
Abstract
Dysregulation of Ras signaling is the major cause of various cancers. Aberrant Ras signaling, however, provides a favorable environment for many viruses, making them suitable candidates as cancer-killing therapeutic agents. Susceptibility of cancer cells to such viruses is mainly due to impaired type I interferon (IFN) response, often as a result of activated Ras/ERK signaling in these cells. In this study, we searched for cellular factors modulated by Ras signaling and their potential involvement in promoting viral oncolysis. We found that upon Ras transformation of NIH-3T3 cells, the N-terminus of Nogo-B (reticulon 4) was proteolytically cleaved. Interestingly, Nogo knockdown (KD) in non-transformed and Ras-transformed cells both enhanced virus-induced IFN response, suggesting that both cleaved and uncleaved Nogo can suppress IFN response. However, pharmacological blockade of Nogo cleavage in Ras-transformed cells significantly enhanced virus-induced IFN response, suggesting that cleaved Nogo contributes to enhanced IFN suppression in these cells. We further showed that IFN suppression associated with Ras-induced Nogo-B cleavage was distinct from but synergistic with that associated with an activated Ras/ERK pathway. Our study therefore reveals an important and novel role of Nogo-B and its cleavage in the suppression of anti-viral immune responses by oncogenic Ras transformation.
Introduction
Ras proteins are small GTP-binding proteins that play crucial roles in diverse biological processes such as cell proliferation, differentiation and survival. Mutations in Ras confer various abnormalities to the cells, ultimately leading to cancer. These mutations have been found in 30–90% of various human cancers,Citation1 making Ras proteins important targets for anti-tumor therapy. Dysregulated Ras signaling has also been exploited by many oncolytic viruses for promoting viral replication, infectivity and dissemination.Citation2-5 For example, Ras mutation upregulates cathepsin lysosomal protesases, thereby facilitating the uncoating step of reovirus during viral entry.Citation6,7 Perhaps the most important aspect of selective preference by oncolytic viruses toward cancer cells is Ras-mediated suppression of antiviral immune response on the part of cancer cells, allowing rapid viral cell-to-cell spread.Citation8,9 Previous studies have demonstrated that a constitutively activated Ras/ERK pathway not only suppresses type I interferon (IFN) production but also inhibits the cell's ability to respond to IFN via down-regulation of STAT2.Citation8,10,11 However, there is evidence that the PI3K/Akt and p38 pathways, which are necessary for IFN response, are also activated by Ras during viral infection.Citation8 It thus appears that in addition to the Ras/ERK pathway, other pathways are likely also involved in IFN impairment.
Nogo (reticulon 4) belongs to the reticulon family of proteins which are membrane-spanning proteins mainly localized to the ER. Nogo protein has 3 major isoforms (Nogo-A, Nogo-B and Nogo-C) representing the 3 alternatively spliced variants.Citation12 While Nogo-B is ubiquitously expressed in various tissues and cells, Nogo-A and Nogo-C are tissue-specific, being present in the brain and muscle, respectively.Citation13 Nogo was firstly identified as neurite outgrowth inhibitor, and Nogo-A, in particular, has been extensively investigated due to its functional role as a CNS-specific inhibitor of axonal regeneration.Citation14-16 Recent studies, however, revealed that Nogo is a multifunctional protein that plays important roles in vascular homeostasis,Citation17-19 angiogenesisCitation20 and Th2-driven lung inflammation.Citation21 In particular, Nogo-B is highly associated with cancer progression and metastasis.Citation22-24 Low expression level of Nogo-B is observed in small cell lung carcinomas and adult T-cell leukemia/lymphoma.Citation23,24 Subsequent studies showed that Nogo-B can induce apoptosis through ER stress,Citation24,25 or reduce anti-apoptotic function of Bcl-2 and Bcl-xL, translocating them from mitochondria to ER.Citation26 In contrast, others have reported that Nogo-B has protective functions against cell death mediated by ER and oxidative stress.Citation25,27,28 Hence, the role of Nogo-B in cancer has yet to be clarified.
In this study, we searched for host factors modulated by Ras and their possible link to the antiviral immune response in Ras-transformed cells. We show that Nogo-B is cleaved in Ras-transformed cells, and that cleaved Nogo-B contributes to the impairment of IFN induction in these cells. Our study therefore reveals a novel connection between Nogo-B modulation by Ras and antiviral immune response in cancer cells.
Results
N-terminal of Nogo-B is cleaved in Ras-transformed cells
It is generally believed that compared to their normal counterparts, cancer cells often display altered surface proteomes that likely play important roles in promoting cancer development and metastasis. However, due to their relatively low abundance and generally hydrophobic nature, identification and isolation of these proteins have been a daunting task. From our oncolytic virus studies, we have shown that NIH-3T3 cells become significantly more permissive to reovirus infection upon Ras-transformation. This is in large part due to attenuation of anti-viral innate immune response by an activated MEK/ERK pathway downstream of Ras, allowing for more efficient virus cell-to-cell spread and facilitating viral dissemination. However, it is unclear if Ras transformation also results in an altered cell surface proteome that may also contribute to suppression of innate immunity. We therefore attempted to compare cell surface proteins between non-transformed and Ras-transformed NIH-3T3 cells, which are well-established systems in reovirus-induced oncolysis.Citation8,29
Cell surface molecules were isolated by labeling the cells with membrane impermeable biotin, followed by pull-down with streptavidin-conjugated magnetic bead. Upon SDS-PAGE/silver staining analysis of the bound proteins, we observed that non-transformed and Ras-transformed cells displayed similar profiles but there were some discernible differences (). Of note, a tight cluster of bands migrating at around the 43 kDa position present in non-transformed cells was absent (or drastically reduced) in Ras-transformed cells, and was replaced by a fainter and faster migrating band at ∼34 kDa. Subsequent peptide sequence analysis by mass spectrometry revealed that the 43 kDa cluster and the 34 kDa band contained sequences belonging to Nogo-related proteins as shown in .
Figure 1 (See previous page). N-terminal of Nogo-(B)is cleaved in Ras-transformed cells. (A) Comparison of cell surface proteins between non-transformed and Ras-transformed NIH-3T3 cells. Cell surface proteins were labeled with membrane impermeable biotin, then isolated using streptavidin-conjugated magnetic beads. Isolated proteins were analyzed by SDS-PAGE/silver nitrate staining. Bands showing different intensity levels were excised and identified by mass spectrometry. Arrows indicate proteins containing Nogo protein peptides. The 43 kDa band(s) represents Nogo-B, and the lower band (˜34 kDa) likely represents its cleavage product. (B) Schematic diagram of Nogo isoforms. Three major isoforms of Nogo (A, B, and C) share C-terminal region (shaded in black). Nogo-A and -B share N-terminal region of 167 a.a. (light gray). Peptide sequences detected by mass spectrometry were shown in Nogo-B structure. Antibodies used for detecting C-terminal and N-terminal ends of Nogo were indicated ('Y' in black and for C-terminus and 'Y' in light gray for N-terminus). (C) Detection of Nogo-B in total cell lysates (left panel) and membrane fractions (right panel) from non- and Ras-transformed cells by western blot using the antibody detecting the C-terminus of Nogo-B. β-actin and Calnexin (ER membrane marker) were used as loading controls. (D) Cell lysate of non- or Ras-transformed cells were fractionated by differential centrifugation (Lys, lysate; Nuc, nucleus; Mit, mitochondrial fraction; Cyt, cytosolic fraction; Mic, microsomal fraction). Western blots were carried out using the anti-C-terminal antibody (upper panel), or the anti-N-terminal antibody (lower panel). Cox IV, Calnexin, and β-actin were used as mitochondrial, ER, and cytosol markers, respectively. (E) mRNA levels of Nogo-B in non-transformed and Ras-transformed cells were compared using RT-PCR with Nogo-B specific primers. GAPDH was used as a loading control. (F) Ras-transformed cells were treated with vehicle control (DMSO), caspase inhibitor (Z-VAD-FMK, 10 μM), proteasome inhibitor (MG-132, 1 μM) or both for 18 hrs. Full-length Nogo-B is indicated as “FL” and a cleavage product as “C.”
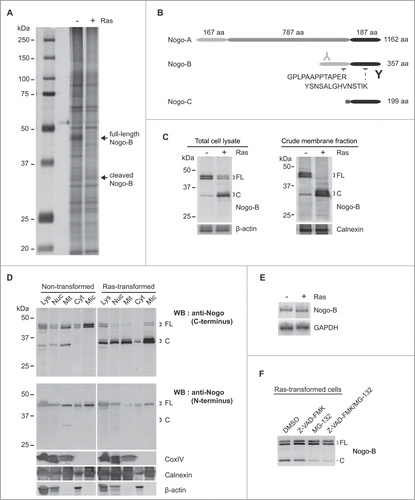
Based on its molecular weight, we deduced that the most likely Nogo isoform was Nogo-B, and that the 34 kDa band was likely its cleavage product (). The 34 kDa protein contained the N-terminal peptide sequence (GPLPAAPPTAPER) of Nogo-B that was not present in the Nogo-C isoform, and its molecular weight was also higher than that of Nogo-C (25 kDa). Furthermore, it has been reported that Nogo-C expression is muscle-cell specific and the endogenous expression of Nogo-C is very low in NIH-3T3 cells.Citation30
The cleavage of Nogo-B was further probed using subcellular fractionation followed by western blot analysis. Two antibodies, recognizing the N-terminus and C-terminus of Nogo-B, respectively (), were used to distinguish the cleavage products. Since Nogo-B is localized in the ER and plasma membrane with the membrane-spanning domains located at the C-terminal region (shaded in black in ), cleavage at a site proximal to the N-terminus would generate a 34 kDa membrane-associated fragment with an intact C-terminal region recognizable by the C-terminal-specific, but not the N-terminal-specific antibody. This was indeed found to be the case: the C-terminal antibody detected uncleaved Nogo-B (43 kDa) as the primary form present in non-transformed cells, and the cleaved 34 kDa protein as the predominant form in Ras-transformed cells (). Both forms were primarily associated with membrane-containing fractions (mitochondria and microsomes), while soluble β-actin remained in the cytosol fraction (, upper panel). In contrast, the N-terminal antibody detected only full-length Nogo-B (43 kDa) but not the 34 kDa band (, lower panel). Our data therefore suggest that approximately 80 amino acid residues of the N-terminus of Nogo-B are removed in Ras-transformed cells. Although we initially compared only cell surface proteins, it appears that Nogo-B cleavage is not restricted to the cell surface since most of the Nogo-B exists in the ER membrane and to some extent in the plasma membrane.Citation30
The Nogo-B mRNA levels were found to be comparable between the 2 cell types (), further supporting the notion that Nogo-B was proteolytically cleaved and not differently regulated at the transcriptional level in Ras-transformed cells. To determine whether the Nogo-B cleavage was mediated by proteasome or caspase-dependent pathway, proteasome inhibitor (MG-132) or caspase inhibitor (Z-VAD-FMK) was added to the Ras-transformed cells. The results () showed that cleavage of Nogo-B was blocked by the proteasome inhibitor but not by the caspase inhibitor, suggesting that Nogo-B cleavage is proteasome-dependent ().
Nogo-B suppresses virus-induced interferon responses
Since virus-induced type I IFN signaling is abrogated in Ras-transformed cells,Citation31 we wished to investigate whether Nogo-B cleavage was associated with suppression of virus-induced type I IFN signaling. Accordingly, mRNA levels of IFN-β, 2′-5′-oligoadenylate synthetase (OAS), or interferon-stimulated gene 56 (ISG56) were compared upon reovirus infection or transfection of a synthetic analog of dsRNA, poly(I:C). As previously observed, IFN-β was highly induced by reovirus infection in non-transformed cells, but not in Ras-transformed cells which showed significant Nogo-B cleavage ().
Figure 2. Nogo-(B) suppresses virus-induced interferon response. (A) Induction of IFN-β response of non-transformed and Ras-transformed NIH-3T3 cells was compared upon reovirus infection. (B) The efficient knockdown of Nogo gene was confirmed by western blot analysis. eGFP shRNA was used as a negative control. Relative protein levels (normalized to β-actin) were compared using ImageJ software (National Institutes of Health, Bethesda, Maryland, USA). (C and D) Effect of Nogo knockdown (KD) on reovirus-induced IFN response was analyzed in non-transformed (C) and Ras-transformed cells (D). Cells were infected with increasing reovirus doses (MOI of 200, 300, 400 based on their titers in L929 cells). (E) Non-transformed cells were transiently transfected 24 hr before viral infection with increasing amounts (0, 3, 6, 9 μg) of mouse Nogo-B expression plasmids. mRNAs were isolated at 24 hr post-infection and subjected to real-time q-PCR. Fold induction of IFN-β or ISG56 was normalized to GAPDH. eGFP shRNA was used as a negative control. Data are presented as mean values with error bars showing the standard deviations.
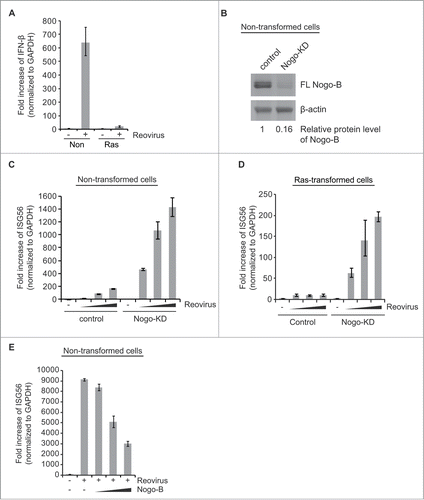
Next, we examined whether Nogo was associated with IFN signaling using knockdown (KD) of Nogo protein or overexpression of Nogo-B. For Nogo-KD, shRNA targeting the C-terminal of Nogo mRNAs, which knocked down all 3 isoforms (A, B and C), was used due to difficulties in targeting Nogo-B specifically. [Of note, Nogo-B is the most ubiquitously expressed and abundant isoform in NIH-3T3 cells, whereas Nogo-A and C are primarily expressed in brain and muscle cells, respectively.Citation13,30 Therefore, the major, if not the sole Nogo isoform that was knocked down was Nogo-B.] We found Nogo-B knockdown using this shRNA to be highly efficient (> 80%) compared to the control knockdown (eGFP shRNA) (). Interestingly, Nogo-KD significantly promoted IFN response in non-transformed cells upon reovirus infection ( and Fig. S1A and S1B). Importantly, Nogo-KD in Ras-transformed cells was also able to restore antiviral IFN response to reovirus infection in a dose-dependent manner ( and Fig. S1C). Furthermore, Nogo-KD in human embryonic kidney (HEK293T) cells significantly enhanced IFN response to the dsRNA analog, poly(I:C) (Fig. 1D), suggesting that suppression of IFN response by Nogo is not restricted to reovirus infection. Meanwhile, overexpression of full-length mouse Nogo-B decreased IFN response dose-dependently in non-transformed cells upon reovirus infection ( and Fig. S2A and B) or poly(I:C) transfection (Fig. S2D). [Dose-dependent overexpression of Nogo-B was confirmed by western blot analysis at the time of infection and no significant Nogo-B cleavage was observed in non-transformed cells (Fig. S2C).] Taken together, our results clearly demonstrate that Nogo-B (both full-length and cleaved) can suppress virus-induced IFN signaling.
Blockade of Nogo-B cleavage restores the impaired IFN production in Ras-transformed cells
We then asked whether cleavage of Nogo-B in Ras-transformed cells was linked to enhanced suppression of IFN signaling in these cells. It would clearly be of interest to see if blocking Nogo-B cleavage in these cells could reverse the suppression of IFN response to reovirus infection. In our search for such reagents, we found that DNA damaging agents (UV, doxorubicin, etoposide and actinomycin D) efficiently blocked Nogo-B cleavage in Ras-transformed cells without changing the overall Nogo-B level (). No significant change in the Nogo-B expression level or cleavage status was observed in non-transformed cells (Fig. S3). Since the tumor suppressor p53 is known to inhibit Ras oncogene-driven transformation,Citation32 reduction of oncogenic transformation by DNA damage-induced p53 expression might result in inhibition of the Nogo-B cleavage in Ras-transformed cells.
Figure 3. Blockade of Nogo-(B) cleavage reverses the impaired IFN response in Ras-transformed cells. (A) Ras-transformed cells were treated with UV, doxorubicin (Dox, 300 nM), etoposide (Etp, 5 μM) or actinomycin D (ActD, 2 nM) for 18 hr. Nogo-B cleavage and p53 levels were analyzed by western blot. (B) Non- or Ras-transformed cells were treated with vehicle (DMSO) or DNA damaging agents (300 nM of doxorubicin, 5 μM of etoposide) during reovirus infection. mRNAs were isolated 24 hr post-infection and subjected to real-time q-PCR. Fold increase of ISG56 was normalized to GAPDH.
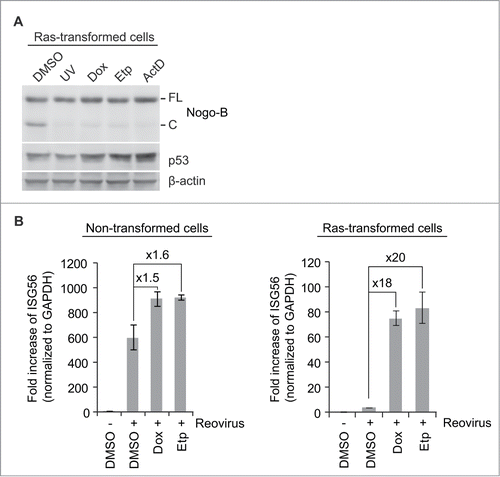
Consistent with the previous data (), reovirus infection alone did not induce IFN response in Ras-transformed cells (, right panel). However, exposure of the cells to doxorubicin or etoposide drastically enhanced (18–20-fold) reovirus-induced IFN response in Ras-transformed cells, compared to control (DMSO-treated) cells. In contrast, IFN response in non-transformed cells increased only slightly (1.5–1.6-fold) (, left panel). This slight increase in non-transformed cells was most likely due to the indirect effects of DNA damage responseCitation33 or p53 induction,Citation34 or to the small amount of cleaved Nogo-B in non-transformed cells as shown in . Altogether, our data support the hypothesis that Ras-induced cleavage of Nogo-B suppresses IFN signaling, at least partially.
IFN suppression by Nogo-B is independent of the Ras/ERK pathway
Next, we investigated a possible cross-talk between Nogo-B and the Ras/ERK pathway by examining the combinatorial effect of Nogo-KD and a MEK inhibitor (U0126). Consistent with previous results (), Nogo-KD alone or U0126 treatment (10 μM) alone was able to induce IFN response in Ras-transformed cells upon viral infection. Importantly, combination of Nogo-KD and MEK inhibition synergistically enhanced IFN response (). These results suggest that both activated Ras/ERK pathway and Nogo-B-mediated pathway are necessary for effective IFN suppression in Ras-transformed cells.
Figure 4. IFN suppression by Nogo is independent of the Ras/ERK pathway. (A and B) Enhancement of IFN response in Ras-transformed cells by MEK inhibitor (U0126) was compared between control and Nogo-KD cells. Cells were infected with increasing doses of reoviruses (MOI of 200 or 400 based on their titers in L929 cells) in the presence or absence of U0126 (10 μM). Data are presented as mean values with error bars showing the standard deviations. (C) Nogo-B cleavage was analyzed by western blot in cells treated with U0126 (10 μM) for 18 hr. (D) Schematic diagram of the proposed model. Ras-transformation suppresses IFN signaling by 2 separate but cooperative mechanisms, with the Ras/ERK and Nogo-B cleavage pathways representing the major and minor IFN inhibitory pathways, respectively.
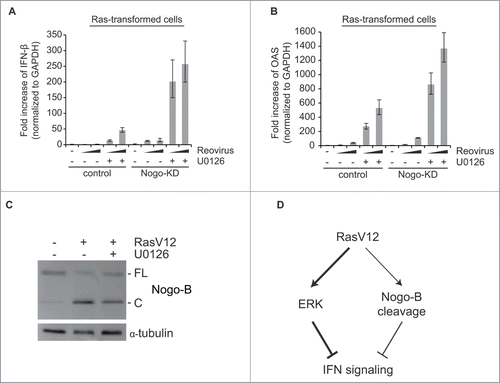
The above observation also suggested that cleavage of Nogo-B in Ras-transformed cells was likely not regulated by the activated Ras/ERK pathway. This was indeed found to be the case, as U0126 treatment did not significantly inhibit Nogo-B cleavage in Ras-transformed cells (). Our data therefore suggests that IFN suppression in Ras-transformed cells is the combined result of an activated Ras/ERK pathway (the major interferon inhibitory pathway), and the cleavage of Nogo-B ().
Discussion
In addition to its roles in apoptosis and ER stress,Citation24,25 Nogo-B also acts as a ligand that binds to its isoform-specific Nogo-B receptor (NgBR), regulating diverse cellular functions such as cell migration and angiogenesis.Citation18-20 In particular, the N-terminal 200 amino acids of Nogo-B (called amNogo-B) harbors the NgBR binding domain, and absence or deletion of this domain reduces the binding affinity to NgBR and NgBR-mediated cell functions.Citation18 Several studies reported post-translational modifications of Nogo-B at the N-terminus. Phosphorylation-dependent cleavage of Nogo-B occurs between AspCitation15 and SerCitation16 by caspase-7 during apoptosis.Citation35 Nogo-B can also be phosphorylated at Ser107 by MAPKAP-K2 [MAPK (mitogen-activated protein kinase)-activated protein kinase-2].Citation36 However, functional roles of such modifications or associations with oncogenic signaling have not yet been established.
Our studies demonstrate for the first time that the N-terminus of Nogo-B (within the functional domain known as amNogo-B) is cleaved upon Ras-induced oncogenic transformation, and this is accompanied by impairment of IFN response. Our observation of the proteasome-dependent cleavage of Nogo-B at a site (approximately 80 amino acids from the N-terminal end) that is different from previously reported sites (SerCitation16 or Ser107) suggests the existence of multiple cleavage sites in Nogo-B. Since cancer cells in general have higher protease activities,Citation37 Nogo-B might be cleaved at multiple sites by dysregulation of specific proteases depending on a myriad of diverse stimuli and signaling pathways. In this regard, it is interesting that NIH-3T3 cells that have been allowed to undergo spontaneously transformation also show Nogo-B cleavage that generates the 34 kDa fragment (our unpublished observation). Such Nogo-B cleavage is therefore likely the result, rather than the cause, of cellular transformation, and may well be involved in the maintenance of the transformed state. Based on previous reports suggesting the pro-apoptotic nature of Nogo-B,Citation24,25 its cleavage may render the cell resistant to apoptotic stimuli, resulting in enhanced proliferation. Yet another study shows that in HeLa cells, over-expression of Nogo-B induces epithelial-mesenchymal transition and promotes cell migration and invasion.Citation22 Whether Nogo-B cleavage is involved in these processes remains to be seen.
Perhaps the most interesting aspect from the present study is the involvement of Nogo-B in type I IFN regulation. We have previously established that activated Ras significantly suppresses virus-induced IFN response via an activated MEK/ERK pathway. The present data shows that uncleaved Nogo-B in non-transformed cells, and cleaved Nogo-B in Ras-transformed cells both have IFN-suppressing properties. Blocking Ras-induced cleavage of Nogo-B restores, at least partially, virus-induced IFN responses. Thus Nogo-B cleavage is associated with enhanced IFN impairment in Ras-transformed cells and, together with an activated MEK/ERK pathway in these cells, synergistically and effectively blocks virus-induced IFN response (). Precisely how Nogo-B and cleaved Nogo-B suppress IFN is unclear at present. Considering its localization, Nogo-B's diversified functions are almost certainly linked to its association with membranous structures. In terms of IFN response, Nogo-B and its cleavage product(s) might affect membrane structures or topology of the mitochondria-ER organelles, where IFN-associated molecules (e.g. RIG-I, TRIF or TBK1) are recruited by MAVS, an adaptor protein localized in the mitochondria,Citation38-40 thereby disrupting the formation or integrity of these complexes. Another possibility is that Nogo-B, and particularly the cleaved form of Nogo-B (either the smaller N-terminal cleaved product or the larger C-terminal product) may interfere with interactions of IFN signaling-associated proteins. It would also be interesting to determine whether these smaller cleavage products can be secreted or delivered outside the cell and function as ligands to modulate cellular physiology of neighboring cells.
While our study clearly demonstrates the induction of Nogo-B cleavage by Ras transformation, whether Nogo-B cleavage plays a role in human cancer, particularly cancers known to harbor Ras mutations, remains to be seen. Furthermore, as neurite outgrowth inhibitors, the Nogo proteins are believed to dictate axonal growth and regeneration. Dysregulation of the Nogo proteins and/or their receptors may lead to, other than cancer, neurological disorders including Alzheimer disease. Additionally, it has been reported that Nogo-B is involved in microglial activation in the CNS and Th2-driven lung inflammation, supporting a possible role of Nogo-B in immune regulation. Overall, the myriad of diverse functions of Nogo-B are only just beginning to be revealed. In view of our present findings, it would clearly be of interest to see whether the Nogo-B cleavage plays any roles in neurological and immune disorders.
There is yet one more intriguing twist to our present finding in terms of reovirus' ability to exploit the Ras-NogoB-IFN nexus to promote its own infection. Type 3 reovirus has been known to manifest specific tropism within the central nervous system in new born mice, infecting neurons and causing lethal encephalitis (reviewed in refs.Citation41). Interesting, Konopka-Anstadt et al.Citation42 recently discovered that reovirus uses the Nogo receptor 1 (NgR1) as an entry receptor in the CNS. Whether reovirus binding to NgR1 and reovirus exploitation of cleaved Nogo-B are separate or linked events is unclear at present but clearly deserves further investigation. It would be interesting, for example, to determine if high levels of Nogo-B have additional effects on reovirus infection in the CNS by occluding availability of NgR1 on target cells. Reciprocally, whether NgR1 binding by reovirus modulates the functions of Nogo-B in IFN regulation also needs to be investigated. Future studies on Nogo-B in neuronal cells should provide insights into these exciting possibilities.
Materials and Methods
Cell lines and viruses
NIH-3T3 cells were purchased from American Type Culture Collection and maintained in DMEM containing 10% newborn calf serum (Thermo Scientific) and HEK293T cells were maintained in DMEM with 10% fetal bovine serum (Gibco). Cells harboring shRNAs were selected by puromycin (2 μg/ml for NIH-3T3 cells, 1 μg/ml for HEK293T cells) or G418 (400 μg/ml). Mammalian reovirus type 3 Dearing was produced in L929 spinner cultures and purified as previously described.Citation43
Reagents and plasmids
Lentiviral vectors including pLKO.1-empty vector, pLKO.1-eGFP shRNA (control), pLKO.1-Nogo shRNA (TRCN0000071688, 5′-GCAGTGTTGATGTGGGTATTT-3′), pGIPZ-empty vector and pGIPZ-Nogo shRNA (V2LMM_33110, 5′-CACATAAACTAGGAAGAGA-3′) plasmids were purchased from Openbiosystems. shRNA sequences for Nogo-KD designed to target C-terminal shared region of transcript variants (Nogo-A, B and C) of mouse Nogo mRNAs. pLKO.1-Nogo shRNA can target both human and mouse Nogo mRNAs. H-RasV12 expressing vector (pLenti-CMV-RasV12-Neo) and vehicle control vector (pLenti-CMV/TO-Neo-DEST) were obtained from Addgene (ID 22259 for RasV12, ID 17292 for vehicle). Retroviral control vector (pBabe-puro) and H-RasV12 expressing vector in pBabe-puro were gifts from Dr. C. Der (University of North Carolina, Chapel Hill, NC). Expression plasmid for mouse Nogo-B was purchased from Origene. MG-132 and Caspase inhibitor (Z-VAD-FMK) were purchased from Calbiochem, MEK1/2 inhibitor (U0126) from Cell Signaling. Antibodies were purchased from Santa Cruz Biotechnology for the detection of N-terminus of Nogo-B (sc-271878) or from Abcam for C-terminal end of Nogo-B (ab65800).
Protein extraction and subcellular fractionation
For Western Blot analysis, cells were lysed using transmembrane protein extraction reagent (FIVEphoton Biochemicals) with protease inhibitor cocktail (Sigma) according to manufacturer's instructions. Subcellular fractionation was performed as previously described.Citation44
Biotinylation and isolation of cell surface protein
For Western Blot analysis, cells surface proteins were biotinylated with membrane impermeable EZ-Link sulfo-NHS-LC-Biotin (succinimidyl-6-[biotinamido]hexanoate) (Thermo scientific) according to the manufacturer's instructions. Cells grown in 15-cm tissue culture dishes were washed 3 times with ice-cold PBS (pH 8.0), harvested and suspended at a concentration of 2.5 × 107 cells/ml in PBS (pH 8.0). 20 mM of EZ-Link sulfo-NHS-LC-Biotin was added to the cell suspension in a final concentration of 2 mM and the cells were incubated at 4°C for 30 min with gentle rotation. The excess biotin reagent was removed by washing 3 times with PBS containing 100 mM glycine. Membrane-containing fractions were prepared as previously described with slight modification.Citation45 Cells were resuspended in lysis buffer containing 15 mM Tris·HCl (pH 7.4), 250 mM sucrose, 0.1 mM phenylmethylsulfonyl fluoride and protease inhibitor cocktail (Sigma) at 4°C and homogenized using a syringe. The cell lysate was centrifuged at 600 × g for 10 min at 4°C to remove nuclei and cell debris. The supernatant was transferred to another tube and centrifuged at 100,000 × g for 1 h at 4°C (Beckman, SW 41 Ti rotor). The pellet containing the membrane fraction was washed sequentially with high salt buffer (10 mM HEPES [pH 7.4], 2 M NaCl, 1 mM EDTA), high pH buffer (0.1 M Na2CO3, 1 mM EDTA, pH 11.3) and PBS, and solubilized in 300 μl of PBS containing 1% NP-40. The solubilized membrane fraction was subjected to pull-down assay.
Analysis of biotinylated cell surface proteins
The resuspended membrane fraction was sonicated briefly at maximum speed to disrupt the membranes and enhance solubilization. Protein concentration in the membrane fraction was quantified and the same amount of protein was used to compare cell surface proteins. The membrane fraction was diluted in PBS (final concentration of NP-40: 0.2%) and incubated with pre-washed Dynabeads M-280 Streptavidin (Invitrogen) for 2 h at 4°C with gentle rotation. Beads were washed 4 times with PBS containing 0.2% NP-40 by gentle pipetting, and resuspended in electrophoresis sample buffer. Captured proteins were dissociated from the beads by boiling for 5 min and resolved on 10 % SDS-PAGE gel (20 cm × 20 cm), followed by staining with silver nitrate. Protein bands of interest were excised from the gel, cut into 3–5 pieces and placed in a microcentrifuge tube where they were rinsed with 100 μl of 0.1 M ammonium bicarbonate followed by 100 μl of acetonitrile. The acetonitrile was removed by evaporation and the dried gel was rehydrated in 10 mM DDT (in 0.1 M ammonium bicarbonate) for 30 min at 56°C in order to reduce disulfides. The tube was cooled to room temperature, the liquid removed and replaced with 100 μl of iodoacetamide followed by incubation for 30 min in the dark at room temperature. The gel was rinsed with acetonitrile, dried and rehydrated using sequencing grade trypsin at a concentration of 12.5 ng/μl. Sufficient volume was added to cover the gel and digestion proceeded at 37°C overnight. Peptide extraction was performed using 20 μl of 0.1 ammonium bicarbonate followed by 2 × 20 μl of 50:50 acetonitrile:water. The extracts were combined in a new tube, evaporated to dryness and resuspended in 5% acetonitrile:water (with 0.1% formic acid). LC-MSMS analysis was performed using an Agilent 1100 capillary LC equipped with a 0.1 mm × 15 cm C18 reverse phase column. A linear gradient of 5% acetonitrile to 35% acetonitrile at a flow rate of 1 μl/min was used to elute the peptides. The column was interfaced to an AB Sciex 4000 QTrap mass spectrometer via a nanoelectrospray source. Mass spectra were collected over a range of 400 to 2000 Th for 1 sec and the 3 most intense peptides subjected to tandem MS analysis, this cycle was repeated for the duration of the chromatographic separation. Spectra were searched against the NCBInr protein database, using a local Mascot server with a Taxonomy of mus musculus, fixed modification of carbamidomethyl cysteine and variable modification of methionine (oxidation) and lysine (biotin EZ link).
Real-time quantitative PCR
Cells were grown on a 6-well plate, then infected with reoviruses. After 18 or 24 hrs post-infection, cells were harvested and RNAs were extracted by TRIzol (Invitrogen) according to the manufacturer's instructions. cDNA was synthesized using Superscript II reverse transcriptase (Invitrogen) and random primers. Real-time qPCR was performed using GoTaq qPCR Master Mix (Promega) with IFN-β, OAS, ISG56 and GAPDH specific primers listed in Table S1. Data are presented as mean values with error bars showing the standard deviations from 3 independent experiments unless otherwise specified.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
1044187_supplement_files.pdf
Download PDF (496.4 KB)Funding
This work was supported by an operating grant from the Canadian Institute of Health Research (CIHR grant #137152) to PWKL, the National Research Council (Canada) Genomics and Health Initiative (DP), Cancer Research Training Program postdoctoral fellowships through the Beatrice Hunter Cancer Research Institute (DA), a Government of Canada Post-Doctoral Research Fellowship (DA), and a CIHR Postdoctoral Fellowship (SG).
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- Bos JL. ras oncogenes in human cancer: a review. Cancer Res 1989; 49: 4682-9; PMID:2547513.
- Cascallo M, Capella G, Mazo A, Alemany R. Ras-dependent oncolysis with an adenovirus VAI mutant. Cancer Res 2003; 63: 5544-50; PMID:14500393.
- Esfandyari T, Tefferi A, Szmidt A, Alain T, Zwolak P, Lasho T, Lee PW, Farassati F. Transcription factors down-stream of Ras as molecular indicators for targeting malignancies with oncolytic herpes virus. Mol Oncol 2009; 3: 464-8; PMID:19766068; http://dx.doi.org/10.1016/j.molonc.2009.07.002
- Strong JE, Coffey MC, Tang D, Sabinin P, Lee PW. The molecular basis of viral oncolysis: usurpation of the Ras signaling pathway by reovirus. EMBO J 1998; 17: 3351-62; PMID:9628872; http://dx.doi.org/10.1093/emboj/17.12.3351
- Balachandran S, Porosnicu M, Barber GN. Oncolytic activity of vesicular stomatitis virus is effective against tumors exhibiting aberrant p53, Ras, or myc function and involves the induction of apoptosis. J Virol 2001; 75: 3474-9; PMID:11238874; http://dx.doi.org/10.1128/JVI.75.7.3474-3479.2001
- Urbanelli L, Trivelli F, Ercolani L, Sementino E, Magini A, Tancini B, Franceschini R, Emiliani C. Cathepsin L increased level upon Ras mutants expression: the role of p38 and p44/42 MAPK signaling pathways. Mol Cell Biochem 2010; 343:49-57; PMID:20524145; http://dx.doi.org/10.1007/s11010-010-0497-3
- Alain T, Kim TS, Lun X, Liacini A, Schiff LA, Senger DL, Forsyth PA. Proteolytic disassembly is a critical determinant for reovirus oncolysis. Mol Ther 2007; 15: 1512-21; PMID:17519890; http://dx.doi.org/10.1038/sj.mt.6300207
- Shmulevitz M, Pan LZ, Garant K, Pan D, Lee PW. Oncogenic Ras promotes reovirus spread by suppressing IFN-β production through negative regulation of RIG-I signaling. Cancer Res 2010; 70: 4912-21; PMID:20501842; http://dx.doi.org/10.1158/0008-5472.CAN-09-4676
- Christian SL, Zu D, Licursi M, Komatsu Y, Pongnopparat T, Codner DA, Hirasawa K. Suppression of IFN-induced transcription underlies IFN defects generated by activated Ras/MEK in human cancer cells. PLoS One 2012; 7: e44267; PMID:22970192; http://dx.doi.org/10.1371/journal.pone.0044267
- Battcock SM, Collier TW, Zu D, Hirasawa K. Negative regulation of the α interferon-induced antiviral response by the Ras/Raf/MEK pathway. J Virol 2006; 80: 4422-30; PMID:16611902; http://dx.doi.org/10.1128/JVI.80.9.4422-4430.2006
- Christian SL, Collier TW, Zu D, Licursi M, Hough CM, Hirasawa K. Activated Ras/MEK inhibits the antiviral response of α interferon by reducing STAT2 levels. J Virol 2009; 83: 6717-26; PMID:19386709; http://dx.doi.org/10.1128/JVI.02213-08
- Oertle T, Klinger M, Stuermer CA, Schwab ME. A reticular rhapsody: phylogenic evolution and nomenclature of the RTN/Nogo gene family. FASEB J 2003; 17: 1238-47; PMID:12832288; http://dx.doi.org/10.1096/fj.02-1166hyp
- Huber AB, Weinmann O, Brosamle C, Oertle T, Schwab ME. Patterns of Nogo mRNA and protein expression in the developing and adult rat and after CNS lesions. J Neurosci 2002; 22: 3553-67; PMID:11978832.
- Wang M, Han Y, Zhang XP, Lu YP. Nogo, a star protein in reticulon family. Neurosci Bull 2006; 22: 183-6; PMID:17704847
- Schwab ME. Functions of Nogo proteins and their receptors in the nervous system. Nat Rev Neurosci 2010; 11: 799-811; PMID:21045861; http://dx.doi.org/10.1038/nrn2936
- Chen MS, Huber AB, van der Haar ME, Frank M, Schnell L, Spillmann AA, Christ F, Schwab ME. Nogo-A is a myelin-associated neurite outgrowth inhibitor and an antigen for monoclonal antibody IN-1. Nature 2000; 403: 434-9; PMID:10667796; http://dx.doi.org/10.1038/35000601
- Sutendra G, Dromparis P, Wright P, Bonnet S, Haromy A, Hao Z, McMurtry MS, Michalak M, Vance JE, Sessa WC, Michelakis ED. The role of Nogo and the mitochondria-endoplasmic reticulum unit in pulmonary hypertension. Sci Transl Med 2011; 3: 88ra55; PMID:21697531; http://dx.doi.org/10.1126/scitranslmed.3002194
- Miao RQ, Gao Y, Harrison KD, Prendergast J, Acevedo LM, Yu J, Hu F, Strittmatter SM, Sessa WC. Identification of a receptor necessary for Nogo-B stimulated chemotaxis and morphogenesis of endothelial cells. Proc Natl Acad Sci U S A 2006; 103: 10997-1002; PMID:16835300; http://dx.doi.org/10.1073/pnas.0602427103
- Acevedo L, Yu J, Erdjument-Bromage H, Miao RQ, Kim JE, Fulton D, Tempst P, Strittmatter SM, Sessa WC. A new role for Nogo as a regulator of vascular remodeling. Nat Med 2004; 10: 382-8; PMID:15034570; http://dx.doi.org/10.1038/nm1020
- Zhao B, Chun C, Liu Z, Horswill MA, Pramanik K, Wilkinson GA, Ramchandran R, Miao RQ. Nogo-B receptor is essential for angiogenesis in zebrafish via Akt pathway. Blood 2010; 116:5423-33; PMID:20813898; http://dx.doi.org/10.1182/blood-2010-02-271577
- Wright PL, Yu J, Di YP, Homer RJ, Chupp G, Elias JA, Cohn L, Sessa WC. Epithelial reticulon 4B (Nogo-B) is an endogenous regulator of Th2-driven lung inflammation. J Exp Med 2010; 207: 2595-607; PMID:20975041; http://dx.doi.org/10.1084/jem.20100786
- Xiao W, Zhou S, Xu H, Li H, He G, Liu Y, Qi Y. Nogo-B promotes the epithelial-mesenchymal transition in HeLa cervical cancer cells via Fibulin-5. Oncol Rep 2013; 29: 109-16; PMID:23042479
- Shimakage M, Inoue N, Ohshima K, Kawahara K, Oka T, Yasui K, Matsumoto K, Inoue H, Watari A, Higashiyama S, Yutsudo M. Downregulation of ASY/Nogo transcription associated with progression of adult T-cell leukemia/lymphoma. Int J Cancer 2006; 119: 1648-53; PMID:16646068; http://dx.doi.org/10.1002/ijc.22011
- Li Q, Qi B, Oka K, Shimakage M, Yoshioka N, Inoue H, Hakura A, Kodama K, Stanbridge EJ, Yutsudo M. Link of a new type of apoptosis-inducing gene ASY/Nogo-B to human cancer. Oncogene 2001; 20: 3929-36; PMID:11494121; http://dx.doi.org/10.1038/sj.onc.1204536
- Kuang E, Wan Q, Li X, Xu H, Zou T, Qi Y. ER stress triggers apoptosis induced by Nogo-B/ASY overexpression. Exp Cell Res 2006; 312: 1983-8; PMID:16687140; http://dx.doi.org/10.1016/j.yexcr.2006.02.024
- Tagami S, Eguchi Y, Kinoshita M, Takeda M, Tsujimoto Y. A novel protein, RTN-XS, interacts with both Bcl-XL and Bcl-2 on endoplasmic reticulum and reduces their anti-apoptotic activity. Oncogene 2000; 19: 5736-46; PMID:11126360; http://dx.doi.org/10.1038/sj.onc.1203948
- Mi YJ, Hou B, Liao QM, Ma Y, Luo Q, Dai YK, Ju G, Jin WL. Amino-Nogo-A antagonizes reactive oxygen species generation and protects immature primary cortical neurons from oxidative toxicity. Cell Death Differ 2012; 19: 1175-86; PMID:22261619; http://dx.doi.org/10.1038/cdd.2011.206
- Teng FY, Tang BL. Nogo/RTN4 isoforms and RTN3 expression protect SH-SY5Y cells against multiple death insults. Mol Cell Biochem 2013; 384: 7-19; PMID:23955438; http://dx.doi.org/10.1007/s11010-013-1776-6
- Marcato P, Shmulevitz M, Pan D, Stoltz D, Lee PW. Ras transformation mediates reovirus oncolysis by enhancing virus uncoating, particle infectivity, apoptosis-dependent release. Mol Ther 2007; 15: 1522-30; PMID:17457318; http://dx.doi.org/10.1038/sj.mt.6300179
- Dodd DA, Niederoest B, Bloechlinger S, Dupuis L, Loeffler JP, Schwab ME. Nogo-A, -B, -C are found on the cell surface and interact together in many different cell types. J Biol Chem 2005; 280: 12494-502; PMID:15640160; http://dx.doi.org/10.1074/jbc.M411827200
- Shmulevitz M, Pan LZ, Garant K, Pan D, Lee PW. Oncogenic Ras promotes reovirus spread by suppressing IFN-β production through negative regulation of RIG-I signaling. Cancer Res 2010; 70: 4912-21; PMID:20501842; http://dx.doi.org/10.1158/0008-5472.CAN-09-4676
- Finlay CA, Hinds PW, Levine AJ. The p53 proto-oncogene can act as a suppressor of transformation. Cell 1989; 57: 1083-93; PMID:2525423; http://dx.doi.org/10.1016/0092-8674(89)90045-7
- Brzostek-Racine S, Gordon C, Van SS, Reich NC. The DNA damage response induces IFN. J Immunol 2011; 187: 5336-45; PMID:22013119; http://dx.doi.org/10.4049/jimmunol.1100040
- Munoz-Fontela C, Macip S, Martinez-Sobrido L, Brown L, Ashour J, Garcia-Sastre A, Lee SW, Aaronson SA. Transcriptional role of p53 in interferon-mediated antiviral immunity. J Exp Med 2008; 205: 1929-38; PMID:18663127; http://dx.doi.org/10.1084/jem.20080383
- Schweigreiter R, Stasyk T, Contarini I, Frauscher S, Oertle T, Klimaschewski L, Huber LA, Bandtlow CE. Phosphorylation-regulated cleavage of the reticulon protein Nogo-B by caspase-7 at a noncanonical recognition site. Proteomics 2007; 7: 4457-67; PMID:18072206; http://dx.doi.org/10.1002/pmic.200700499
- Rousseau S, Peggie M, Campbell DG, Nebreda AR, Cohen P. Nogo-B is a new physiological substrate for MAPKAP-K2. Biochem J 2005; 391: 433-40; PMID:16095439; http://dx.doi.org/10.1042/BJ20050935
- Choi KY, Swierczewska M, Lee S, Chen X. Protease-activated drug development. Theranostics 2012; 2: 156-178; PMID:22400063; http://dx.doi.org/10.7150/thno.4068
- Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, Ishii KJ, Takeuchi O, Akira S. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol 2005; 6: 981-8; PMID:16127453; http://dx.doi.org/10.1038/ni1243
- Xu LG, Wang YY, Han KJ, Li LY, Zhai Z, Shu HB. VISA is an adapter protein required for virus-triggered IFN-β signaling. Mol Cell 2005; 19: 727-40; PMID:16153868; http://dx.doi.org/10.1016/j.molcel.2005.08.014
- Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 2005; 122: 669-82; PMID:16125763; http://dx.doi.org/10.1016/j.cell.2005.08.012
- Tyler KL, Fields BN. Reoviruses. In Fields BN, Knipe DM, Howley PM, eds.. Virology. Raven Press: New York, 1996; 1597-624.
- Konopka-Anstadt JL, Mainou BA, SutherlandDM, Sekine Y, Strittmatter SM, Dermody TS. The Nogo receptor NgR1 mediates infection by mammalian reovirus. Cell Host Microbe 2014; 15:681-91.
- Mendez II, Hermann LL, Hazelton PR, Coombs KM. A comparative analysis of freon substitutes in the purification of reovirus and calicivirus. J Virol Methods 2000; 90:59-67.
- Samali A, Cai J, Zhivotovsky B, Jones DP, Orrenius S. Presence of a pre-apoptotic complex of pro-caspase-3, Hsp60 and Hsp10 in the mitochondrial fraction of jurkat cells. EMBO J 1999; 18:2040-48.
- Xiong X, Huang S, Zhang H, Li J, Shen J, Xiong J, Lin Y, Jiang L, Wang X, Liang S. Enrichment and proteomic analysis of plasma membrane from rat dorsal root ganglions. Proteome Sci 2009; 7:41.