
Abstract
Prostate cancer (PCa) is the second leading cause of cancer-related death in males in the United States. Majority of prostate cancers are originally androgen-dependent and sensitive to androgen-deprivation therapy (ADT), however, most of them eventually relapse and progress into incurable castration-resistant prostate cancer (CRPC). Of note, the activity of androgen receptor (AR) is still required in CRPC stage. The mitotic kinase polo-like kinase 1 (Plk1) is significantly elevated in PCa and its expression correlates with tumor grade. In this study, we assess the effects of Plk1 on AR signaling in both androgen-dependent and androgen-independent PCa cells. We demonstrate that the expression level of Plk1 correlated with tumorigenicity and that inhibition of Plk1 caused reduction of AR expression and AR activity. Furthermore, Plk1 inhibitor BI2536 down-regulated SREBP-dependent expression of enzymes involved in androgen biosynthesis. Of interest, Plk1 level was also reduced when AR activity was inhibited by the antagonist MDV3100. Finally, we show that BI2536 treatment significantly inhibited tumor growth in LNCaP CRPC xenografts. Overall, our data support the concept that Plk1 inhibitor such as BI2536 prevents AR signaling pathway and might have therapeutic potential for CRPC patients.
Introduction
Androgen receptor (AR) plays a critical role in initiation and progression of prostate cancer (PCa). In PCa cells, AR is activated by androgen and then translocates from the cytoplasm to the nucleus, acting as a transcription factor that promotes many downstream proteins involved in PCa progression, such as ETS-related gene (ERG) and prostate-specific antigen (PSA).Citation1,2 Until recently, chemical castration and surgical castration (orchiectomy) are both widely used as androgen-deprivation therapy (ADT) for PCa.Citation3 Unfortunately, the remissions caused by castration are temporary and PCa eventually progresses to castration-resistant prostate cancer (CRPC). However, it has been established that AR signaling continues to be essential for CRPC progression.
Multiple mechanisms are responsible for PCa cells to escape from androgen deprivation.Citation4 For example, amplification of AR gene increases the expression level of AR protein.Citation5 Mutations occur in AR gene and confer broader ligand specificity or oncogenic property to AR.Citation6 Some cross-talk signal transduction pathways, such as EGF pathway and IGF pathway, are upregulated and can activate AR in an androgen-independent manner.Citation7,8 Moreover, PCa cells can also increase expression of steroidogenic enzymes, produce androgen through de novo steroidogenesis and no longer need exogenous androgen supply.Citation9
The serine/threonine polo-like kinase 1 (Plk1) is a regulator of many cell cycle events, such as mitotic entry, bipolar spindle formation and cytokinesis.Citation10 The elevation of Plk1 expression begins in G2 phase and reaches the peak at mitosis. Plk1 is overexpressed in a wide range of cancer cells, including prostate, breast, pancreatic and melanoma. Inhibition of Plk1 has been proposed a hopeful strategy for enhancement of cancer therapy.Citation10 Plk1 inhibitors such as BI2536, BI6727 (Volasertib), NMS-P937 have been tested in phase I or II clinical studies for patients with various cancers.Citation11-14 BI2536 is an ATP-competitive Plk1 kinase inhibitor and has been used in phase II clinical studies in breast cancer, endometrial cancer, head and neck cancer, melanoma, ovarian cancer and sarcoma.Citation15 In prostate cancer, Plk1 knockdown by RNAi causes induction of mitotic catastrophe,Citation16 suggesting that Plk1 might be a target and BI2536 might be a potential drug of PCa treatment.
In the present study, we show that Plk1 expression correlates with PCa cell proliferation and has a positive effect on AR signaling. We have examined whether Plk1 inhibitor BI2536 can suppress AR expression and activity in PCa cells. Meanwhile, BI2536 can also restrain de novo steroidogenesis through the SREBP pathway. Finally, we find that, in the CRPC xenograft model, castrated mice treated with BI2536 experienced a significant delay in tumor growth.
Results
Plk1 level in PCa cells is correlated with tumorigenicity
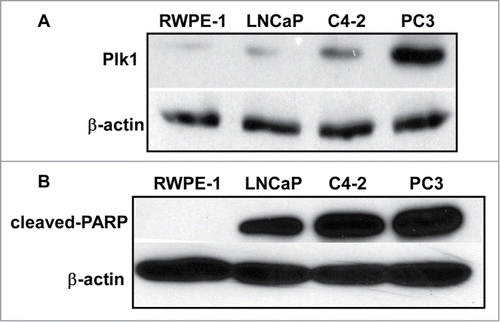
Plk1 expression is correlated to carcinogenesis and overexpression of Plk1 has been found in many cancer cell lines, including PCa cells.Citation17 We speculated that Plk1 level is associated with tumorigenicity in PCa. To test this hypothesis, we examined Plk1 protein level by western blotting in non-transformed human prostate epithelium and a series of human PCa cell lines, with various metastasis grade and androgen dependence ( and ). RWPE-1 cell is an immortal cell line originated from normal human prostate epithelium and it could not form any solid tumor when implanted into the nude mice. While LNCaP is androgen dependent, C4-2 is derived from LNCaP but androgen independent. Finally, PC3 is AR negative thus androgen insensitive. As indicated in , RWPE-1 has a basal level of Plk1, but the Plk1 levels of LNCaP and C4-2 cells are clearly elevated. As expected, PC3 cells have the highest level of Plk1, suggesting that Plk1 level was correlated with tumorigenicity. Furthermore, cell lines mentioned above were treated with Plk1 inhibitor BI2536 and their apoptosis status was detected. Here cleaved-PARP (poly ADP-ribose polymerase) was chosen as a marker of cells undergoing apoptosis (). We previously showed that normal cells are insensitive to Plk1 depletion. To support, RWPE-1 cells were resistant to BI2536, but all 3 transformed cell lines showed significantly elevated levels of cleaved-PARP upon BI2536 treatment, supporting the notion Plk1 is a valid target for PCa therapy.
Figure 1. Plk1 level is correlated with tumorigenicity in different prostate cell lines. (A) Immunoblotting of Plk1 in RWPE-1, LNCaP, C4–2 and PC3 cells. (B) RWPE-1, LNCaP, C4-2 and PC3 cells were treated with 20 nmol/L BI2536 for 24 hours and harvested.
Plk1 affects AR expression level and AR activity in PCa cells
It has been established that AR remains to be transcriptionally active in CRPC. Thus, it is of clinical significance to dissect the underlying mechanisms responsible for reactivation of AR after castration. Toward that end, we tested whether Plk1 is involved in AR activation in CRPC. Using lentivirus-based shRNA to deplete Plk1 in 2 CRPC cell lines, C4-2 and 22Rv1, we found that depletion of Plk1 caused a reduction of the level of AR protein (). In 22Rv1 cells, multiple alternatively spliced AR variants have been identified besides full-length AR, displaying a 110-kDa band (full-length) and an 80-kDa band (variants) after immunoblotting.Citation18 As indicated, both full-length AR and AR variants were reduced when Plk1 was depleted in 22Rv1 cells (). We also detected the levels of AR transcripts in PCa cells treated with BI2536. When Plk1 activity was inhibited by BI2536, AR mRNA level was downregulated in LNCaP, C4-2 and 22Rv1 cells (). Of significance, transcription levels of 2 AR variants, AR-V1 and AR-V7, were decreased upon BI2536 treatment as well (). This is important as increasing expression of AR-variants is one documented mechanism for AR reactivation after castration and acquisition of resistance to drugs targeting AR signaling.
Figure 2. Plk1 affects AR signaling in PCa cells. (A) Depletion of Plk1 reduced AR level in PCa cells. C4-2 and 22Rv-1 cells were infected with lentivirus to deplete Plk1 for 36 hours and harvested. (B) BI2536 inhibits AR expression in PCa cells. LNCaP, C4-2 and 22Rv-1 cells were treated with or without 200 nmol/L BI2536 and harvested for Real-Time qPCR to measure the mRNA levels of AR and AR variants. (C) AR activity is inhibited by BI2536 in LNCaP cells. LNCaP cells were cultured in RPMI-1640+ 5% CSS medium for 48hrs, then treated with or without 200 nmol/L BI2536 or 1 nmol/L R1881 for 4 hours, and harvested for Real-Time qPCR to measure the mRNA levels of ERG and PSA.
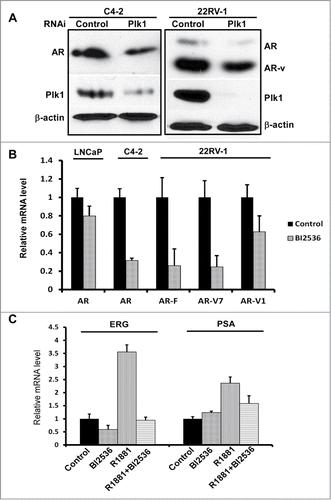
Considering that AR mRNA in LNCaP was reduced less significant than those in C4–2 and 22Rv1 cells when cells were treated with BI2536, we then asked whether AR activity was inhibited upon BI2536 treatment. Primers toward PSA and ERG, 2 target genes directly activated by AR, were prepared and utilized for real-time PCR analysis. As indicated in , when LNCaP cells were cultured in androgen-deprived medium (RPMI-1640+5% CSS), inhibition of Plk1 hardly affects AR activity. However, when these cells were stimulated by R1881, a synthetic androgen, BI2536 addition significantly attenuated the stimulation effect toward AR ().
Inhibition of Plk1 negatively regulates de novo steroidogenesis
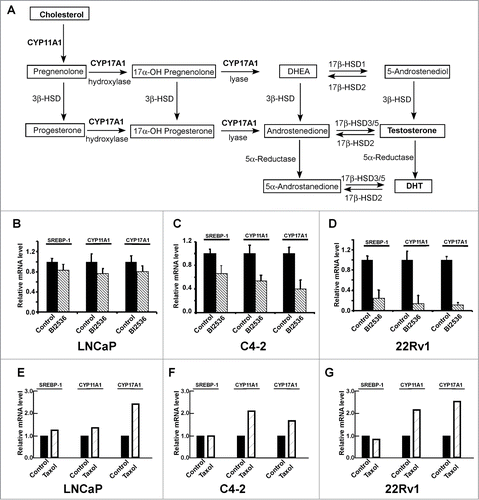
Given that CRPC cells are able to produce androgen through de novo steroidogenesis, thus activating AR signaling, we asked whether Plk1 affects androgen synthesis pathway (). It has been generally accepted that 2 enzymes CYP17A1 (Cytochrome P450 17A1 or steroid 17-α-monooxygenase) and CYP11A1 (Cytochrome P450 11A1 or cholesterol side-chain cleavage enzyme) play important roles during de novo steroidogenesis and that both enzymes are under the control of the SREBP pathway.Citation19 Therefore, we examined transcription levels of SREBP1, CYP17A1 and CYP11A1 upon BI2536 treatment in PCa cells. As shown in , inhibition of Plk1 with BI2536 leads to reduced expression levels of SREBP1 and its downstream targets, CYP17A1 and CYP11A1, suggesting that inhibition of Plk1 did interfere with the SREBP pathway and androgen biosynthesis. Similarly to the transcription of AR, in androgen-independent PCa cell lines C4-2 and 22Rv1, the SREBP pathway was much more sensitive to BI2536 than that in androgen-dependent LNCaP cells. Interestingly, treatment of cells with taxol actually partially activates the androgen biosynthesis pathway (), suggesting that CRPC patients likely benefit from a combination of taxol and AR antagonists.
Figure 3. Inhibition of Plk1 negatively regulates de novo steroidogenesis. (A) The classical pathway of androgen biosynthesis, modified from ref. Citation28,29 The substrate cholesterol was converted to precursor pregnenolone by CYP11A1 and finally converted to testosterone and DHT by a series of reactions involving the activity of CYP17A1 and other enzymes. DHT, dehydrotestosterone. DHEA, dehydroepiandrosterone. HSD, hydroxysteroid dehydrogenase. (B–D) LNCaP, C4-2 and 22Rv1 cells were treated with or without 200 nmol/L BI2536 for 4 hours, and harvested for Real-Time qPCR to measure the mRNA levels of SREBP-1, CYP11A1 and CYP17A1. E-G, LNCaP, C4-2 and 22Rv1 cells were treated with or without 1 μmol/L taxol for 12 hours, and harvested.
AR inhibition caused Plk1 reduction in PCa in different stages
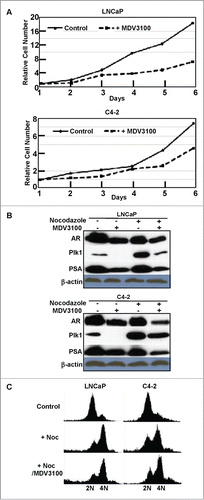
As expected, treatment of AR-positive PCa cells, LNCaP and C4-2, with AR antagonist MDV3100 (also known as enzalutamide) leads to inhibition of cell proliferation (). Considering that AR is a transcription factor, we next asked whether AR also affects Plk1 expression. Accordingly, LNCaP and C4-2 cells were treated with or without MDV3100 and harvested for Plk1 immunoblotting, with PSA level as a marker of AR activity. As indicated, AR inhibition caused Plk1 reduction in PCa cells when the cells were either randomly growing or treated with nocodazole to arrest at G2/M phase (), suggesting that AR also affects the level of Plk1 in PCa cells. Because Plk1 is mitotic marker, we thus asked whether AR inhibition-associated Plk1 down-regulation is a secondary effect of cell cycle arrest. For that purpose, cells were arrested at late G1 with thymidine block, released into nocodazole-containing medium in the presence or absence of MDV3100 for 12 hours. FACS (fluorescence-activated cell sorting) analysis indicated addition of MDV3100 did not significantly affect the cell cycle progression (). So, reduced cell numbers in the presence of MDV3100 might be due to increased cell death.
Figure 4. Inhibition of AR reduced Plk1 level in LNCaP and C4–2 cells. (A) Cell proliferation was inhibited by 10 μmol/L MDV3100 treatment. (B) Cells were treated with or without 10 μmol/L MDV3100 to inhibit AR activity or not, then treated with or without 1 μmol/L nocodazole to arrest cells in G2/M phase or randomly growing, and harvested for immunoblotting. (C) Cells were treated with thymidine overnight to arrest at G1 phase, released into nocodazole-containing medium ± 10 μmol/L MDV3100 for 12 hours, and harvested for FACS analysis.
Plk1 inhibitor BI2536 suppresses tumor growth of CRPC xenograft
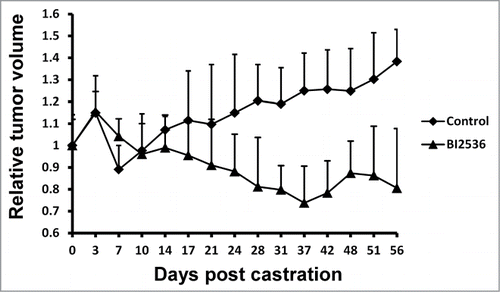
Finally, we tested the anti-tumor effect of BI2536 by using the LNCaP CRPC xenograft model. Tumor sizes were measured 3 times a week and blood was collected weekly during the study. We observed a significant difference between 2 groups treated with or without BI2536 (). When mice carrying LNCaP xenograft tumors were castrated, tumor volumes declined within a week because of androgen deprivation. After that time point, tumor cells possessed castration resistance and tumors without BI2536 treatment continued to grow, but tumors of BI2536-injected group were dramatically suppressed ().
Figure 5. BI2536 inhibits LNCaP CRPC xenografts. LNCaP cells were inoculated into nude mice, which were castrated when tumors reach 200–300 mm3. Since the day of castration, mice were intravenously injected twice per week with BI2536 (25 mg/kg body weight) or saline solution as control, and followed for additional 8 weeks. Relative tumor volumes were calculated and tumor growth curve was generated after measurement of tumor volumes.
Discussion
AR plays a critical role in PCa progression as a transcription factor that controls many downstream targets such as PSA. Castration and chemical blockade against AR are both effective initial therapies for PCa treatment. However, it has been accepted that the AR signaling somehow is reactivated and continues to be essential for CRPC. To support, AR antagonists such as abiraterone and enzalutamide are 2 recently FDA-approved drugs to treat CRPC. Unfortunately, overall patient survival was only improved by 2 to 5 months with these inhibitors.Citation20 Therefore, it is urgently needed to identify new targets and develop novel approaches for treatment of CRPC patients. Toward that end, we first found that Plk1 level was correlated to PCa cell growth rate and androgen-independent PCa cells were more sensitive to Plk1 inhibition than androgen-dependent PCa cells (), suggesting a gradually increased role of Plk1 in cell survival as PCa progresses from androgen dependent to independent stage.
The major transcript derived from AR gene encodes a 110-kDa protein with 4 functional domains, including N-terminal transactivation domain (NTD), a DNA-binding domain (DBD), a flexible Hinge region, and a C-terminal ligand-binding domain (LBD). While NTD is required for transcriptional activity, LBD is responsible for binding to androgens and other agonists. A series of AR variants lacking of LBD has been reported either in PCa cells such as 22Rv1 or in CRPC tissues from patients.Citation18,21 Such ARΔLBD isoforms are functionally effective without androgen stimulation, thus resulting in androgen-independent PCa cell growth. AR antagonist drugs cannot target AR variants due to loss of ligand binding sites. In this study, we found that BI2536 inhibited the transcription of both full-length AR and 2 AR variants lacking of LBD, suggesting that Plk1 is a potential drug target in CRPC cells expressing AR variants.
We further analyzed the mutual regulation between Plk1 and AR signaling in PCa cells. Enough evidence supports the concept that the PI3K/AKT/mTOR pathway is frequently activated in PCa due to loss of function of PTEN.Citation22 The PI3K/AKT/mTOR and AR signaling pathways regulate each other through complex reciprocal feedback mechanisms.Citation23 Recently, we reported that Plk1 is involved in oxidative stress-induced AR elevation through activation of the PI3K/AKT/mTOR pathway.Citation24 In this study, we provide evidence that Plk1 elevation leads to activation of the AR signaling in the absence of external oxidative stress. Moreover, to function as a transcription factor, AR has to translocate from the cytoplasm to the nucleus after stimulation by androgen. This translocation relies on microtubule dynamics, which are controlled by 2 Plk1 substrates, CLIP-170 and p150Glued.Citation25 Failed AR nucleus translocation could be the reason why Plk1 inhibition caused decreased AR activity in LNCaP cells.
Upon androgen deprivation therapy, PCa cells gradually gain the ability to synthesize androgens through de novo steroidogenesis and develop to CRPC. SREBP transcription factors are responsible for coordinately regulating the enzymes required for synthesis of cholesterol and steroidogenesis.Citation26 Cytochrome p450 family members CYP11A1 and CYP17A1 are 2 key enzymes involved in de novo steroidogenesis and under control of SREBP. Results of our study suggest that Plk1 inhibition reduces mRNA level of SREBP1, CYP17A1 and CYP11A1, thereby inhibiting androgen biosynthesis ().
Although AR is a transcription factor and Plk1 level was downregulated upon AR inhibition, we cannot affirm that the transcription of Plk1 is directly promoted by AR. A ChIP-on-chip analysis predicted all putative AR binding sites among the human genome sequence in LNCaP cells.Citation27 According to the database (http://research.dfci.harvard.edu/brownlab/datasets/), no putative AR binding sites locate close to PLK1 gene, but other M-phase cell cycle genes are upregulated by AR especially in androgen independent PCa cells.Citation27 It is possible that AR increases Plk1 expression indirectly through promoting other Plk1 regulators.
In summary, we have revealed that Plk1 is positively involved in AR signaling and de novo steroidogenesis in PCa cells. Plk1 inhibitor BI2536 significantly reduces the tumor growth rate of CRPC xenograft. This work suggests that targeted inhibition of Plk1 might be beneficial for CRPC patients.
Materials and Methods
Cell culture
LNCaP, C4-2 (derived from LNCaP, but androgen independent), 22Rv1 (androgen independent) cells were cultured in RPMI-1640 (Sigma) supplemented with 10% FBS (v/v), 100 U/mL penicillin, and 100 U/mL streptomycin at 37°C in 5% CO2. PC3 cells were cultured in F12K (ATCC) supplemented with 10% FBS (v/v), 100 U/mL penicillin and 100 U/mL streptomycin at 37°C in 5% CO2. RWPE-1 cells were cultured in keratinocyte serum-free medium (K-SFM, Invitrogen) supplemented with 50 mg/mL bovine pituitary extract (BPE), 5 ng/mL epidermal growth factor (EGF, Human Recombinant), 100 U/mL penicillin, and 100 U/mL streptomycin at 37°C in 5% CO2. MDV3100 was purchased from Selleckchem.
Quantitative Real-Time PCR
Total RNA was extracted using TRIzol (Invitrogen) and reverse transcribed using the iScript™ cDNA Synthesis Kit (Bio-Rad). cDNA product from 1 μg total RNA was amplified using Faststart Universal SYBR Green Master (ROX) (Roche) and analyzed by Applied Biosystems 7300 Real-Time PCR System. PCR primers used in this study were shown in .
Table 1. Sequences for primers used to conduct quantitative PCR
Table 2. Prostate cells used in this study
Western blotting
Cells were harvested and lysed in TBSN buffer (20 mmol/L Tris, pH 8.0, 150 mmol/L NaCl, 1.5 mmol/L EDTA, 5 mmol/L EGTA, 0.5% NP-40, and 0.5 mmol/L Na3VO4) supplemented with proteinase inhibitors. The lysates were resolved by SDS-PAGE and transferred to PVDF membrane (EMD Millipore), followed by incubation with antibodies against Plk1 (Santa Cruz Biotechnology; sc-17783), AR (Santa Cruz Biotechnology; sc-7305), cleaved-PARP (EMD Millipore; AB3620), PSA (Cell Signaling Technology; 5365), and β-actin (Sigma; A5441).
Depletion and overexpression of Plk1
HEK293T cells in 10-cm dishes were co-transfected with 4 µg of pHR‘-CMV-△8.20vpr, 2 µg of pHR’-CMV-VSV-G, and 4 µg of pLKO.1-Plk1 plasmids for depletion of Plk1. Supernatants were collected every 12 hours after 24 hours post-transfection. Viruses were filtered through a 0.45-µm poresize filter, concentrated by spin at 20,000 rpm for 2 hours, re-suspended in TNE buffer (50 mmol/L Tris-HCl, pH7.8, 130 mmol/L NaCl, 1 mmol/L EDTA), and rotated overnight at 4°C. Infections were carried out in the presence of 10 µg/ml of polybrene and 10 mmol/L of HEPES, followed by selection with 1 µg/ml of puromycin for at least 36 hours.
Xenograft model
LNCaP cells were harvested, pelleted and co-inoculated with 100 μl of Matrigel (1:1 volume ratio). Six-week old male athymic nude mice (BALB/c strain) were implanted with 5 × 106 LNCaP cells for each mouse at right flank. Tumor volume was calculated as length × width2/2. Six weeks after inoculation, when tumors reached 200–300 mm3, all the mice were castrated. BI2536 (Symansis) was dissolved in 0.1 mol/L HCl and then diluted in 0.9% NaCl (6 mg/ml) for injection. Since the day of castration, mice were intravenously injected twice per week with BI2536 (25 mg/kg body weight) or saline solution as control, and followed for additional 8 weeks.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
Z Zhang was financially supported by China Scholarship Council (CSC). This work was supported by NIH grants R01CA157429 (XL), R01 AR059130 (NA), R01 CA176748 (NA) and ACS grant RSG-13-073 (XL).
Related Research Data
References
- Bastus NC, Boyd LK, Mao X, Stankiewicz E, Kudahetti SC, Oliver RT, Berney DM, Lu YJ. Androgen-induced TMPRSS2:ERG fusion in nonmalignant prostate epithelial cells. Cancer Res 2010; 70(23):9544-8; PMID:20947519; http://dx.doi.org/10.1158/0008-5472.CAN-10-1638
- Liu X, Choi RY, Jawad SM, Arnold JT. Androgen-induced PSA expression requires not only activation of AR but also endogenous IGF-I or IGF-I/PI3K/Akt signaling in human prostate cancer epithelial cells. Prostate 2011; 71(7):766-77; PMID:21031436; http://dx.doi.org/10.1002/pros.21293
- Fitzpatrick JM, Bellmunt J, Fizazi K, Heidenreich A, Sternberg CN, Tombal B, Alcaraz A, Bahl A, Bracarda S, Di Lorenzo G, et al. Optimal management of metastatic castration-resistant prostate cancer: highlights from a European Expert Consensus Panel. Eur J Cancer 2014; 50(9):1617-27; PMID:24703899; http://dx.doi.org/10.1016/j.ejca.2014.03.010
- Attar RM, Takimoto CH, Gottardis MM. Castration-resistant prostate cancer: locking up the molecular escape routes. Clin Cancer Res 2009; 15(10):3251-5; PMID:19447877; http://dx.doi.org/10.1158/1078-0432.CCR-08-1171
- Ford OH 3rd, Gregory CW, Kim D, Smitherman AB, Mohler JL. Androgen receptor gene amplification and protein expression in recurrent prostate cancer. J Urol 2003; 170(5):1817-21; PMID:14532783; http://dx.doi.org/10.1097/01.ju.0000091873.09677.f4
- Hara T, Miyazaki J, Araki H, Yamaoka M, Kanzaki N, Kusaka M, Miyamoto M. Novel mutations of androgen receptor: a possible mechanism of bicalutamide withdrawal syndrome. Cancer Res 2003; 63(1):149-53; PMID:12517791
- Aaronson DS, Muller M, Neves SR, Chung WC, Jayaram G, Iyengar R, Ram PT. An androgen-IL-6-Stat3 autocrine loop re-routes EGF signal in prostate cancer cells. Mol Cell Endocrinol 2007; 270(1–2):50-6; PMID:17374439; http://dx.doi.org/10.1016/j.mce.2007.02.006
- Krueckl SL, Sikes RA, Edlund NM, Bell RH, Hurtado-Coll A, Fazli L, Gleave ME, Cox ME. Increased insulin-like growth factor I receptor expression and signaling are components of androgen-independent progression in a lineage-derived prostate cancer progression model. Cancer Res 2004; 64(23):8620-9; PMID:15574769; http://dx.doi.org/10.1158/0008-5472.CAN-04-2446
- Chen Y, Clegg NJ, Scher HI. Anti-androgens and androgen-depleting therapies in prostate cancer: new agents for an established target. Lancet Oncol 2009; 10(10):981-91; PMID:19796750; http://dx.doi.org/10.1016/S1470-2045(09)70229-3
- Strebhardt K. Multifaceted polo-like kinases: drug targets and antitargets for cancer therapy. Nat Rev Drug Discov 2010; 9(8):643-60; PMID:20671765; http://dx.doi.org/10.1038/nrd3184
- Lenart P, Petronczki M, Steegmaier M, Di Fiore B, Lipp JJ, Hoffmann M, Rettig WJ, Kraut N, Peters JM. The small-molecule inhibitor BI 2536 reveals novel insights into mitotic roles of polo-like kinase 1. Curr Biol 2007; 17(4):304-15; PMID:17291761; http://dx.doi.org/10.1016/j.cub.2006.12.046
- Triscott J, Lee C, Foster C, Manoranjan B, Pambid MR, Berns R, Fotovati A, Venugopal C, O'Halloran K, Narendran A, et al. Personalizing the treatment of pediatric medulloblastoma: polo-like kinase 1 as a molecular target in high-risk children. Cancer Res 2013; 73(22):6734-44; PMID:24019381; http://dx.doi.org/10.1158/0008-5472.CAN-12-4331
- Valsasina B, Beria I, Alli C, Alzani R, Avanzi N, Ballinari D, Cappella P, Caruso M, Casolaro A, Ciavolella A, et al. NMS-P937, an orally available, specific small-molecule polo-like kinase 1 inhibitor with antitumor activity in solid and hematologic malignancies. Mol Cancer Ther 2012; 11(4):1006-16; PMID:22319201; http://dx.doi.org/10.1158/1535-7163.MCT-11-0765
- Markant SL, Esparza LA, Sun J, Barton KL, McCoig LM, Grant GA, Crawford JR, Levy ML, Northcott PA, Shih D, et al. Targeting sonic hedgehog-associated medulloblastoma through inhibition of Aurora and Polo-like kinases. Cancer Res 2013; 73(20):6310-22; PMID:24067506; http://dx.doi.org/10.1158/0008-5472.CAN-12-4258
- Schoffski P, Blay JY, De Greve J, Brain E, Machiels JP, Soria JC, Sleijfer S, Wolter P, Ray-Coquard I, Fontaine C, et al. Multicentric parallel phase II trial of the polo-like kinase 1 inhibitor BI 2536 in patients with advanced head and neck cancer, breast cancer, ovarian cancer, soft tissue sarcoma and melanoma. The first protocol of the European Organization for Research and Treatment of Cancer (EORTC) Network Of Core Institutes (NOCI). Eur J Cancer 2010; 46(12):2206-15; PMID:20471824; http://dx.doi.org/10.1016/j.ejca.2010.03.039
- Liu XS, Song B, Elzey BD, Ratliff TL, Konieczny SF, Cheng L, Ahmad N, Liu X. Polo-like kinase 1 facilitates loss of Pten tumor suppressor-induced prostate cancer formation. J Biol Chem 2011; 286(41):35795-800; PMID:21890624; http://dx.doi.org/10.1074/jbc.C111.269050
- Deeraksa A, Pan J, Sha Y, Liu XD, Eissa NT, Lin SH, Yu-Lee LY. Plk1 is upregulated in androgen-insensitive prostate cancer cells and its inhibition leads to necroptosis. Oncogene 2013; 32(24):2973-83; PMID:22890325; http://dx.doi.org/10.1038/onc.2012.309
- Hu R, Dunn TA, Wei S, Isharwal S, Veltri RW, Humphreys E, Han M, Partin AW, Vessella RL, Isaacs WB, et al. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res 2009; 69(1):16-22; PMID:19117982; http://dx.doi.org/10.1158/0008-5472.CAN-08-2764
- Ozbay T, Rowan A, Leon A, Patel P, Sewer MB. Cyclic adenosine 5'-monophosphate-dependent sphingosine-1-phosphate biosynthesis induces human CYP17 gene transcription by activating cleavage of sterol regulatory element binding protein 1. Endocrinology 2006; 147(3):1427-37; PMID:16306078; http://dx.doi.org/10.1210/en.2005-1091
- Froehner M, Wirth MP. Enzalutamide in metastatic prostate cancer before chemotherapy. N Engl J Med 2014; 371(18):1755; PMID:25354111; http://dx.doi.org/10.1056/NEJMc1410239
- Haile S, Sadar MD. Androgen receptor and its splice variants in prostate cancer. Cell Mol Life Sci 2011; 68(24):3971-81; PMID:21748469; http://dx.doi.org/10.1007/s00018-011-0766-7
- Sarker D, Reid AH, Yap TA, de Bono JS. Targeting the PI3K/AKT pathway for the treatment of prostate cancer. Clin Cancer Res 2009; 15(15):4799-805; PMID:19638457; http://dx.doi.org/10.1158/1078-0432.CCR-08-0125
- Carver BS, Chapinski C, Wongvipat J, Hieronymus H, Chen Y, Chandarlapaty S, Arora VK, Le C, Koutcher J, Scher H, et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell 2011; 19(5):575-86; PMID:21575859; http://dx.doi.org/10.1016/j.ccr.2011.04.008
- Zhang Z, Hou X, Shao C, Li J, Cheng JX, Kuang S, Ahmad N, Ratliff T, Liu X. Plk1 inhibition enhances the efficacy of androgen signaling blockade in castration-resistant prostate cancer. Cancer Res 2014; 74(22):6635-47; PMID:25252916; http://dx.doi.org/10.1158/0008-5472.CAN-14-1916
- Hou X, Li Z, Huang W, Li J, Staiger C, Kuang S, Ratliff T, Liu X. Plk1-dependent microtubule dynamics promotes androgen receptor signaling in prostate cancer. Prostate 2013; 73(12):1352-63; PMID:23661607; http://dx.doi.org/10.1002/pros.22683
- Cai C, Chen S, Ng P, Bubley GJ, Nelson PS, Mostaghel EA, Marck B, Matsumoto AM, Simon NI, Wang H, et al. Intratumoral de novo steroid synthesis activates androgen receptor in castration-resistant prostate cancer and is upregulated by treatment with CYP17A1 inhibitors. Cancer Res 2011; 71(20):6503-13; PMID:21868758; http://dx.doi.org/10.1158/0008-5472.CAN-11-0532
- Wang Q, Li W, Zhang Y, Yuan X, Xu K, Yu J, Chen Z, Beroukhim R, Wang H, Lupien M, et al. Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell 2009; 138(2):245-56; PMID:19632176; http://dx.doi.org/10.1016/j.cell.2009.04.056
- Mostaghel EA, Marck BT, Plymate SR, Vessella RL, Balk S, Matsumoto AM, Nelson PS, Montgomery RB. Resistance to CYP17A1 inhibition with abiraterone in castration-resistant prostate cancer: induction of steroidogenesis and androgen receptor splice variants. Clin Cancer Res 2011; 17(18):5913-25; PMID:21807635; http://dx.doi.org/10.1158/1078-0432.CCR-11-0728
- Yin L, Hu Q. CYP17 inhibitors–abiraterone, C17,20-lyase inhibitors and multi-targeting agents. Nat Rev Urol 2014; 11(1):32-42; PMID:24276076; http://dx.doi.org/10.1038/nrurol.2013.274