.
Materials and Methods
Materials
All chemicals used were of analytical grade and were purchased from Sigma-Aldrich (St Louis, Missouri) unless otherwise stated. The following antibodies were used: anti-p-PERK (#3179), anti-PERK (#3192) (Cell Signaling Technology Inc., Danvers, Massachusetts); anti-IRS-1 (#sc-559), anti-pY20 (#sc-508), anti-p-Akt (#sc-33437), anti-Akt (#sc-8312), anti-p-eIF2α (#sc-101670), anti-eIF2α (#sc-11386), anti-TNFR1(#sc-8436), anti-TNFR2(#sc-7862) and anti-GAPDH (#sc-365062) (Santa Cruz Biotechnology Inc., Santa Cruz, California).
Preparation of recombinant mouse PGRN (rmPGRN)
Mouse PGRN cDNA clone, MGC Image clone, was purchased from Invitrogen. pCAGIPuro-FLAGmPGRN was constructed by subcloning the insert encoding the mouse PGRN without signal peptide (amino acid 18 to 589), into pCAGIPuro-FLAG. To prepare CHO-K1 cells stably expressing the FLAG-tagged mouse PGRN, CHO-K1 cells were transfected with pCAGIPuro-FLAG-mPGRN construct by electroporation. Stably expressed cells were maintained in CD OptiCHO medium (Invitrogen) supplemented with 10 μg/ml puromycin (SIGMA), 4 mM GlutaMAX (Invitrogen), 1 × HT supplement (Invitrogen) and 10 μg/ml insulin (SIGMA). Culture supernatants were collected and subjected to anti-FLAG M1 agarose affinity gel (SIGMA) column. The column was washed with 50 mM Tris-HCl (pH 7.5), 150 mM NaCl and 1 mM CaCl2, and then eluted with 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, and 2 mM EDTA. The eluted proteins were dialyzed against PBS. The eluted proteins were dialyzed against PBS. Purified PGRN was made endotoxin-free using the Detoxi-Gel endotoxin removing column (Pierce) as recommended by the manufacturer. Because of the remote possibility of low levels of endotoxin contamination, an “ultrapure” batch of PGRN was prepared using an additional gel filtration column (Superdex-75, SMART system; Amersham, Arlington Heights, IL).
Animals
Male C57BL/6J mice (4-week old) were group-housed at 22–24°C on a 12-h light/dark cycle with the lights on at 8:00 AM and had free access to food and water. All animals were fed with standard rodent chow (Harlan Teklad, Madison, WI). Mice were distributed in 4 groups (12–15/per group): (1) vehicle (saline solution intraperitoneally i.p.); (2) rmPGRN (i.p.20 μg/day, 6 p.m.); (3) phenylbutyric acid (PBA) (orally, 1 g/Kg of body weight); (4) rmPGRN (i.p.20 μg/day, 6 p.m.)+PBA (orally, 1 g/Kg of body weight). At the end of the 3-week study period, mice received an ip injection of insulin at a dosage of 2 IU/Kg for insulin signaling; 15 minutes after the injection, all mice were euthanized, and their blood, livers, omental adipose tissue, and gastrocnemius muscle were obtained and stored at −80°C for subsequent analysis. Body weight, food intake, and tail blood glucose were monitored every other day. Blood glucose levels were measured by Antsense III glucose analyzer (Bayer Ya kuhin, Osaka, Japan) and serum insulin levels were measured by an ultrasensitive mouse-specific ELISA kit (Crystal Chem, Downers Grove, Illinois) with intra- and interassay coefficients of variation of 3.9% to 6.6% and 5.6% to 5.8%, respectively. Peripheral serum was subject to ELISA using standard kits (R&D Systems, Inc., Minneapolis, MN, USA) for PGRN. Animal care and experimental procedures performed in this study were approved by the Institutional Animal Ethics Committee for the Care and Use of Laboratory Animals.
Metabolic tests
Glucose tolerance testing (GTT) was performed after the mice were fasted overnight. A total of 2 g/Kg glucose was administrated through an ip injection, and blood glucose was measured at the indicated time points. Insulin tolerance testing (ITT) was performed after the animals had fasted for 4 hours. Then, 0.75 U/Kg insulin was administered via ip injection and blood glucose was measured at the indicated time points. Free fatty acid and triglyceride levels were determined in duplicate using an enzyme colorimetric assay (BioVision, Mountain View, California).
Physiological measurements
To measure food intake, mice were individually housed in metabolic cages and fed ad libitum. Food consumption was determined by weighing the powdered diet before and after a 24-hour period. Energy expenditure parameters were measured using a 6-chamber Oxymax system (Columbus Instruments, Columbus, Ohio).
Hyperinsulinemic-euglycemic clamp
Seven days prior to the hyperinsulinemic-euglycemic clamp studies, indwelling catheters were placed into the right internal jugular vein. After overnight fast, [3−3H]-glucose (HPLC purified; PerkinElmer) was infused at a rate of 0.05 µCi per min for 2 h to assess the basal glucose turnover. Following the basal period, hyperinsulinemic-euglycemic clamp was conducted for 140 min with a primed/continuous infusion of human insulin (126 pmol per kg during priming, 18 pmol per kg per min during infusion) (Eli Lilly), while plasma glucose was maintained at a basal concentration (˜6.7 mM) as described.Citation41 Throughout the clamps, [3−3H]-glucose was infused at a rate of 0.1 µCi per min to estimate insulin-stimulated whole-body glucose flux, and 2-deoxy-D-[1−14C]-glucose (2-[14C]-DG; PerkinElmer) was injected as a bolus at the 85th minute of the clamp to estimate the rate of insulin-stimulated tissue glucose uptake. Blood samples (10 µl) were taken at the end of the basal period and during the last 45 min of the clamp for the measurement of plasma 3H and 14C activities.
Glucose flux calculation
To determine plasma 3H and 14C activities, plasma was deproteinized with ZnSO4 and Ba(OH)2, dried to remove 3H2O, resuspended in water and counted in scintillation fluid (Ultima gold, PerkinElmer) using a Beckman scintillation counter. The rates of basal and insulin-stimulated whole-body glucose flux were determined as the ratio of the [3−3H]-glucose infusion rate (disintegrations per minute [DPM]) to the specific activity of plasma glucose (DPM per mg) at the end of the basal period and during the final 30 min of the clamp experiment, respectively. Hepatic glucose production (HGP) was determined by subtracting the glucose infusion rate from the total glucose appearance rate. The plasma concentration of 3H2O was determined by the difference between 3H counts with and without drying. Whole-body glycolysis was calculated from the rate of the increase in plasma 3H2O concentration divided by the specific activity of plasma 3H-glucose, as previously described. Whole-body glycogen synthesis was estimated by subtracting whole-body glycolysis from whole-body glucose uptake, assuming that glycolysis and glycogen synthesis account for the majority of insulin-stimulated glucose uptake. To determine individual tissue glucose uptake, tissue samples were homogenized, and the supernatants were subjected to an ion-exchange column to separate tissue 14C-2-DG-6-phosphate (2-DG-6-P) from 2-DG. Tissue glucose uptake was calculated from the area under the curve of plasma 14C-2-DG profile for the last 45 min of the clamp and muscle 14C-2-DG-6-P content, as previously described.
In vivo fatty acid oxidation
In vivo fatty acid oxidation rate was determined by the rate of 14CO2 production after infusion of 3 μCi of [1−14C]-oleic acid (PerkinElmer) as previously described.Citation63,64 Briefly, 300 μg of cold oleic acid (Sigma) conjugated to fatty acid-free BSA (Sigma) was injected intraperitoneally into mice with [1−14C]-oleic acid (3μCi) at 9 AM. Mice were put in metabolic chambers connected with 1 N NaOH trap to capture expired 14CO2. 14C radioactivity from NaOH trap was counted at 30-min interval over the next 4h, and the slope for the initial 2 h was plotted because captured radioactivity is saturated after 2 h.
Mitochondria histomorphometry
Superficial gastrocnemius muscles were fixed in 4% parafor-maldehyde-2% glutaraldehyde-0.1 M sodium cacodylate, pH 7.3, postfixed in 1% osmium tetroxide, and embedded in epoxyresin (Epon). Ultrathin sections (80 nm) were stained with aque-ous uranyl acetate and lead citrate and examined with a JEOL 2000FX transmission electron microscope (JEOL, Peabody, Massachusetts). Sixteen electron micrographs per mouse were digitized and the area and number of clearly distinguishable mitochondria were analyzed using OsteoMeasure software (OsteoMetrics, Decatur, Georgia). Mitochondria in liver and adipose tissue were observed by the same methods.
Liver and muscle histomorphometry
Liver and skeletal muscle samples (5 × 5 × 5 mm) were fixed in paraformaldehyde 4%–1×PBS, washed 3 times with 1×PBS, and equilibrated in 20% sucrose-1 × PBS, before being embedded in OCT (optimal cutting temperature) compound. Samples were next sectioned at 10 nm using a cryostat. The sections were air-dried, postfixed in formalin, rinsed with 60% isopropanol, stained with Oil Red O (in 60% isopropanol), and counter-stained with hematoxylin. The Oil Red O–positive area over the total area was quantified using Quantity One software.
Cell culture and treatment
BNL-CL.2, 3T3-L1 cells, L6 cells were purchased from the American Type Culture Collection (ATCC, Manassas, VA) and cultured in DMEM with 10% fetal bovine serum (FBS) (HyClone, Thermo Fisher Scientific Inc.. Logan, UT). 3T3-L1 cells were induced to differentiate mature fat cells with induction media (isobutylmethylxanthine 0.5mM, dexamethasone 1 μM, insulin 10 μg/ml and DMEM with 10% FBS) and insulin media in turn every 2 d Four days later, the media were changed to 10% FBS/DMEM. Cells were then fed with 10% FBS/DMEM every 2 d Full differentiation is usually achieved on the 8th day. For the effect of PGRN, cells were treated with 100 ng/ml of PGRN at the indicated times. For insulin signaling, cells were stimulated with 10 nM of insulin for 10 min. Prior to each experiment, cells were washed and starved for 4 h in DMEM with 1% FBS.
Gene silencing
Gene silencing was carried out as manufacturers described. Cells were transfected with a silencing RNA (siRNA) targeted for mouse PGRN (sc-39262), PERK (sc-36214), TNFR1 (sc-36688) and TNFR2 (sc-36690) using Lipofectamine 2000 (Invitrogen). A siRNA consisting of a scrambled sequence of similar length was used as a control. One day before transfection, cells were plated in 500 μl of growth medium without antibiotics such that they were 30–50% confluent at the time of transfection. The transfected cells were cultured in DMEM containing 10% FBS for 72 h after transfection.
mRNA isolation and analysis by real-time PCR
Total RNA was extracted from liver, adipose tissues or muscle tissues with TRIzol reagent (Invitrogen) and converted into cDNA using a cDNA synthesis kit (Applied Biosystems). Quantitative real-time PCR was performed using SYBR Green in 7300 Real-Time PCR System (Applied Biosystems). Quantitative real-time PCR analysis was performed as described previously.Citation65 Primers are as follows (name, sense, and antisense primer): sXBP-1, 5′-GAACCAGGAGTTAAGAACACG-3′ and 5′-AGGCAACAGTGTCAGAGTCC-3′; ATF6, 5′- TGCTAGGACTGGAGGCCAGGCTCAA-3′ and 5′-CATGTCTATGAACCCAGCCTCGAAGT-3′; TNFR1, 5′-CTCTTGGTGACCGGGAGAAG-3′ and 5′-GGTTCCTTTGTGGCACTTGGT-3′; TNFR2, 5′-CATCCCTGTGTCCTTGGG-3′ and 5′-CCCGTGATGCTTGGTTCA-3′.The level of target gene expression was normalized against β-actin.
Western blot and immunoprecipitation (IP)
Tissues and cells under various treatments were lysed in lysis buffer containing 25 mM Tris HCl (pH 6.8), 2% SDS, 6% glycerol, 1% 2-mercaptoethanol, 2 mM phenylmethylsulfonyl fluoride, 0.2% bromophenol blue, and a protease inhibitor cocktail for 20 min. Nuclear extracts were obtained using a nuclear extraction kit (Sigma, St. Louis, MO). Western blotting was performed by utilizing a standard protocol as described.
IP was performed as described in the previous study.Citation44 200 μg of cytoplasmic lysate were incubated for 2 h at 4°C with the corresponding antibodies coupled to 20 μl of packed protein A+G sepharose beads (Beyotime). Immunocomplexes were resolved by means of SDS-PAGE and immuno-blotted with the indicated antibodies. The relative quantity of proteins was analyzed using the Image J software.
Statistics analysis
Data were analyzed by IBM SPSS 20.0 software. Statistical analysis between the 2 groups was performed using an unpaired, 2-tailed Student t test. Multiple comparisons of quantitative variables among groups were made using one-way ANOVA with the least significant difference post hoc test. P < 0.05 was considered to be significant.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
1041686_supplemental_files.zip
Download Zip (8.1 MB)Acknowledgment
We appreciate the technical support and materials from the electron microscope center of Xi'an Jiaotong University.
Funding
National Natural Science Foundation of China (General Program nos. 30971392, 81170741, 81071440 and 81222026), National Excellent Young Scientist Program (no.81222026) and the New Century Excellent Talents in University from the Ministry of Education, China (NCET-08–0435, NCET-11–0437).
Related Research Data
References
- Wu S, Zang W, Li X, Sun H. Proepithelin stimulates growth plate chondrogenesis via nuclear factor-kappaB-p65-dependent mechanisms. J Biol Chem 2011; 286:24057-67; PMID:21566130; http://dx.doi.org/10.1074/jbc.M110.201368
- Gijselinck I, van der Zee J, Engelborghs S, Goossens D, Peeters K, Mattheijssens M, Corsmit E, Del-Favero J, De Deyn PP, Van Broeckhoven C, et al. Progranulin locus deletion in frontotemporal dementia. Hum Mutat 2008; 29:53-8; PMID:18157829; http://dx.doi.org/10.1002/humu.20651
- Cenik B, Sephton CF, Kutluk Cenik B, Herz J, Yu G. Progranulin: a proteolytically processed protein at the crossroads of inflammation and neurodegeneration. J Biol Chem 2012; 287:32298-306; PMID:22859297; http://dx.doi.org/10.1074/jbc.R112.399170
- Nicholson AM, Gass J, Petrucelli L, Rademakers R. Progranulin axis and recent developments in frontotemporal lobar degeneration. Alz Res Ther 2012; 4:4; PMID:22277331; http://dx.doi.org/10.1186/alzrt102
- He Z, Bateman A. Progranulin (granulin-epithelin precursor, PC-cell-derived growth factor, acrogranin) mediates tissue repair and tumorigenesis. J Mol Med 2003; 81:600-12; PMID:12928786; http://dx.doi.org/10.1007/s00109-003-0474-3
- Daniel R, Daniels E, He Z, Bateman A. Progranulin (acrogranin/PC cell-derived growth factor/granulin-epithelin precursor) is expressed in the placenta, epidermis, microvasculature, and brain during murine development. Dev Dynam: Off Pub Am Assoc Anatomists 2003; 227:593-9; PMID:12889069; http://dx.doi.org/10.1002/dvdy.10341
- Matsubara T, Mita A, Minami K, Hosooka T, Kitazawa S, Takahashi K, Tamori Y, Yokoi N, Watanabe M, Matsuo E, et al. PGRN is a key adipokine mediating high fat diet-induced insulin resistance and obesity through IL-6 in adipose tissue. Cell Metab 2012; 15:38-50; PMID:22225875; http://dx.doi.org/10.1016/j.cmet.2011.12.002
- Bossu P, Salani F, Alberici A, Archetti S, Bellelli G, Galimberti D, Scarpini E, Spalletta G, Caltagirone C, Padovani A, et al. Loss of function mutations in the progranulin gene are related to pro-inflammatory cytokine dysregulation in frontotemporal lobar degeneration patients. J Neuroinflammation 2011; 8:65; PMID:21645364; http://dx.doi.org/10.1186/1742-2094-8-65
- Qu H, Deng H, Hu Z. Plasma progranulin concentrations are increased in patients with type 2 diabetes and obesity and correlated with insulin resistance. Mediat Inflammat 2013; 2013:360190; PMID:23476101; http://dx.doi.org/10.1155/2013/360190
- Richter J, Focke D, Ebert T, Kovacs P, Bachmann A, Lossner U, Kralisch S, Kratzsch J, Beige J, Anders M, et al. Serum levels of the adipokine progranulin depend on renal function. Diabetes Care 2013; 36:410-4; PMID:23033238; http://dx.doi.org/10.2337/dc12-0220
- Youn BS, Bang SI, Kloting N, Park JW, Lee N, Oh JE, Pi KB, Lee TH, Ruschke K, Fasshauer M, et al. Serum progranulin concentrations may be associated with macrophage infiltration into omental adipose tissue. Diabetes 2009; 58:627-36; PMID:19056610; http://dx.doi.org/10.2337/db08-1147
- Tonjes A, Fasshauer M, Kratzsch J, Stumvoll M, Bluher M. Adipokine pattern in subjects with impaired fasting glucose and impaired glucose tolerance in comparison to normal glucose tolerance and diabetes. PloS One 2010; 5:e13911; PMID:21085476; http://dx.doi.org/10.1371/journal.pone.0013911
- Kloting N, Fasshauer M, Dietrich A, Kovacs P, Schon MR, Kern M, Stumvoll M, Bluher M. Insulin-sensitive obesity. Am J Physiol Endocrinol Metab 2010; 299:E506-15; PMID:20570822; http://dx.doi.org/10.1152/ajpendo.00586.2009
- Yoo HJ, Hwang SY, Hong HC, Choi HY, Yang SJ, Choi DS, Baik SH, Bluher M, Youn BS, Choi KM. Implication of progranulin and C1q/TNF-related protein-3 (CTRP3) on inflammation and atherosclerosis in subjects with or without metabolic syndrome. PloS One 2013; 8:e55744; PMID:23409033; http://dx.doi.org/10.1371/journal.pone.0055744
- Richter J, Ebert T, Stolzenburg JU, Dietel A, Hopf L, Hindricks J, Kralisch S, Kratzsch J, Fasshauer M. Response to comment on: Richter et al. Serum levels of the adipokine progranulin depend on renal function. Diabetes Care 2013;36:410-414. Diabetes care 2013; 36:e84; PMID:23033238; http://dx.doi.org/10.2337/dc12-0220
- Feng JQ, Guo FJ, Jiang BC, Zhang Y, Frenkel S, Wang DW, Tang W, Xie Y, Liu CJ. Granulin epithelin precursor: a bone morphogenic protein 2-inducible growth factor that activates Erk1/2 signaling and JunB transcription factor in chondrogenesis. FASEB J: Off Pub Federat Am Soc Exp Biol 2010; 24:1879-92; PMID:20124436; http://dx.doi.org/10.1096/fj.09-144659
- Guo FJ, Liu Y, Zhou J, Luo S, Zhao W, Li X, Liu C. XBP1S protects cells from ER stress-induced apoptosis through Erk1/2 signaling pathway involving CHOP. Histochem Cell Biol 2012; 138:447-60; PMID:22669460; http://dx.doi.org/10.1007/s00418-012-0967-7
- Masuda M, Miyazaki-Anzai S, Levi M, Ting TC, Miyazaki M. PERK-eIF2alpha-ATF4-CHOP signaling contributes to TNFalpha-induced vascular calcification. J Am Heart Assoc 2013; 2:e000238; PMID:24008080; http://dx.doi.org/10.1161/JAHA.113.000238
- Hu P, Han Z, Couvillon AD, Kaufman RJ, Exton JH. Autocrine tumor necrosis factor α links endoplasmic reticulum stress to the membrane death receptor pathway through IRE1alpha-mediated NF-kappaB activation and down-regulation of TRAF2 expression. Mol Cell Biol 2006; 26:3071-84; PMID:16581782; http://dx.doi.org/10.1128/MCB.26.8.3071-3084.2006
- Uysal KT, Wiesbrock SM, Hotamisligil GS. Functional analysis of tumor necrosis factor (TNF) receptors in TNF-α-mediated insulin resistance in genetic obesity. Endocrinology 1998; 139:4832-8; PMID:9832419
- Greene MW, Ruhoff MS, Burrington CM, Garofalo RS, Orena SJ. TNFalpha activation of PKCdelta, mediated by NFkappaB and ER stress, cross-talks with the insulin signaling cascade. Cell Signalling 2010; 22:274-84; PMID:19782747; http://dx.doi.org/10.1016/j.cellsig.2009.09.029
- Peraldi P, Hotamisligil GS, Buurman WA, White MF, Spiegelman BM. Tumor necrosis factor (TNF)-α inhibits insulin signaling through stimulation of the p55 TNF receptor and activation of sphingomyelinase. J Biol Chem 1996; 271:13018-22; PMID:8662983; http://dx.doi.org/10.1074/jbc.271.22.13018
- Ito D, Yagi T, Ikawa M, Suzuki N. Characterization of inclusion bodies with cytoprotective properties formed by seipinopathy-linked mutant seipin. Hum Mol Genet 2012; 21:635-46; PMID:22045697; http://dx.doi.org/10.1093/hmg/ddr497
- Judy ME, Nakamura A, Huang A, Grant H, McCurdy H, Weiberth KF, Gao F, Coppola G, Kenyon C, Kao AW. A shift to organismal stress resistance in programmed cell death mutants. PLoS Genet 2013; 9:e1003714; PMID:24068943; http://dx.doi.org/10.1371/journal.pgen.1003714
- Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO, Gorgun CZ, Hotamisligil GS. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 2006; 313:1137-40; PMID:16931765; http://dx.doi.org/10.1126/science.1128294
- Mayer CM, Belsham DD. Palmitate attenuates insulin signaling and induces endoplasmic reticulum stress and apoptosis in hypothalamic neurons: rescue of resistance and apoptosis through adenosine 5' monophosphate-activated protein kinase activation. Endocrinology 2010; 151:576-85; PMID:19952270; http://dx.doi.org/10.1210/en.2009-1122
- Peng G, Li L, Liu Y, Pu J, Zhang S, Yu J, Zhao J, Liu P. Oleate blocks palmitate-induced abnormal lipid distribution, endoplasmic reticulum expansion and stress, and insulin resistance in skeletal muscle. Endocrinology 2011; 152:2206-18; PMID:21505048; http://dx.doi.org/10.1210/en.2010-1369
- Guo W, Wong S, Xie W, Lei T, Luo Z. Palmitate modulates intracellular signaling, induces endoplasmic reticulum stress, and causes apoptosis in mouse 3T3-L1 and rat primary preadipocytes. Am J Physiol Endocrinol Metab 2007; 293:E576-86; PMID:17519282; http://dx.doi.org/10.1152/ajpendo.00523.2006
- Tang W, Lu Y, Tian QY, Zhang Y, Guo FJ, Liu GY, Syed NM, Lai Y, Lin EA, Kong L, et al. The growth factor progranulin binds to TNF receptors and is therapeutic against inflammatory arthritis in mice. Science 2011; 332:478-84; PMID:21393509; http://dx.doi.org/10.1126/science.1199214
- Dupuis L, Petersen A, Weydt P. Progranulin bridges energy homeostasis and fronto-temporal dementia. Cell Metab 2012; 15:269-70; author reply 70; PMID:22405061; http://dx.doi.org/10.1016/j.cmet.2012.02.003
- Hossein-Nezhad A, Mirzaei K, Ansar H, Emam-Gholipour S, Tootee A, Keshavarz SA. Obesity, inflammation and resting energy expenditure: possible mechanism of progranulin in this pathway. Minerva Endocrinologica 2012; 37:255-66; PMID:22766892
- Kim HK, Shin MS, Youn BS, Namkoong C, Gil SY, Kang GM, Yu JH, Kim MS. Involvement of progranulin in hypothalamic glucose sensing and feeding regulation. Endocrinology 2011; 152:4672-82; PMID:21933869; http://dx.doi.org/10.1210/en.2011-1221
- Guilherme A, Virbasius JV, Puri V, Czech MP. Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat Rev Mol Cell Biol 2008; 9:367-77; PMID:18401346; http://dx.doi.org/10.1038/nrm2391
- Bateman A, Belcourt D, Bennett H, Lazure C, Solomon S. Granulins, a novel class of peptide from leukocytes. Biochem Biophy Res Commun 1990; 173:1161-8; PMID:2268320; http://dx.doi.org/10.1016/S0006-291X(05)80908-8
- Kawasaki N, Asada R, Saito A, Kanemoto S, Imaizumi K. Obesity-induced endoplasmic reticulum stress causes chronic inflammation in adipose tissue. Sci Rep 2012; 2:799; PMID:23150771; http://dx.doi.org/10.1038/srep00799
- Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, Tuncman G, Gorgun C, Glimcher LH, Hotamisligil GS. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 2004; 306:457-61; PMID:15486293; http://dx.doi.org/10.1126/science.1103160
- Park B, Buti L, Lee S, Matsuwaki T, Spooner E, Brinkmann MM, Nishihara M, Ploegh HL. Granulin is a soluble cofactor for toll-like receptor 9 signaling. Immunity 2011; 34:505-13; PMID:21497117; http://dx.doi.org/10.1016/j.immuni.2011.01.018
- Zhu J, Nathan C, Jin W, Sim D, Ashcroft GS, Wahl SM, Lacomis L, Erdjument-Bromage H, Tempst P, Wright CD, et al. Conversion of proepithelin to epithelins: roles of SLPI and elastase in host defense and wound repair. Cell 2002; 111:867-78; PMID:12526812; http://dx.doi.org/10.1016/S0092-8674(02)01141-8
- Tanaka A, Tsukamoto H, Mitoma H, Kiyohara C, Ueda N, Ayano M, Ohta S, Inoue Y, Arinobu Y, Niiro H, et al. Serum progranulin levels are elevated in patients with systemic lupus erythematosus, reflecting disease activity. Arthrit Res Ther 2012; 14:R244; PMID:23140401; http://dx.doi.org/10.1186/ar4087
- Li H, Zhou B, Xu L, Liu J, Zang W, Wu S, Sun H. Circulating PGRN is significantly associated with systemic insulin sensitivity and autophagic activity in metabolic syndrome. Endocrinology 2014; 155:3493-507; PMID:24971611; http://dx.doi.org/10.1210/en.2014-1058
- Yang L, Li P, Fu S, Calay ES, Hotamisligil GS. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab 2010; 11:467-78; PMID:20519119; http://dx.doi.org/10.1016/j.cmet.2010.04.005
- Qin L, Wang Z, Tao L, Wang Y. ER stress negatively regulates AKT/TSC/mTOR pathway to enhance autophagy. Autophagy 2010; 6:239-47; PMID:20104019; http://dx.doi.org/10.4161/auto.6.2.11062
- Ding WX, Ni HM, Gao W, Yoshimori T, Stolz DB, Ron D, Yin XM. Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am J Pathol 2007; 171:513-24; PMID:17620365; http://dx.doi.org/10.2353/ajpath.2007.070188
- Zhou B, Li H, Xu L, Zang W, Wu S, Sun H. Osteocalcin reverses endoplasmic reticulum stress and improves impaired insulin sensitivity secondary to diet-induced obesity through nuclear factor-kappaB signaling pathway. Endocrinology 2013; 154:1055-68; PMID:23407450; http://dx.doi.org/10.1210/en.2012-2144
- Deng J, Lu PD, Zhang Y, Scheuner D, Kaufman RJ, Sonenberg N, Harding HP, Ron D. Translational repression mediates activation of nuclear factor kappa B by phosphorylated translation initiation factor 2. Mol Cell Biol 2004; 24:10161-8; PMID:15542827; http://dx.doi.org/10.1128/MCB.24.23.10161-10168.2004
- Yamazaki H, Hiramatsu N, Hayakawa K, Tagawa Y, Okamura M, Ogata R, Huang T, Nakajima S, Yao J, Paton AW, et al. Activation of the Akt-NF-kappaB pathway by subtilase cytotoxin through the ATF6 branch of the unfolded protein response. J Immunol 2009; 183:1480-7; PMID:19561103; http://dx.doi.org/10.4049/jimmunol.0900017
- Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest 2006; 116:1793-801; PMID:16823477; http://dx.doi.org/10.1172/JCI29069
- Senn JJ, Klover PJ, Nowak IA, Mooney RA. Interleukin-6 induces cellular insulin resistance in hepatocytes. Diabetes 2002; 51:3391-9; PMID:12453891; http://dx.doi.org/10.2337/diabetes.51.12.3391
- Chen X, Chang J, Deng Q, Xu J, Nguyen TA, Martens LH, Cenik B, Taylor G, Hudson KF, Chung J, et al. Progranulin does not bind tumor necrosis factor (TNF) receptors and is not a direct regulator of TNF-dependent signaling or bioactivity in immune or neuronal cells. J Neurosci: Off J Soc Neurosci 2013; 33:9202-13; PMID:23699531; http://dx.doi.org/10.1523/JNEUROSCI.5336-12.2013
- Etemadi N, Webb A, Bankovacki A, Silke J, Nachbur U. Progranulin does not inhibit TNF and lymphotoxin-α signalling through TNF receptor 1. Immunol Cell Biol 2013; 91:661-4; PMID:24100384; http://dx.doi.org/10.1038/icb.2013.53
- Liu CJ, Bosch X. Progranulin: a growth factor, a novel TNFR ligand and a drug target. Pharmacol Therap 2012; 133:124-32; PMID:22008260; http://dx.doi.org/10.1016/j.pharmthera.2011.10.003
- Liang H, Yin B, Zhang H, Zhang S, Zeng Q, Wang J, Jiang X, Yuan L, Wang CY, Li Z. Blockade of tumor necrosis factor (TNF) receptor type 1-mediated TNF-α signaling protected Wistar rats from diet-induced obesity and insulin resistance. Endocrinology 2008; 149:2943-51; PMID:18339717; http://dx.doi.org/10.1210/en.2007-0978
- Li M, Liu Y, Xia F, Wu Z, Deng L, Jiang R, Guo FJ. Progranulin is required for proper ER stress response and inhibits ER stress-mediated apoptosis through TNFR2. Cell Signalling 2014; 26:1539-48; PMID:24703938; http://dx.doi.org/10.1016/j.cellsig.2014.03.026
- Hu Y, Xiao H, Shi T, Oppenheim JJ, Chen X. Progranulin promotes tumour necrosis factor-induced proliferation of suppressive mouse CD4(+) Foxp3(+) regulatory T cells. Immunology 2014; 142:193-201; PMID:24383743; http://dx.doi.org/10.1111/imm.12241
- Jian J, Zhao S, Tian Q, Gonzalez-Gugel E, Mundra JJ, Uddin SM, Liu B, Richbourgh B, Brunetti R, Liu CJ. Progranulin directly binds to the CRD2 and CRD3 of TNFR extracellular domains. FEBS Lett 2013; 587:3428-36; PMID:24070898; http://dx.doi.org/10.1016/j.febslet.2013.09.024
- Liu C, Li XX, Gao W, Liu W, Liu DS. Progranulin-derived Atsttrin directly binds to TNFRSF25 (DR3) and inhibits TNF-like ligand 1A (TL1A) activity. PloS One 2014; 9:e92743
- Bluml S, Binder NB, Niederreiter B, Polzer K, Hayer S, Tauber S, Schett G, Scheinecker C, Kollias G, Selzer E, et al. Antiinflammatory effects of tumor necrosis factor on hematopoietic cells in a murine model of erosive arthritis. Arthritis Rheum 2010; 62:1608-19; PMID:20155834; http://dx.doi.org/10.1002/art.27399
- Faustman D, Davis M. TNF receptor 2 pathway: drug target for autoimmune diseases. Nat Rev Drug Discov 2010; 9:482-93; PMID:20489699; http://dx.doi.org/10.1038/nrd3030
- Kessenbrock K, Frohlich L, Sixt M, Lammermann T, Pfister H, Bateman A, Belaaouaj A, Ring J, Ollert M, Fassler R, et al. Proteinase 3 and neutrophil elastase enhance inflammation in mice by inactivating antiinflammatory progranulin. J Clin Invest 2008; 118:2438-47; PMID:18568075
- Wu H, Siegel RM. Medicine. Progranulin resolves inflammation. Science 2011; 332:427-8; PMID:21512023; http://dx.doi.org/10.1126/science.1205992
- Tian Q, Zhao S, Liu C. A solid-phase assay for studying direct binding of progranulin to TNFR and progranulin antagonism of TNF/TNFR interactions. Methods Mol Biol 2014; 1155:163-72; PMID:24788181; http://dx.doi.org/10.1007/978-1-4939-0669-7_14
- Tian QY, Zhao YP, Liu CJ. Modified yeast-two-hybrid system to identify proteins interacting with the growth factor progranulin. J Visualized Exp: JoVE 2012; pii:3562; PMID:22297851; http://dx.doi.org/10.3791/3562
- Cha SH, Hu Z, Chohnan S, Lane MD. Inhibition of hypothalamic fatty acid synthase triggers rapid activation of fatty acid oxidation in skeletal muscle. Proc Natl Acad Sci U S A 2005; 102:14557-62; PMID:16203972; http://dx.doi.org/10.1073/pnas.0507300102
- Kim KH, Jeong YT, Oh H, Kim SH, Cho JM, Kim YN, Kim SS, Kim do H, Hur KY, Kim HK, et al. Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nat Med 2013; 19:83-92; PMID:23202295; http://dx.doi.org/10.1038/nm.3014
- Zhou B, Li H, Liu J, Xu L, Zang W, Wu S, Sun H. Intermittent injections of osteocalcin reverse autophagic dysfunction and endoplasmic reticulum stress resulting from diet-induced obesity in the vascular tissue via the NFkappaB-p65-dependent mechanism. Cell Cycle 2013; 12:1901-13; PMID:23708521; http://dx.doi.org/10.4161/cc.24929