
Abstract
Defects in the maintenance of protein homeostasis, or proteostasis, has emerged as an underlying feature of a variety of human pathologies, including aging-related diseases. Proteostasis is achieved through the coordinated action of cellular systems overseeing amino acid availability, mRNA translation, protein folding, secretion, and degradation. The regulation of these distinct systems must be integrated at various points to attain a proper balance. In a recent study, we found that the mechanistic target of rapamycin (mTOR) complex 1 (mTORC1) pathway, well known to enhance the protein synthesis capacity of cells while concordantly inhibiting autophagy, promotes the production of more proteasomes. Activation of mTORC1 genetically, through loss of the tuberous sclerosis complex (TSC) tumor suppressors, or physiologically, through growth factors or feeding, stimulates a transcriptional program involving the sterol-regulatory element binding protein 1 (SREBP1) and nuclear factor erythroid-derived 2-related factor 1 (NRF1; also known as NFE2L1) transcription factors leading to an increase in cellular proteasome content. As discussed here, our findings suggest that this increase in proteasome levels facilitates both the maintenance of proteostasis and the recovery of amino acids in the face of an increased protein load consequent to mTORC1 activation. We also consider the physiological and pathological implications of this unexpected new downstream branch of mTORC1 signaling.
Introduction
The mechanistic target of rapamycin (mTOR) complex 1 (mTORC1) senses and integrates cellular growth signals and stimulates key anabolic processes that promote the production of biomass and cell growth.Citation1 The protein kinase mTOR forms 2 physically and functionally distinct protein complexes, mTORC1 and mTORC2. mTORC2 is activated by extracellular growth factors and regulates, among other things, cell survival pathways.Citation2 mTORC1 is also activated by growth factor signaling pathways, but only when there is sufficient intracellular nutrients and energy present. This property of mTORC1 allows it to integrate cell intrinsic signals with tissue-specific or organismal signals to properly sense and respond to changes in local and systemic growth conditions.
Once activated, mTORC1 regulates many downstream cellular processes, stimulating the anabolic synthesis of proteins, lipids, and nucleotidesCitation3 and inhibiting the catabolic process of autophagy, which recycles macromolecules into their nutrient components under conditions of nutrient depletion.Citation4 mTORC1 controls these processes through distinct downstream targets leading to transcriptional, translational, or post-translational regulatory mechanisms that differ in their temporal nature. mTORC1 rapidly induces specific aspects of protein and nucleotide synthesis and acutely blocks the onset of autophagy through direct phosphorylation of key regulatory proteins and enzymes.Citation5-8 On the other hand, mTORC1 stimulates lipid synthesis with more delayed kinetics through the sterol-regulatory element binding protein family of transcription factors (SREBP1a, 1c, and 2), which induce the expression of nearly all genes encoding the enzymes of de novo fatty acid and sterol synthesis.Citation9-11
In a recent study, we uncovered a surprising new function of mTORC1 signaling in its activation of a transcriptional program leading to the production of more proteasomes, thereby increasing the cellular capacity for protein degradation.Citation12 Here, we discuss the molecular mechanism underlying this seemingly paradoxical role for mTORC1 and how it fits in with its more well-established anabolic functions, such as promoting protein synthesis.
mTORC1 increases cellular proteasome content by inducing NRF1
In an attempt to understand the effects of mTORC1 signaling on protein homeostasis (proteostasis), we compared rates of protein synthesis and degradation in cells with or without activation of mTORC1.Citation12 We were surprised to find that cells with activated mTORC1 signaling not only produced protein at increased rates but also turned over protein more efficiently than cells with prolonged inactivation of mTORC1. This enhanced rate of protein degradation downstream of mTORC1 was found to be dependent on the proteasome, but not the lysosome, and occurred independent of mTORC1s regulation of autophagy. Cells and tissues with activated mTORC1 had increased expression of nearly all proteasome genes (PSM genes), including those encoding subunits of both the 20S core particle and the 19S regulatory complex, and displayed elevated levels of intact proteasomes. Recent independent studies of mice treated with the mTORC1 inhibitor rapamycin have also demonstrated a significant decrease in the expression of PSM genes and proteasome activity in the liver upon mTORC1 inhibition.Citation13,14
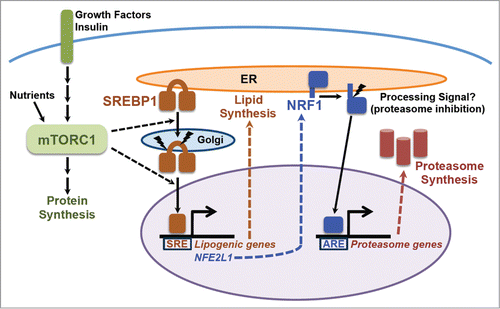
Previous studies had identified a transcription factor called nuclear factor erythroid-derived 2-related factor 1 (NRF1; also known as NFE2L1 or TCF11) as being a global regulator of proteasome gene expression.Citation15,16 Note: this NRF1 should not be confused with nuclear respiratory factor 1, which goes by the same name. We found that the increase in PSM gene expression, proteasome levels, and enhanced rate of protein turnover upon mTORC1 activation were all dependent on NRF1, but not the closely related NRF2. Genetic or physiological activation of mTORC1 lead to increased NRF1 protein levels in cells and tissues, including the liver and brain. Another surprise came from our finding that this increase in NRF1 levels results from the mTORC1-mediated activation of SREBP1, which directly induces NRF1 gene expressionCitation12 ().
Figure 1. The mTORC1-SREBP-NRF1-Proteasome pathway. Growth factors and nutrients activate mTORC1, which promotes an increase in cellular protein synthesis. mTORC1 also stimulates activation of the SREBP1 transcription factor by promoting its processing and nuclear accumulation, which requires its trafficking to the Golgi, where it is proteolytically cleaved by 2 proteases, resulting in release of the N-terminus encompassing the mature active transcription factor. Mature SREBP1 binds to SRE sequences in the promoters of genes, including the enzymes of de novo lipid synthesis and NFE2L1, encoding the NRF1 transcription factor. NRF1 is synthesized as an ER transmembrane protein and must be processed to release the active transcription factor. Proteasome inhibitors stimulate this processing, but the nature of the physiological signal is currently unknown. Once activated, NRF1 goes to the nucleus and turns on a subset of genes containing AREs in their promoter, including those encoding all, or nearly all, subunits of the proteasome, leading to an increase in cellular proteasome content. See text for more details.
NRF1 and its physiological activation by mTORC1 and SREBP1
NRF1 belongs to a family of 4 related transcription factors that also includes the more widely studied NRF2 (NFE2L2), which is known for its role in the cellular adaptive response to oxidative stress. Like NRF2, NRF1 regulates gene expression through antioxidant response elements (AREs) in a broad range of gene promoters.Citation17,18 However, NRF1 and NRF2 are regulated in distinct manners and have non-redundant functions, with NRF1, but not NRF2, being essential for embryonic development.Citation19,20 The functional differences between NRF1 and NRF2 appear to be due to these transcription factors targeting distinct subsets of ARE-containing genes.Citation21 Interestingly, all 32 proteasome subunit genes from humans have putative AREs in their promoters, and increasing evidence indicates that these genes are primarily regulated by NRF1, and not NRF2.Citation15,16,22 Likewise, in our study we found that mTORC1 signaling induces the expression of proteasome genes exclusively through NRF1.Citation12
The nematode C. elegans encodes just one distant ortholog of NRF1/NRF2/NRF3, dubbed SKN-1, which regulates a large number of genes that overlap with the collective targets identified for the vertebrate family members, including oxidative and xenobiotic stress response and PSM genes.Citation23-25 Interestingly, genetic studies in C. elegans have indicated that SKN-1 is epistatic to both TORC1 and TORC2 for their influence on longevity, with the genetic relationship pointing to negative regulation of SKN-1 by both TOR complexes.Citation26 The apparent discrepancy between these findings and our study indicating that mTORC1 signaling positively regulates NRF1,Citation12 but not NRF2, perhaps reflects the evolutionary pressures that led to the split of SKN-1 into multiple family members with distinct subsets of gene targets. Even SKN-1 has been shown to selectively regulate different sets of genes upon exposure to specific stresses, leading to a specialized response to a given stress.Citation24,27 The split of SKN-1 into multiple separate genes and gene products in higher metazoans further facilitates both differential stress sensing and the channeling of the stress response toward the most appropriate ARE-containing gene targets for that stress.
The factors that control NRF1 activation are not fully known. Like SREBP isoforms, which are also activated in response to mTORC1 signaling,Citation9-11 NRF1 is synthesized as a transmembrane protein residing at the endoplasmic reticulum (ER) and must be proteolytically processed to release the active transcription factor.Citation15,16,22,28,29 NRF1 processing and activation is greatly enhanced by treatment with chemical inhibitors of the proteasome, thereby triggering a proteasome recovery mechanism leading to the production of more proteasomes.Citation15,16 Studies on the activation of NRF1 have been largely restricted to its response to proteasome inhibitors, with the physiological signals that regulate NRF1 processing currently unknown, but perhaps reflecting changes in cellular proteasome levels or activity. Importantly, we found no evidence that mTORC1 signaling influences the processing of NRF1 per se, and the activating effects of proteasome inhibitors on NRF1 are independent of mTORC1.Citation12 Instead, genetic or growth factor-stimulated activation of mTORC1 results in a robust increase in full-length NRF1 protein that also results in the presence of more processed, active NRF1 and subsequent expression of NRF1 gene targets. Our evidence indicates that mTORC1 induces NRF1 primarily at the transcript level through the post-translational activation of SREBP1 (). The NFE2L1 gene, which encodes NRF1, has 4 consensus SREs near its predicted transcriptional start site that are conserved in mammals and are not found in NFE2L2, encoding NRF2.Citation12 An analysis of data from independent genome-wide studies geared at identifying transcriptional targets of SREBP further support our finding that NRF1 is a direct target of SREBP1.Citation30-32
On its surface, the discovery that mTORC1 activates NRF1, and ultimately an increase in proteasome levels, through SREBP1 is surprising. The SREBP family of transcription factors are well established drivers of de novo lipid synthesis.Citation33 It has become clear that the SREBPs are a major transcriptional effector of mTORC1 signaling in multiple cellular settings.Citation9-11,34,35 Lipids serve as a major source of stored energy that can be mobilized quickly, and growing cells have an increased demand for lipids for the production of new membranes. mTORC1 plays a central role in the control of cell growth, and evidence indicate that the activation of SREBP-dependent lipid synthesis is one essential downstream factor in its promotion of growth.Citation9,10 Recent studies have further suggested that, in parallel to its induction of protein synthesis, mTORC1 must also activate SREBP and de novo lipid synthesis in order to prevent ER stress,Citation36,37 which results from the accumulation of mis-folded proteins in the ER. The essential nature of SREBP in the context of mTORC1 activation underlies a need to balance an increase in protein load in the ER and elsewhere with the expansion of ER and other cellular membranes through de novo lipid synthesis. Importantly, like depletion of SREBP, loss of NRF1 can also cause ER stress.Citation38 ER-associated protein degradation, or ERAD, plays an essential role in maintaining protein homeostasis by alleviating the protein burden of the ER,Citation39 and transcriptional targets of NRF1, including the proteasome and VCP/p97, are required for ERAD and the prevention of ER stress.Citation15,16,22,38 Thus, the placement of both lipid synthesis and NRF1 downstream of mTORC1's activation of SREBP1 might reflect selective pressures to couple enhanced protein synthesis upon mTORC1 activation with membrane expansion and an enhanced capacity for protein turnover, both of which protect from proteotoxic stress, including ER stress (). Finally, it is worth noting that liver-specific deletion of Nfe2l1 in mice disrupts lipid homeostasis, resulting in hepatic lipid accumulation, and NRF1 has been proposed to control the expression of a subset of enzymes involved in lipid metabolism.Citation38,40,41 These findings suggest additional underlying functions of NRF1 that might also account for its control by SREBP1.
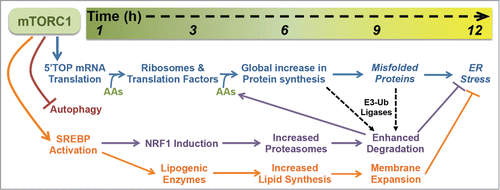
Figure 2. The temporal response to mTORC1 activation. Within the first hour of mTORC1 activation, it stimulates the translation of 5′TOP mRNAs, inhibits autophagy, and promotes SREBP activation, initiating a cascade of cellular events that promote cell growth while protecting from proteotoxic stress. A key feature of this program is the delayed production of proteasomes by NRF1 induction, which enhances the efficiency of protein degradation following the global increase in protein synthesis, thereby facilitating the clearance of proteins and misfolded proteins that have been independently targeted for degradation by target-specific E3-ubiquitin (Ub) ligases. Downstream of SREBP transcriptional targets, the activation of lipid synthesis and expansion of cellular membranes combined with the NRF1-dependent increase in proteasomes and protein turnover protects cells with activated mTORC1 signaling from proteotoxic stress, including ER stress. The time scale on the top provides a rough estimate of the kinetics of this response, with the precise timing likely to vary in different cell and tissue settings. See text for more details.
Temporal influence of mTORC1 signaling on protein homeostasis
A key feature of our findings that mTORC1 induces the synthesis of new proteasomes, thereby enhancing the degradative capacity of cells, lies in the timing of this regulation (). Several cellular events are induced within minutes of mTORC1 activation.Citation1,42 Relevant to our discussion here, are the acute induction of mRNA translation, inhibition of autophagy, and stimulation of SREBP. The activation of mTORC1 leads to the immediate and robust initiation of translation of a specific class of mRNAs that have 5′-terminal oligopyrimidine (5′TOP) or 5′TOP-like motifs, which encode ribosomal proteins and translation factors.Citation43-45 As such, mTORC1 initiates a program to increase the global protein synthesis capacity of the cell. It is particularly important to suppress protein degradation during this early response to mTORC1 activation, such that the newly generated ribosomes and translation initiation and elongation complexes are maintained. The acute inhibition of autophagy is one mechanism by which mTORC1 blocks the turnover of protein and other macromolecules during this phase.Citation46-49 While the precise molecular mechanism is currently unknown, mTORC1 also acutely stimulates the processing and nuclear accumulation of the SREBP transcription factors, with subsequent induction of all of the major enzymes of de novo lipid synthesis.Citation9-11,35 The result is a measureable increase in rates of lipid synthesis within 6 hours of mTORC1 activation; for instance, as observed in primary hepatocytes stimulated with insulin.Citation34 As described above, the activation of SREBP also leads to the transcriptional activation of NRF1, resulting in a substantial increase in its abundance in cells and tissue by 6 hours.12 NRF1 accumulation and its subsequent processing from the ER and translocation to the nucleus lead to the global induction of PSM gene expression and an increase in intact 26S proteasomes in cells several hours after the initial stimulation of mTORC1 ().
While all of these processes downstream of mTORC1 are initiated at the same time, they impact the cell with different kinetics and under different intracellular states. The fact that mTORC1's induction of proteasome synthesis requires 2 rounds of transcription means that cellular proteasome content rises following the initial wave of mRNA translation that drives ribosome biogenesis and a global increase in protein synthesis (). We believe that having more proteasomes allows cells to better deal with the resulting elevated rates of translation and increased protein load stemming from mTORC1 activation. Besides the delayed timing, what prevents this from creating a futile cycle of protein synthesis and degradation is the essential role of E3-ubiquitin ligases for targeting specific proteins, or nascent polypeptides, for proteasomal degradation.Citation50 While we cannot completely rule out a role for mTORC1 in the selection of specific proteins for degradation, our data are consistent with a model whereby mTORC1 signaling does not influence which proteins get targeted, but rather makes the degradation of those independently targeted proteins more efficient through the NRF1-mediated increase in cellular proteasome levels.
It is evident that cells with higher rates of protein synthesis and protein content and that are larger in size have an increased demand for proteasomes. The importance of coupling these events is underscored by their placement as distinct downstream branches of mTORC1 signaling, with the induction of proteasome synthesis representing a preprogrammed adaptive response. This downstream function of mTORC1 is not a classical adaptive stress response, as it is not initiated as a result of the increase in protein load per se, but rather in anticipation of the increase in mRNA translation that follows mTORC1 activation.
Relationship between the proteasome and autophagy in the control of amino acid and protein homeostasis downstream of mTORC1
Numerous cellular pathways and processes are tied together through the fundamental need to maintain intracellular amino acid and protein homeostasis. For instance, amino acid levels influence translation, both directly as a limiting substrate and indirectly through amino acid sensing signaling pathways that control translation initiation, such as mTORC1 and GCN2.Citation51,52 While the mechanisms are less well understood, there are also critical points of cross-talk between the pathways controlling protein synthesis and degradation, and imbalances between these processes can be detrimental (e.g. Refs. Citation53,54). In addition to the need for quality control during protein synthesis and folding, global rates of proteasome-mediated protein turnover influence the pool of amino acids available for the synthesis of new proteins. Consistent with this idea, we found that the NRF1-proteasome branch of mTORC1 signaling facilitates the maintenance of adequate pools of amino acids needed to sustain protein synthesis under conditions of mTORC1 activation.Citation12 We believe that this mechanism of amino acid recovery following prolonged stimulation of protein synthesis is particularly important given the strong inhibitory effect of mTORC1 signaling on autophagy, another major source of amino acids.Citation4,46-49
It is important to note that a reciprocal relationship between autophagy and the proteasome has been recognized. For instance, prolonged inhibition of the proteasome can induce autophagy,Citation55 perhaps through amino acid depletion and inhibition of mTORC1. However, we found that the influence of mTORC1 on long-term rates of protein turnover was separable from its effects on autophagy, as mTORC1 inhibition slowed protein degradation to an even greater extent in autophagy-deficient cells.Citation12 Given the differential control of autophagy and the proteasome by growth conditions that respectively inhibit or activate mTORC1 signaling, it is apparent that the amino acids produced from these different forms of protein turnover are made available to the cell under very different cellular states. As autophagy is induced acutely under starvation conditions where mTORC1 and protein synthesis are inhibited, it is likely that the bulk of autophagy-derived amino acids are catabolized for energy. In contrast, the fate of those amino acids produced from protein degradation in proteasomes, which are elevated several hours after mTORC1 is initially activated, will depend on whether mTORC1 signaling, which is inherently transient in nature, is sustained or not. Therefore, by reciprocal control of these 2 modes of protein turnover, cellular conditions that activate or inhibit mTORC1 can differentially and temporally influence whether the amino acid products are used for new protein synthesis, catabolized, or perhaps released for use by other tissues.
Implications in health, aging and disease
Moving forward, it is essential to understand the ramifications of the new mTORC1-NRF1-proteasome pathway in different physiological and pathological contexts. Defects in the coordination of cellular processes controlling proteostasis, including protein synthesis, folding, and turnover, are believed to underlie a diverse array of human diseases.Citation56 Likewise, dysregulation of mTORC1 signaling is associated with an overlapping set of pathological states, including cancer, aging, and aging-related diseases.Citation42,57,58 In model organisms from yeast to mice, the mTORC1 inhibitor rapamycin has been shown to promote longevity, leading to intense interest in the use of mTORC1 inhibitors for specific aging-related diseases, especially those with underlying defects in proteostasis, such as neurodegenerative diseases.Citation58,59 For instance, mTORC1 signaling is elevated in the brains of patients with Alzheimer disease (AD), and rapamycin is effective in AD mouse models, where it is believed that the resulting induction of autophagy facilitates the clearance of protein aggregates.Citation60,61 A question that arises from our recent findingsCitation12 is whether the reciprocal control of autophagy and proteasome levels by mTORC1 is fully maintained as cells, tissues, and organisms age. One might imagine that a break down in this balance could tip the scales toward the accumulation of misfolded proteins in one setting (e.g.,, neurodegeneration) or toward an increase in protein degradation in another (e.g., sarcopenia or muscle wasting). Future studies aimed at defining the net effect of protein synthesis and breakdown mechanisms downstream of mTORC1 will be particularly interesting in skeletal muscle, where resistance exercise can stimulate both processes, with a greater ratio of synthesis to degradation when exercise is followed by amino acid or protein ingestion.Citation62 mTORC1 also plays a key role in neurocognition and behavior, including autism, which is thought to be through effects on synaptic plasticity,Citation59 a process that requires dynamic control of protein synthesis and degradation. Importantly, we found that mTORC1 activation in cortical neurons leads to the accumulation of NRF1 and expression of PSM genes, suggesting that mTORC1 regulates the proteasome content of neurons, which could influence its role in both neurodegeneration and synaptic plasticity.Citation12
Aberrant activation of mTORC1 is a common feature in genetic tumor syndromes and sporadic cancers.57 Tuberous sclerosis complex (TSC) is a genetic disorder caused by loss of TSC1 or TSC2, the gene products of which form a protein complex that is a key negative regulator of mTORC1.Citation63 Loss of the TSC genes leads to chronically elevated mTORC1 signaling, which underlies the major clinical features of TSC, including the development of widespread tumors, epilepsy, and autism spectrum disorders. We found that cell and mouse models of TSC display increased NRF1 and proteasome levels, suggesting that this branch of mTORC1 signaling might contribute to some of the manifestations of this disease.Citation12 Importantly, NFE2L1 and PSM genes are overexpressed and proteasome levels are elevated in a number of different cancers (e.g., Refs. Citation64–66 and gene expression analyses at Oncomine.org and cBioPortal.orgCitation67). Neither the molecular mechanism nor functional consequences of this increase are currently known. However, given the frequent activation of mTORC1 in human cancers, across nearly all lineages,Citation57 future studies should be aimed at determining the role of mTORC1 in driving an increase in proteasome content in tumor cells. It will also be important to determine whether the mTORC1-NRF1 pathway influences the sensitivity of different cancers to compounds targeting the ubiquitin-proteasome system, including proteasome inhibitors already in clinical use such as bortezomib/velcade.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
funding
This work was supported by the NIH (R01-CA122617 and P01-CA120964), DOD (TSCRP grant W81XWH-10-1-0861), and the Ellison Medical Foundation.
Related Research Data
References
- Dibble CC, Manning BD. Signal integration by mTORC1 coordinates nutrient input with biosynthetic output. Nat Cell Biol 2013; 15:555-64; PMID:23728461; http://dx.doi.org/10.1038/ncb2763
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005; 307:1098-101; PMID:15718470; http://dx.doi.org/10.1126/science.1106148
- Howell JJ, Ricoult SJ, Ben-Sahra I, Manning BD. A growing role for mTOR in promoting anabolic metabolism. Biochem Soc Trans 2013; 41:906-12; PMID:23863154; http://dx.doi.org/10.1042/BST20130041
- Singh R, Cuervo AM. Autophagy in the cellular energetic balance. Cell Metab 2011; 13:495-504; PMID:21531332; http://dx.doi.org/10.1016/j.cmet.2011.04.004
- Ma XM, Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol 2009 10:307-18; PMID:19339977; http://dx.doi.org/10.1038/nrm2672
- Ben-Sahra I, Howell JJ, Asara JM, Manning BD. Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science 2013; 339:1323-8; PMID:23429703; http://dx.doi.org/10.1126/science.1228792
- Robitaille AM, Christen S, Shimobayashi M, Cornu M, Fava LL, Moes S, Prescianotto-Baschong C, Sauer U, Jenoe P, Hall MN. Quantitative phosphoproteomics reveal mTORC1 activates de novo pyrimidine synthesis. Science 2013; 339:1320-3; PMID:23429704; http://dx.doi.org/10.1126/science.1228771
- Russell RC, Yuan HX, Guan KL. Autophagy regulation by nutrient signaling. Cell Res 2014; 24:42-57; PMID:24343578; http://dx.doi.org/10.1038/cr.2013.166
- Porstmann T, Santos CR, Griffiths B, Cully M, Wu M, Leevers S, Griffiths JR, Chung YL, Schulze A. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab 2008; 8:224-36; PMID:18762023; http://dx.doi.org/10.1016/j.cmet.2008.07.007
- Duvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, Triantafellow E, Ma Q, Gorski R, Cleaver S, et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell 2010; 39:171-83; PMID:20670887; http://dx.doi.org/10.1016/j.molcel.2010.06.022
- Owen JL, Zhang Y, Bae SH, Farooqi MS, Liang G, Hammer RE, Goldstein JL, Brown MS. Insulin stimulation of SREBP-1c processing in transgenic rat hepatocytes requires p70 S6-kinase. Proc Natl Acad Sci U S A 2012; 109:16184-9; PMID:22927400; http://dx.doi.org/10.1073/pnas.1213343109
- Zhang Y, Nicholatos J, Dreier JR, Ricoult SJ, Widenmaier SB, Hotamisligil GS, Kwiatkowski DJ, Manning BD. Coordinated regulation of protein synthesis and degradation by mTORC1. Nature 2014; 513:440-3; PMID:25043031; http://dx.doi.org/10.1038/nature13492
- Fok WC, Chen Y, Bokov A, Zhang Y, Salmon AB, Diaz V, Javors M, Wood WH 3rd, Zhang Y, Becker KG, et al. Mice fed rapamycin have an increase in lifespan associated with major changes in the liver transcriptome. PLoS One 2014; 9:e83988; PMID:24409289; http://dx.doi.org/10.1371/journal.pone.0083988
- Zhang Y, Bokov A, Gelfond J, Soto V, Ikeno Y, Hubbard G, Diaz V, Sloane L, Maslin K, Treaster S, et al. Rapamycin extends life and health in C57BL/6 mice. J Gerontol A Biol Sci Med Sci 2014; 69:119-30; PMID:23682161; http://dx.doi.org/10.1093/gerona/glt056
- Radhakrishnan SK, Lee CS, Young P, Beskow A, Chan JY, Deshaies RJ. Transcription factor Nrf1 mediates the proteasome recovery pathway after proteasome inhibition in mammalian cells. Mol Cell 2010; 38:17-28; PMID:20385086; http://dx.doi.org/10.1016/j.molcel.2010.02.029
- Steffen J, Seeger M, Koch A, Kruger E. Proteasomal degradation is transcriptionally controlled by TCF11 via an ERAD-dependent feedback loop. Mol Cell 2010; 40:147-58; PMID:20932482; http://dx.doi.org/10.1016/j.molcel.2010.09.012
- Johnsen O, Murphy P, Prydz H, Kolsto, AB. Interaction of the CNC-bZIP factor TCF11/LCR-F1/Nrf1 with MafG: binding-site selection and regulation of transcription. Nucleic Acids Res 1998; 26:512-20; PMID:9421508; http://dx.doi.org/10.1093/nar/26.2.512
- Biswas M, Chan JY. Role of Nrf1 in antioxidant response element-mediated gene expression and beyond. Toxicol Appl Pharmacol 2010; 244:16-20; PMID:19665035; http://dx.doi.org/10.1016/j.taap.2009.07.034
- Farmer SC, Sun CW, Winnier GE, Hogan BL, Townes TM. The bZIP transcription factor LCR-F1 is essential for mesoderm formation in mouse development. Genes Dev 1997; 11:786-98; PMID:9087432; http://dx.doi.org/10.1101/gad.11.6.786
- Chan JY, Kwong M, Lu R, Chang J, Wang B, Yen TS, Kan YW. Targeted disruption of the ubiquitous CNC-bZIP transcription factor, Nrf-1, results in anemia and embryonic lethality in mice. EMBO J 1998; 17:1779-87; PMID:9501099; http://dx.doi.org/10.1093/emboj/17.6.1779
- Ohtsuji M, Katsuoka F, Kobayashi A, Aburatani H, Hayes JD, Yamamoto M. Nrf1 and Nrf2 play distinct roles in activation of antioxidant response element-dependent genes. J Biol Chem 2008; 283:33554-62; PMID:18826952; http://dx.doi.org/10.1074/jbc.M804597200
- Sha Z, Goldberg AL. Proteasome-mediated processing of Nrf1 is essential for coordinate induction of all proteasome subunits and p97. Curr Biol 2014; 24:1573-83; PMID:24998528; http://dx.doi.org/10.1016/j.cub.2014.06.004
- Niu W, Lu ZJ, Zhong M, Sarov M, Murray JI, Brdlik CM, Janette J, Chen C, Alves P, Preston E. Diverse transcription factor binding features revealed by genome-wide ChIP-seq in C. elegans. Genome Res 2011; 21:245-54; PMID:21177963; http://dx.doi.org/10.1101/gr.114587.110
- Oliveira RP, Porter Abate J, Dilks K, Landis J, Ashraf J, Murphy CT, Blackwell TK. Condition-adapted stress and longevity gene regulation by Caenorhabditis elegans SKN-1/Nrf. Aging Cell 8:524-41 2009; PMID:19575768; http://dx.doi.org/10.1111/j.1474-9726.2009.00501.x
- Park SK, Tedesco PM, Johnson TE. Oxidative stress and longevity in Caenorhabditis elegans as mediated by SKN-1. Aging Cell 2009; 8:258-69
- Robida-Stubbs S, Glover-Cutter K, Lamming DW, Mizunuma M, Narasimhan SD, Neumann-Haefelin E, Sabatini DM, Blackwell TK. TOR signaling and rapamycin influence longevity by regulating SKN-1/Nrf and DAF-16/FoxO. Cell Metab 2012; 15:713-24; PMID:22560223; http://dx.doi.org/10.1016/j.cmet.2012.04.007
- Li X, Matilainen O, Jin C, Glover-Cutter KM, Holmberg CI, Blackwell TK. Specific SKN-1/Nrf stress responses to perturbations in translation elongation and proteasome activity. PLoS genetics 2011; 7:e1002119; PMID:21695230; http://dx.doi.org/10.1371/journal.pgen.1002119
- Wang W, Chan JY. Nrf1 is targeted to the endoplasmic reticulum membrane by an N-terminal transmembrane domain. Inhibition of nuclear translocation and transacting function. J Biol Chem 2006; 281:19676-87; PMID:16687406; http://dx.doi.org/10.1074/jbc.M602802200
- Radhakrishnan SK, den Besten W, Deshaies RJ. p97-dependent retrotranslocation and proteolytic processing govern formation of active Nrf1 upon proteasome inhibition. Elife 2014; 3:e01856; PMID:24448410; http://dx.doi.org/10.7554/eLife.01856
- Lewis CA, Brault C, Peck B, Bensaad K, Griffiths B, Mitter R, Chakravarty P, East P, Dankworth B, Alibhai D, et al. SREBP maintains lipid biosynthesis and viability of cancer cells under lipid- and oxygen-deprived conditions and defines a gene signature associated with poor survival in glioblastoma multiforme. Oncogene 2015; http://dx.doi.org/10.1038/onc.2014.439onc2014439
- Reed BD, Charos AE, Szekely AM, Weissman SM, Snyder M. Genome-wide occupancy of SREBP1 and its partners NFY and SP1 reveals novel functional roles and combinatorial regulation of distinct classes of genes. PLoS genetics 2008; 4:e1000133; PMID:18654640; http://dx.doi.org/10.1371/journal.pgen.1000133
- Rome S, Lecomte V, Meugnier E, Rieusset J, Debard C, Euthine V, Vidal H, Lefai E. Microarray analyses of SREBP-1a and SREBP-1c target genes identify new regulatory pathways in muscle. Physiol Genomics 2008; 34:327-37; PMID:18559965; http://dx.doi.org/10.1152/physiolgenomics.90211.2008
- Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest 2002; 109:1125-31; PMID:11994399; http://dx.doi.org/10.1172/JCI0215593
- Yecies JL, Zhang HH, Menon S, Liu S, Yecies D, Lipovsky AI, Gorgun C, Kwiatkowski DJ, Hotamisligil GS, Lee CH, et al. Akt stimulates hepatic SREBP1c and lipogenesis through parallel mTORC1-dependent and independent pathways. Cell Metab 2011; 14:21-32; PMID:21723501; http://dx.doi.org/10.1016/j.cmet.2011.06.002
- Peterson TR, Sengupta SS, Harris TE, Carmack AE, Kang SA, Balderas E, Guertin DA, Madden KL, Carpenter AE, Finck BN, et al. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell 2011; 146:408-20; PMID:21816276; http://dx.doi.org/10.1016/j.cell.2011.06.034
- Griffiths B, Lewis CA, Bensaad K, Ros S, Zhang Q, Ferber EC, Konisti S, Peck B, Miess H, East P, et al. Sterol regulatory element binding protein-dependent regulation of lipid synthesis supports cell survival and tumor growth. Cancer Metab 2013; 1:3, doi:10.1186/2049-3002-1-3; PMID:24280005; http://dx.doi.org/10.1186/2049-3002-1-3
- Young RM, Ackerman D, Quinn ZL, Mancuso A, Gruber M, Liu L, Giannoukos DN, Bobrovnikova-Marjon E, Diehl JA, Keith B, et al. Dysregulated mTORC1 renders cells critically dependent on desaturated lipids for survival under tumor-like stress. Genes Dev 2013; 27:1115-31; PMID:23699409; http://dx.doi.org/10.1101/gad.198630.112
- Lee CS, Ho DV, Chan JY. Nuclear factor-erythroid 2-related factor 1 regulates expression of proteasome genes in hepatocytes and protects against endoplasmic reticulum stress and steatosis in mice. FEBS J 2013; 280:3609-20; PMID:23702335; http://dx.doi.org/10.1111/febs.12350
- Smith MH, Ploegh HL, Weissman JS. Road to ruin: targeting proteins for degradation in the endoplasmic reticulum. Science 2011; 334:1086-90; PMID:22116878; http://dx.doi.org/10.1126/science.1209235
- Xu Z, Chen L, Leung L, Yen TS, Lee C, Chan JY. Liver-specific inactivation of the Nrf1 gene in adult mouse leads to nonalcoholic steatohepatitis and hepatic neoplasia. Proc Natl Acad Sci U S A 2005; 102:4120-5; PMID:15738389; http://dx.doi.org/10.1073/pnas.0500660102
- Hirotsu Y, Hataya N, Katsuoka F, Yamamoto M. NF-E2-related factor 1 (Nrf1) serves as a novel regulator of hepatic lipid metabolism through regulation of the Lipin1 and PGC-1beta genes. Mol Cell Biol 2012; 32:2760-70; PMID:22586274; http://dx.doi.org/10.1128/MCB.06706-11
- Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell 2012; 149:274-93; PMID:22500797; http://dx.doi.org/10.1016/j.cell.2012.03.017
- Hsieh AC, Liu Y, Edlind MP, Ingolia NT, Janes MR, Sher A, Shi EY, Stumpf CR, Christensen C, Bonham MJ, et al. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature 2012; 485:55-61; PMID:22367541; http://dx.doi.org/10.1038/nature10912
- Jefferies HB, Reinhard C, Kozma SC, Thomas G. Rapamycin selectively represses translation of the "polypyrimidine tract" mRNA family. Proc Natl Acad Sci U S A 1994; 91:4441-5; PMID:8183928; http://dx.doi.org/10.1073/pnas.91.10.4441
- Thoreen CC, Chantranupong L, Keys HR, Wang T, Gray NS, Sabatini DM. A unifying model for mTORC1-mediated regulation of mRNA translation. Nature 2012; 485:109-13; PMID:22552098; http://dx.doi.org/10.1038/nature11083
- Ganley IG, Lam du H, Wang J, Ding X, Chen S, Jiang X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem 2009; 284:12297-305; PMID:19258318
- Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, Iemura S, Natsume T, Takehana K, Yamada N, et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell 2009; 20:1981-91; PMID:19211835; http://dx.doi.org/10.1091/mbc.E08-12-1248
- Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, Kundu M, Kim DH. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell 2009; 20:1992-2003; PMID:19225151; http://dx.doi.org/10.1091/mbc.E08-12-1249
- Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 2011; 13:132-41; PMID:21258367; http://dx.doi.org/10.1038/ncb2152
- Petroski MD, Deshaies RJ. Function and regulation of cullin-RING ubiquitin ligases. Nat Rev Mol Cell Biol 2005; 6:9-20; PMID:15688063; http://dx.doi.org/10.1038/nrm1547
- Gallinetti J, Harputlugil E, Mitchell JR. Amino acid sensing in dietary-restriction-mediated longevity: roles of signal-transducing kinases GCN2 and TOR. Biochem J 2013; 449:1-10; PMID:23216249; http://dx.doi.org/10.1042/BJ20121098
- Sonenberg N, Hinnebusch AG. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell 2009; 136:731-45; PMID:19239892; http://dx.doi.org/10.1016/j.cell.2009.01.042
- Fonseca R, Vabulas RM, Hartl FU, Bonhoeffer T, Nagerl UV. A balance of protein synthesis and proteasome-dependent degradation determines the maintenance of LTP. Neuron 2006; 52:239-45; PMID:17046687; http://dx.doi.org/10.1016/j.neuron.2006.08.015
- Suraweera A, Munch C, Hanssum A, Bertolotti A. Failure of amino acid homeostasis causes cell death following proteasome inhibition. Mol Cell 2012; 48:242-53; PMID:22959274; http://dx.doi.org/10.1016/j.molcel.2012.08.003
- Ding Q, Dimayuga E, Martin S, Bruce-Keller AJ, Nukala V, Cuervo AM, Keller JN. Characterization of chronic low-level proteasome inhibition on neural homeostasis. J Neurochem 2003; 86:489-97; PMID:12871590; http://dx.doi.org/10.1046/j.1471-4159.2003.01885.x
- Labbadia J, Morimoto RI. The Biology of Proteostasis in Aging and Disease. Annu Rev Biochem 2015; 84:32.1-32.30; PMID:25784053; http://dx.doi.org/10.1146/annurev-biochem-060614-033955
- Menon S, Manning BD. Common corruption of the mTOR signaling network in human tumors. Oncogene 2009; 27:S43-S51; http://dx.doi.org/10.1038/onc.2009.352
- Johnson SC, Rabinovitch PS, Kaeberlein M. mTOR is a key modulator of ageing and age-related disease. Nature 2013; 493:338-45; PMID:23325216; http://dx.doi.org/10.1038/nature11861
- Lipton JO, Sahin M. The neurology of mTOR. Neuron 2014; 84:275-91; PMID:25374355; http://dx.doi.org/10.1016/j.neuron.2014.09.034
- Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med 2013; 19:983-97; PMID:23921753; http://dx.doi.org/10.1038/nm.3232
- Perluigi M, Di Domenico F, Butterfield DA. mTOR Signaling in Aging and Neurodegeneration: At the Crossroad between Metabolism dysfunction and Impairment of autophagy. Neurobiol Dis 2015; http://dx.doi.org/S0969-9961(15)00086-8;pii10.1016/j.nbd.2015.03.014
- Kumar V, Atherton P, Smith K, Rennie MJ. Human muscle protein synthesis and breakdown during and after exercise. J Appl Physiol 1985 2009; 106:2026-39; PMID:19164770; http://dx.doi.org/10.1152/japplphysiol.91481.2008
- Huang J, Manning BD. The TSC1-TSC2 complex: a molecular switchboard controlling cell growth. Biochem J 2008; 412:179-90; PMID:18466115; http://dx.doi.org/10.1042/BJ20080281
- Kumatori A, Tanaka K, Inamura N, Sone S, Ogura T, Matsumoto T, Tachikawa T, Shin S, Ichihara A, et al. Abnormally high expression of proteasomes in human leukemic cells. Proc Natl Acad Sci U S A 1990; 87:7071-5; PMID:2205851; http://dx.doi.org/10.1073/pnas.87.18.7071
- Pilarsky C, Wenzig M, Specht T, Saeger HD, Grutzmann R. Identification and validation of commonly overexpressed genes in solid tumors by comparison of microarray data. Neoplasia New York, N.Y 2004; 6:744-50; PMID:15720800; http://dx.doi.org/10.1593/neo.04277
- Chen L, Madura K. Increased proteasome activity, ubiquitin-conjugating enzymes, and eEF1A translation factor detected in breast cancer tissue. Cancer Res 2005; 65:5599-606; PMID:15994932; http://dx.doi.org/10.1158/0008-5472.CAN-05-0201
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013; 6:pl1; PMID:23550210; http://dx.doi.org/10.1126/scisignal.2004088