
Abstract
Calreticulin (CRT) is an endoplasmic reticulum (ER) resident calcium binding protein that is involved in several cellular activities. Transcriptome analyses in CRT knockdown HepG2 cells revealed 253 altered unique genes and subsequent in silico protein-protein interaction network and MCODE clustering identified 34 significant clusters, of which p53 occupied the central hub node in the highest node-rich cluster. Toward validation, we show that CRT knockdown leads to inhibition of p53 protein levels. Both, CRT and p53 siRNA promote hepatic lipid accumulation and this was accompanied by elevated SREBP-1c and FAS levels. p53 was identified to bind at −219 bp on the SREBP-1c promoter and in the presence of CRT siRNA, there was decreased occupancy of p53 on this binding element. This was associated with increased SREBP-1c promoter activity and both, mutation in this binding site or p53 over-expression antagonised the effects of CRT knockdown. We, therefore, identify a negatively regulating p53 binding site on the SREBP-1c promoter that is critical during hepatic lipid accumulation. These results were validated in mouse primary hepatocytes and toward a physiological relevance, we report that while the levels of CRT and p53 are reduced in the fatty livers of diabetic db/db mice, SREBP-1c levels are significantly elevated. Our results suggest that decreased CRT levels might be involved in the development of a fatty liver by preventing p53 occupancy on the SREBP-1c promoter and thereby facilitating SREBP-1c up-regulation and consequently, lipid accumulation.
Keywords:
Introduction
CRT is multifunctional calcium (Ca2+) buffering chaperone in the endoplasmic reticulum (ER) that participates in diverse cellular functions that include apoptosis, angiogenesis, cell adhesion, transcription and immune responses.Citation1 Such vital functions rendered by CRT and other ER chaperones makes the ER critical in cellular signaling and communication. Calreticulin binds to newly synthesized glycoproteins, preventing their aggregation and assisting them in their proper folding.Citation2 Together with calnexin and ERp57, CRT forms a cycle that is critical in proper protein folding and maintaining them in their correct native conformation. As a modulator of Ca2+ homeostasis, calreticulin binds Ca2+ and consequently directly impacts the Ca2+capacity of the ER. All these indicate that regulation of cellular CRT levels is critical for normal functioning of the cell.
In a previous report, we had shown that CRT levels are regulated by elevated levels of the proinflammatory cytokine, tumor necrosis factor- α (TNFα).Citation3 Although identified for its notable role in inflammation and inflammatory diseases, TNFα is now frequently associated with metabolic disorders like obesity, diabetes and insulin resistance.Citation4 Insulin resistance occurs due to interference at one or more points of the insulin signaling cascadeCitation5-7 and CRT has been reported to interfere with the stabilization and modulation of the human insulin receptor (hIR) suggesting its possible correlation in insulin signaling.Citation8 Our previous study demonstrated that TNFα promotes transcriptional inhibition of CRT by decreasing the binding of C/EBPα to its cognate element on the CRT promoter and overexpression of CRT prevents TNFα mediated attenuation of insulin signaling in HepG2 cells.Citation3,9
Abrogation of hepatic insulin signaling is associated with lipid accumulation in the liver as seen during diabetes. The mechanisms involved in promoting such lipid accumulation are multifactorial, yet are not completely understood. A very well accepted factor is the altered bioavailability and altered retention of lipids within the hepatocytes that is associated with insulin resistance.Citation10 In fact, a strong relationship exists between whole body insulin resistance and fat accumulation in the liver in humans.Citation11-13 An increase in the hepatic fat content is an evident feature encountered with the occurrence of insulin resistance and Type 2 Diabetes.Citation10,14
In the present study, we sought to assess the effects of CRT inhibition in the liver. Using gene expression microarray and in silico protein- protein interaction (PPI) network analysis, we show that p53 is a central hub node among the set of genes altered by CRT inhibition. Further, using HepG2 cells and primary hepatocytes, we evaluate the effect of CRT and p53 on hepatic lipid accumulation and our results suggest that CRT inhibition promotes lipid accumulation by down regulating p53 protein levels and its occupancy on the SREBP-1c promoter, thereby up-regulating SREBP-1c and fatty acid synthase (FAS) levels.
Results
Microarray
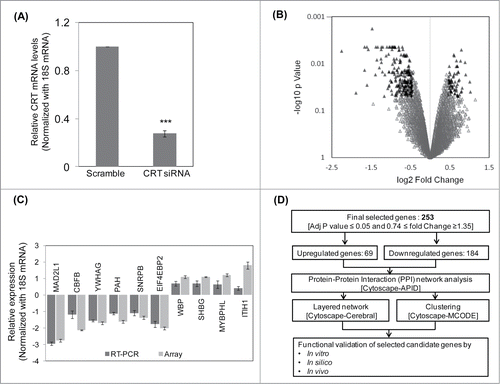
To assess the global effect of CRT knockdown, HepG2 cells were transfected with scramble siRNA and CRT siRNA (10 nM). This dose of CRT siRNA was chosen as described in a previous report from our laboratory.Citation9 As shown in , there was a marked inhibition in the transcript levels of CRT in the presence of the siRNA as determined by qRT-PCR. RNA from scramble and CRT siRNA transfected HepG2 cells were subjected to microarray analysis using the Illumina HumanHT-12 v3 Expression BeadChip arrays and data were analyzed using Lumi package (R package for illumina Microarray). The selection criteria included adjusted p-value ≤0.05 and fold Change ≥1.35. A total of 286 genes were altered; 212 genes were down regulated and 74 genes were upregulated (Table S1). The volcano plot of the microarray experiment is shown in where black triangles represent the significantly altered genes with up and down- regulated genes being shown on either side of the central axis. The microarray experiment was done according to the MIAME guidelines and data have been deposited in GEO (GSE57261). The microarray experiment was validated by qRT-PCR and the all genes depicted a similar pattern of alteration as in the microarray experiment. While MAD2L1, CBFB, YWHAG, PAH, SNRPB and EIF4EBP2 were down-regulated, WBP, SHBG, MYBPHL and ITIH1 were up-regulated ().
Figure 1. Microarray, qRT-PCR validation and the pipeline of the experimental strategy. (A) HepG2 cells were transfected with the scramble or CRT siRNA (10 nM) for 24 h. On termination of incubation, CRT levels were estimated by qRT-PCR using CRT specific primers. (B) RNA as in “A” was subjected to microarray analyses using Illumina Human HT-12 V3 expression Bead chip arrays. Genes differentially regulated (p < 0.5) are represented as black triangles in a volcano plot with the up- and downregulated genes being represented on either side of the central axis. (C) RNA from scramble and CRT siRNA treated HepG2 cells were subjected to qRT-PCR toward validation of differentially altered genes as obtained from the microarray. All fold changes were significant at p < 0.05. (D) Computational and experimental pipeline of the strategy followed for the altered genes. Values presented are means ±SEM of 3 experiments. ***p < 0.001 as compared to scramble transfected cells.
Predicting interacting partners using a PPI Network
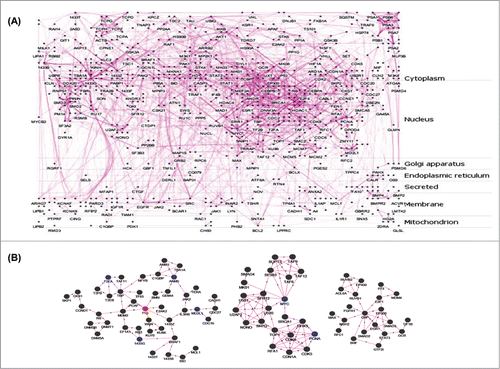
To functionally correlate this set of altered genes with hepatic metabolism, we used a protein-protein interaction (PPI) network for further analysis. A flow chart of the subsequent analysis protocol is given in . The 286 differentially expressed genes (DEGs) from the microarray analysis were mapped to corresponding HGNC ids and filtered as described in ‘Material and Methods’ section resulting in a final unique set of 253 genes. The interacting partners of the 253 DEGs were identified using plug-in APID2NETCitation15 in Cytoscape.Citation16 The PPI network was built considering only those interactions that have been validated in at least 2 experiments based on BioGrid, DIP, HPRD, IncAct and MINT databases.Citation17 Single isolated nodes and small components of the PPI network were removed resulting in a final PPI network composed of 818 nodes and 2794 edges. From this PPI network, a layered network based on the sub-cellular localization information of 818 proteins using “Cerebral” pluginCitation18 in Cytoscape was obtained. The layered network is shown in . Nodes in the PPI network are labeled by different colors to indicate the source of proteins. Blue nodes represent proteins encoded by DEGs and dark gray nodes represent interacting proteins from APID2NET. Further, another cytoscape plugin “MCODE”Citation19 was layered on the PPI to identify sub-networks. This resulted in 34 clusters and these are given as Table S2. Among these, the top 3 clusters based on node density are shown in and p53, MYC and CBP occupied the central nodes in the respective clusters.
Figure 2. Protein-protein interaction (PPI) networks and sub-networks formed by the altered genes. (A) The set of 253 unique differentially expressed genes were assessed for their interacting partners using the APID2NET plugin in Cytoscape. The resultant PPI network composed of 818 nodes and 2794 edges and was used to generate a layered network based on subcellular localization of the 818 proteins. Blue colored nodes are representative of proteins encoded by the differentially expressed genes while the gray ones are the interacting partners. (B) The PPI as in ‘A’ was used to identify sub-networks using the MCODE clustering algorithm and the top 3 node-dense networks are shown.
CRT inhibition downregulates p53 and promotes lipid accumulation
The above results demonstrated that p53 occupied the central position in the topmost node-dense cluster among the interactome maps of DEGs. This suggested that p53 might be central to the cellular effects of CRT inhibition and therefore, we sought to evaluate the effects of CRT inhibition on p53 levels. HepG2 cells transfected with CRT siRNA demonstrated significant inhibition of p53 levels (). Previous studies from our laboratory have shown that TNFα promotes a decrease in CRT levels by altering its promoter occupancy by C/EBPα and CRT overexpresion rescues TNFα mediated inhibition of insulin signalling.Citation3,9 Decreased insulin signaling is usually manifested as the development of a fatty liver and therefore, we sought to evaluate whether CRT and p53 inhibition could regulate hepatic lipid accumulation.
Figure 3. Calreticulin (CRT) inhibition down-regulates p53 and promotes lipid accumulation through up-regulation of lipogenic factors in HepG2 cells. (A) HepG2 cells were transfected with the scramble or CRTsiRNA (10 nM) for 48 h and on termination of incubation, cells were collected, lysed and lysates (30 µg) were assessed for the levels of p53 by western blot analysis. β-actin was taken as the loading control. (B) HepG2 cells were transfected with the scramble or p53 siRNA (10 nM) for 24 h. On termination of incubation, p53 levels were evaluated by qRT-PCR and normalized to the levels of 18S rRNA. (C) HepG2 cells transfected with either the scramble or p53 siRNA or CRT siRNA (10 nM) for 48 h. Cells were then fixed and stained with DAPI and Bodipy and viewed under a fluorescent microscope (63X). Bodipy fluorescence was quantified using the ImageJ software and these are given in the right panel of image (a.u). All experiments were done in triplicate and values in the histograms are means ±SEM. (D) Scramble, CRT siRNA and p53 siRNA transfected cells were assessed for the levels of the lipogenic genes: FAS and SREBP-1c by Western Blot analyses. Vinculin was taken as the loading control. Lower panel represents the densitometric analyses of respective blots normalized with loading control. Values are means ± SEM of 3 independent experiments. ***p < 0.001, **p < 0.01 and *p<0.05 as compared to scramble transfected cells.
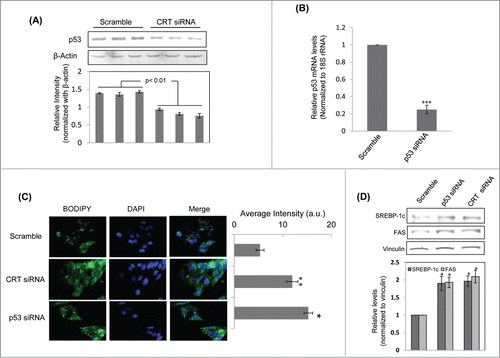
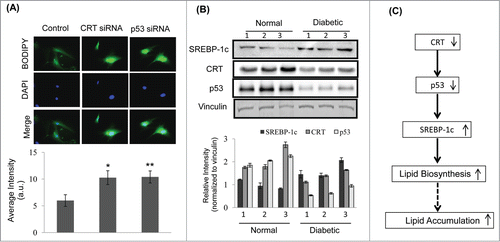
Transfection of HepG2 cells with p53 siRNA led to a significant decrease in p53 transcript levels (). Consequently, lipid accumulation was evaluated in CRT and p53 siRNA treated HepG2 cells by BODIPY staining. There was significant increase in lipid accumulation in cells treated with CRT siRNA and p53 siRNA as compared to scramble treated cells (). This was associated with significant upregulation of the lipogenic gene, fatty acid synthase (FAS) and its transcriptional regulator, SREBP-1c (), both in the presence of CRT siRNA and p53 siRNA. These suggest that in the presence of inhibited levels of both CRT and p53, there is significant up-regulation of lipogenic factors that are critical during lipid accumulation.
CRT inhibition decreases the occupancy of p53 on the SREBP-1c promoter
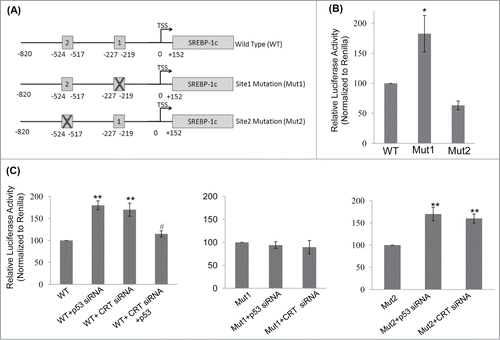
To study the mechanism of SREBP-1c upregulation in the presence of CRT siRNA and p53 siRNA, the SREBP-1c promoter was scanned for p53 binding sites using the bioinformatic tool Lasanga. The SREBP-1c promoter harbours 2 p53 binding sites at −219 (site1; 5′GGCTCGGCTTGCCCGCGT3′) and −517 (site2; 5′CACAGGTCATGCTCCCCTCC3′) from the TSS as shown in . Luciferase reporter SREBP-1c promoter constructs for the wild type and mutant (for both the p53 binding sites; underlined sequences were substituted with AAGCTT (site 1) and TCTAGA (site 2)) promoters were analyzed and as shown in , mutation at the p53 binding site1 (Mut 1) showed a significant increase in the luciferase activity as compared to the wild type (WT) construct. However, mutation in site2 showed a modest decrease in luciferase activity suggesting that occupation of site1 by p53 is presumably the major determinant during negative regulation of SREBP-1c levels. Both p53 and CRT siRNAs significantly increased the luciferase activity of the wild type construct () and this increase was abolished in the construct harbouring the site1 mutation but not in the one with mutation on site2 (). Interestingly, overexpression of p53 rescued CRT siRNA facilitated increase in SREBP-1c promoter activity suggesting a mediatory role of p53 in the CRT effect. These indicate that site1 is critical in regulating the levels of SREBP-1c by p53 and mutation at this site prevents binding of p53 to this region and presumably facilitates the occupation of active transcriptional machinery that stimulates SREBP-1c transcription.
Figure 4. p53 binding site on the SREBP-1c promoter and its occupancy regulates SREBP-1c promoter activity. (A) The wild type (WT) SREBP-1c promoter harbouring 2 binding sites for p53 (site 1 at −219 bp and site 2 at −517bp) is shown. Mutations in both these sites were done independently by base substitution at 4 positions within the binding site (site 1 mutation: Mut1; site 2 mutation: Mut 2). (B) HepG2 cells were transfected with either the wild type SREBP-1c promoter construct (WT) or the mutated constructs (Mut1 or Mut2) and luciferase activity was assessed using the Dual luciferase assay kit. Renilla luciferase (30 ng) was cotransfected and used as the normalization control. (C) HepG2 cells were transfected with the WT or mutated SREBP-1c promoter constructs as in ‘B’ and also co-transfected with either the scramble or p53 siRNA or CRT siRNA or with CRT siRNA and the p53 cDNA clone (1 μg). After 48 h, cells were lysed and luciferase activities were determined. All experiments were done thrice and values are means ± SEM. **p < 0.01 and *p < 0.05 as compared to WT or Mut2 and #p < 0.05 as compared to CRT siRNA alone.
CRT inhibition decreases p53 binding on the SREBP-1c promoter and over expression of p53 rescues SREBP-1c expression
As described above, mutation in the p53 binding site (site1) on the SREBP-1c promoter abrogated the stimulatory effects on the SREBP-1c promoter in the presence of p53 or CRT siRNAs, we further assessed the binding of p53 on this site of the promoter by EMSA and ChIP. Labeled oligonucleotides corresponding to the site1 cognate binding element on the SREBP-1c promoter was used for EMSA and as shown in , there was significant binding of p53 on this site that was markedly decreased by a cold-chase with the unlabelled oligonucleotide. Subsequently, ChIP using a p53 antibody showed significant reduction in the levels of p53 bound to this site of the promoter in the presence of CRT siRNA () thereby emphasizing the role of this site during regulation of SREBP-1c levels. Further, to confirm the specific role of CRT or p53, HepG2 cells were transfected with CRT siRNA alone or together with a p53 cDNA clone. As shown in , over-expression of p53 rescued the increase in SREBP-1c levels induced by CRT inhibition. These indicate that CRT inhibition decreases p53 that up-regulate SREBP-1c levels.
Figure 5. Binding of p53 to the SREBP-1c promoter and its decrease by CRT inhibition. (A) Double stranded oligonucleotides spanning the p53 binding site on the SREBP-1c promoter (site1) were labeled with γ-32P and incubated with purified p53 at 37°C for 30 min. On termination of incubation, samples were separated on a 5% polyacrylamide gel, dried and autoradiographed. A cold chase with unlabelled oligonucleotides at dilutions of 1:1 and 1:10 was done to check the specificity of the binding. (B) HepG2 cells were transfected with scramble or CRT siRNA (10 nM) for 48 h and the chromatin was isolated, sheared, immunoprecipitated with IgG or anti-p53 antibody and pulled down using protein A-sepharose beads. Immunoprecipitated DNA was quantified (qRT-PCR) for p53 enrichment on site1 using site specific primers as given in . Data was normalized to the IgG pulldown DNA. Experiments were done thrice and values are mean ±SEM. (C) HepG2 cells were transfected with CRT siRNA and co-transfected with p53 cDNA clone and incubated for 48h. On termination of incubations, cells were lysed and the levels of SREBP-1c were evaluated by protein gel blot analysis. β-Actin was taken as the loading control. Densitometric analyses are given in the panel below. (D) HepG2 cells were incubated in the absence or presence of palmitate or oleate for 24h and total RNA was isolated. CRT transcript levels were evaluated by qRT-PCR and data was normalized to 18S rRNA. All values are means±SEM and derived from 3 independent experiments. *p < 0.05 as compared to free probe (A); ***p < 0.001 as compared to scramble (B); ###p < 0.001 and #p < 0.05 as compared to control cells; ap < 0.05 as compared to CRT siRNA transfected cells.
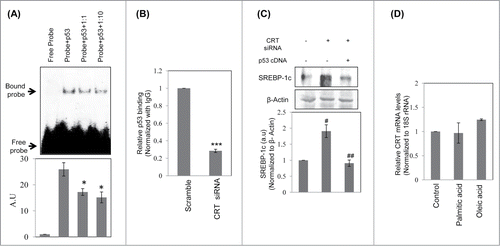
Table 1. List of primers
Since circulatory free fatty acids are also reported to be elevated in obese subjects, we sought to assess if fatty acids, in turn, might affect CRT levels. HepG2 cells were incubated with palmitic or oleic acid and CRT transcript levels were quantified by qRT-PCR. As shown in , neither palmitic acid nor oleic acid had any effect on CRT mRNA levels indicting that altered fatty acids do not alter the levels of CRT.
CRT inhibtion promotes lipid accumulation in mouse primary hepatocytes and its hepatic levels are decreased in the diabetic db/db mice
To validate the above events in primary hepatocytes, cells were transfected with the scramble or CRT siRNA or p53 siRNA and the lipid accumulation was assessed by Bodipy staining. As shown in , there was a significant increase in lipid accumulation in the presence of both, CRT and p53 siRNA as compared to scramble transfected cells. To assess the significance of the status of these proteins in vivo, it was observed that hepatic levels of CRT and p53 are significantly downregulated in diabetic db/db mice with a parallel increase in the levels of SREBP-1c (). These suggest that as in in-vitro, CRT inhibition through p53 down-regulation might be one of the contributory factors toward development of a fatty liver in db/db mice. The scheme of such a potential pathway is shown in where inhibited levels of CRT promote a decrease in p53 levels with a consequent increase in SREBP-1c, lipid biosynthesis and accumulation.
Figure 6. CRT downregulation promotes lipid accumulation in mice primary hepatocytes and its levels are inhibited in the db/db mice liver (A) Mice primary hepatocytes were transfected with either the scramble or CRT siRNA or p53 siRNA and stained with Bodipy and DAPI. Bodipy fluorescence was quantified and the data are given below. (B) 30 µg protein from normal and db/db mice (n = 3 ) liver were run on SDS-PAGE, transferred to nitrocellulose membranes and probed with antibodies against SREBP-1c, CRT and p53. Vinculin was used as the loading control and densitometric analyses of the same are given below. Experiment from each animal was run thrice and a representative blot is shown. (C) Schema demonstrating that CRT inhibition leads to p53 downregulation, that due to its decreased occupancy of the SREBP-1c promoter, facilitates its upregulation and subsequently promotes lipid synthesis and accumulation. All values are means±SEM from 3 independent experiments. **p < 0.01 and *p < 0.05 as compared to scramble transfected cells.
Discussion
Previous reports from our laboratory had demonstrated that TNFα inhibits hepatic CRT levels by decreasing the occupancy of C/EBPα on the CRT promoter.Citation3,9 In the present study, we show that such decrease in CRT levels promotes hepatic lipid accumulation that might contribute toward the development of fatty liver.
CRT is a multifunctional Ca2+ binding protein of the ER and its levels are tightly regulated by a complex interplay of several transcription factors.Citation20 Diverse unique functions have been ascertained for CRT, including apoptosis, angiogenesis, wound healing, embryonic development, adipogenesis etc.Citation21 In dedifferentiated liposarcoma cells, calreticulin deficiency has been shown promote to adipogenesis and intracytoplasmic lipid droplet accumulation.Citation22 The current study unravels a critical role of CRT in modulating hepatic lipid metabolism that might be co-relatable to diverse pathophysiologies associated with the fatty liver. Our results suggest that CRT knockdown facilitates lipid accumulation through increased de novo lipogenesis in hepatocytes.
Using whole genome microarray and subsequent protein-protein interaction and network analysis, we show that a cluster node harbouring p53 is the most enriched in the interacting network of genes altered by CRT siRNA. Apart from being a tumor suppressor, p53 functions in DNA repair, apoptosis and cell cycle arrest.Citation23 Moreover, p53 has also been shown to prevent the shift to glycolysis as seen in the cancerous cells and thus help cells to survive states of metabolic stress.Citation24 p53 regulates fatty acid oxidation (FAO) in glucose deprived human cultured cells through induction of guanidinoacetate N-methyltransferase (GAMT).Citation25 In another study p53 was reported to enhance FAO and inhibit fatty acid synthesis through activation of lipin1 (LPIN 1).Citation26,27 Depletion of p53 promotes lipid accumulation in mouse embryonic fibroblasts and mouse liver by directly binding to glucose-6-phosphate dehydrogenase (G6PD) and inhibiting the PPP pathwayCitation23,28 and in p53−/− mice, high fat diet promotes obesity and hepatic steatosis.Citation23
Here, we show a novel mechanism of hepatic lipid accumulation during CRT deficiency that involves p53. Our results show that CRT knockdown leads to significant inhibition of p53 levels and a possible mechanism could be as shown by Mesaeli et al. in CRT−/− MEF cells. In the absence of CRT, p53 inhibition occurs through increased MDM2 activation followed by ubiquitin mediated degradation of p53.Citation29 Here, we demonstrate that such CRT inhibition-mediated p53 downregulation is associated with increased lipid accumulation and elevated levels of the critical lipogenic enzyme - fatty acid synthase (FAS) and its transcriptional regulator, SREBP-1c.
FAS is a multi-enzyme protein catalyzing a critical step during de novo fatty acid synthesis and it is transcriptionally regulated by the transcription factor, SREBP-1c.Citation30 There are 2 potential binding sites for SREBP-1c on the FAS promoterCitation31 that suggests its regulatory role in FAS expression. On the other hand, SREBP-1c regulation, by itself is highly complex. Nuclear hormone receptors RXR and LXR, heterodimerise to activate SREBP-1c in response to cholesterol overloading presumably for cholesterol esterification and storage.Citation31 Interestingly, SREBP-1c and SREBP-2 transcription is stimulated by SREBPs themselves in a sterol regulation element (SRE) sequence dependent manner.Citation32 The SREBP-1c promoter has been reported to harbour binding sites for NF-Y, Sp1 and compose of an E-box and SRE-3 elements.Citation33
Our data show that in addition to these, the SREBP-1c promoter possesses binding sites for p53, one at-219 bp (site1) and other at −517 bp (site2) upstream to the TSS. As stated above, CRT inhibition caused significant p53 down-regulation, this together with the identification that SREBP-1c promoter harbours p53 binding sites and there is decreased p53 occupancy in the absence of CRT suggests that this decreased p53 binding on the SREBP-1c promoter might be responsible for its up regulation. Yahegi et al.Citation34 have demonstrated that elevated p53 levels in the adipocytes of ob/ob mice negatively regulate SREBP-1c levels and overexpression of p53 suppresses SREBP-1c promoter activity. Our data demonstrate that there is a negative correlation between p53 occupancy at site 1 on the SREBP-1c promoter and the promoter activity. Mutation at this site prevents binding of p53 and consequently increases SREBP-1c promoter activity, possibly by allowing an easy access to the active transcription machinery.
To conclude, our results show that CRT inhibition promotes significant downregulation of p53 levels. Because of a negative correlation with SREBP-1c levels, the decrease in p53 levels and its decreased occupancy on the SREBP-1c promoter leads to increased SREBP-1c and FAS levels and consequently facilitates lipid accumulation within the hepatocytes. This reveals new means of hepatic lipid accumulation and assumes importance since it unravels mechanisms critical to the development of fatty liver and puts forth a novel aspect of regulation of this hepatic pathophysiology.
Materials and Methods
Cell culture and transfections
Experiments were performed in HepG2 cells obtained from National Center for Cell Science, Pune, India. HepG2 cells that are used to study hepatic physiology possess several properties and specific functions of the normal human liver including synthesis of lipoprotein and albumin that are important to study several aspects of hepatic function and metabolism remain preserved in these cells and they are considered an in vitro human hepatic model system.Citation35,36 These cells were cultured in a humidified atmosphere of 5% CO2 at 37°C in DMEM medium (Sigma Chemical Co., St. Louis, MO, USA) supplemented with 10% Fetal bovine serum (GIBCO Laboratory, NY) and 1% antibiotic–antimycotic (100 units/ml penicillin, 0.1 mg/ml streptomycin and 0.25 µg/ml amphotericinB). For siRNA tranfections, cells were grown to 80% confluency and reverse transfected with the respective siRNAs (either scramble or CRT siRNA or p53 siRNA) using Lipofectamine RNAiMax reagent (Invitrogen, CA, USA) as per the manufacturer's instructions.
RNA isolation and microarray
Total RNA was isolated from HepG2 cells transfected with control siRNA (scramble) and CRT siRNA (10 nM) using the mirVana™ miRNA Isolation Kit (Ambion, CA, USA) and quantified using Nanodrop-1000 (Thermo Fischer Scientific, MA, USA). Total RNA (500 ng each) was used to prepare cRNA using the Illumina® TotalPrep™ RNA Amplification Kit (Ambion) as per the manufacturer's instructions. Quantitation of cRNA was performed using Nanodrop-1000 and 750 ng cRNA of each sample was hybridized to the Illumina HumanHT-12 v3 Expression BeadChip arrays, containing approximately 48,000 probes representing more than 25,000 annotated genes. Hybridizations and washings were performed according to the manufacturer's protocol. The arrays were scanned and read using the Illumina iScan System, and the data extraction, average normalization and downstream analysis performed using Lumi package under R statistical programming language.Citation37
Quantitative real time PCR
Quantitative Real Time PCR (qRT-PCR) was done for validation of microarray analysis. Genes with the highest fold of up- and or down-regulation were taken for RT-PCR validation. HepG2 cells were transfected with either the scramble or CRT siRNA and total RNA was isolated as described above. RNA was reverse transcribed using High capacity cDNA Reverse transcription kit (Applied Biosystems) according to the manufacturer's instructions and qRT-PCR was done in Step One PLUS Real time PCR system (Applied Biosystems) using gene specific primers (). 18S rRNA was used as endogenous control. Each incubation was performed thrice and relative quantification was performed using comparative CT method.Citation38 For experiments with p53 siRNA, total RNA was isolated from HepG2 cells transfected with either the scramble or p53 siRNA (10 nM) and levels of p53 were quantified by qRT-PCR using specific primers and normalized with 18S rRNA. To evaluate the effects of free fatty acids on calreticulin levels, HepG2 cells were incubated in the presence of palmitic acid and oleic acid (Sigma, USA). For this, HepG2 cells grown to 80% confluence were serum starved for 12 h and incubated in the absence or presence of palmitic acid and oleic acid (0.66 mM) in DMEM with 1% bovine serum albumin for 24 h as described by Ricchi et al.Citation39 On termination of incubation, total RNA was isolated and levels of CRT were analyzed by qRT-PCR using CRT specific primers ().
Protein-protein interaction and network analysis
Protein-protein interaction (PPI) networks of differentially expressed genes (DEGs) were created to identify their direct interacting partners and network analyses were done to classify the functional modules by clustering of PPI network. Briefly, the 286 DEGs from the whole genome microarray analysis were mapped to their corresponding HGNC (HUGO Gene Nomenclature Committee) ids and filtered by excluding duplicate genes, pseudogenes, genes with ‘no match’ and open reading frames (ORFs). This led to identification of 253 genes. Uniprot ids of these genes were retrieved using uniprot id mapping tool (http://www.uniprot.org)Citation17 and used as input to build a PPI network using plugin APID2NETCitation15 in Cytoscape version 2.8.1.Citation16 The APID2NET directly accesses the Agile Protein Interaction Database (APID) server to create PPI network of user-provided proteins using literature-curated protein interaction information from various databases such as BIND (Biomolecular Interaction Network Database), BioGRID (The General Repository for Interaction Datasets), DIP (Database of Interacting Proteins), HPRD (Human Protein Reference Database), IntAct (Database system and analysis tools for protein interaction data) and MINT (Molecular Interactions Database). At least 2 experimental validations were considered in order to minimize false-positive interactions.Citation15 Two Cytoscape tools viz CerebralCitation18 and MCODECitation19 were then applied for PPI network modeling. Isolated nodes were also manually removed from the final PPI network. Protein sub-cellular localization information for 787 proteins was retrieved from uniprot databaseCitation17 and the remaining 31 genes from human protein atlas database.Citation40 Then Cytoscape plugin “Cerebral” v.2.8.2 was applied to the final network to layout all nodes according to their sub-cellular localization such as membrane, cytoplasm, nucleus, golgi apparatus, endoplasmic reticulum (ER), secreted and mitochondria. To elucidate the clusters of interest, the Cytoscape plugin MCODE was run on the network using a degree cutoff of 2, a node score cutoff of 0.2, a K-Core of 2, and a max depth of 100.Citation19
Bodipy staining
HepG2 cells were grown on sterile cover-slips placed in 6-well plates and transfected with either scramble or CRT siRNA (10 nM) or p53 siRNA (10 nM) for 48 hours using Lipofectamine RNAiMax reagent (Invitrogen). Cells were then washed with PBS (phosphate buffered saline) and fixed with 1% formaldehyde (Merck, Germany) for 1 hour. Fixed cells were washed and stained with Bodipy (Invitrogen) dye dissolved in DMSO (1:10000) for 15 minutes. Cells were washed thoroughly and counter stained with DAPI (Invitrogen). Stained cells were viewed in a fluorescent microscope at 63× (Leica DMI 6000, Germany). Quatifications of green fluorescence were done using the ImageJ software (http://imagej.nih.gov/ij/)
Western blot analysis
Cells were transfected with either CRT siRNA (10 nM) or p53 siRNA (10 nM) for 48 h and lysed with RIPA lysis buffer (Sigma Chemical Co., St. Louis, MO, USA) containing protease inhibitors. Protein lysates were quantified by BCA assay (Pierce, Thermoscientific, IL, USA). 30 µg of protein lysate from each incubation was resolved on SDS-PAGE, transferred to nitrocellulose membranes and probed with primary antibodies (anti-CRT, anti-p53, anti-FAS, anti-SREBP-1c, anti-Vinculin and anti-β Actin) for overnight at 4°C. Membranes were washed with TBST (Tris Buffered saline-0.05% Tween20) and probed with HRP linked secondary antibodies followed by detection with the ECL western blotting Kit (Pierce, Thermo Scientific, Rockford, IL, USA).
Cloning of SREBP-1c promoter and luciferase assays
Human SREBP-1c promoter sequence from −820 to +152 was extracted from the NCBI database and scanned for p53 binding sites on it using the bioinformatic tool, Lasagna (www.biogrid-head.engr.uconn.edu/lasagna_search/). The promoter sequence was amplified using Phusion High-Fidelity DNA Polymerase (Fermentas, Lithuania, USA) and specific primers () and cloned into the pGL3 Basic vector (Promega, USA). Positive clones were confirmed by restriction digestion and sequencing. Mutations (substitution at 6 positions underlined in primer sequences,) in the putative p53 binding site on SREBP1c promoter construct were created by using mutation primers for both sites () and QuickChange II XL Site-Directed Mutagenesis Kit (Agilent, CA, USA) as per manufacturer's instructions and mutations were confirmed by sequencing. Further, the above promoter clones were cotransfected with 30 ng of Renilla luciferase plasmid and siRNAs of CRT (10 nM) and p53 (10 nM) in HepG2 cells using Lipofectamine RNAiMax reagent (Invitrogen) as mentioned above. After 48 h, cells were lysed and analyzed for luciferase activity using the Dual Luciferase assay kit (Promega, WI, USA) as per the manufacturer's instructions.
Cloning and protein purification
Human p53 cDNA was cloned in pET28-His10-Smt3 expression vector. Total Human RNA was purchased from M/S. Biochain Life Sciences (Newark, CA, USA) and used for cDNA preparation by high capacity reverse transcription cDNA synthesis kit (Applied Biosystems, CA, USA) as per manufacturer's instructions. The cDNA was PCR amplified using p53 specific primers () and the amplified product was purified with Qiaquick PCR purification kit (Qiagen, Venlo, Limburg, Netherlands). The purified product was cloned in the pET28-His10-Smt3 expression vector and clones were identified by digestion and sequencing. Plasmids were isolated and transformed in E.Coli BL21 strain for protein expression. Protein expression was induced by 100 µM IPTG and the expressed protein was confirmed by protein gel blot using anti-p53 antibody. Protein was purified as described by Mishra et al.Citation41 Human p53 cDNA was then subcloned into pcDNA 3.1-His A (Invitrogen) for its overexpression in HepG2 cells. To evaluate the involvement of p53 on the effects of CRT on SREBP-1c luciferase activity and levels, HepG2 cells were co-transfected with CRT siRNA and the p53 cDNA clone (1 μg) for 48 h. On termination of incubation, cells were lysed and evaluated for SREBP-1c promoter luciferase activity or for SREBP-1c levels by Western Blot as described above.
Electrophoretic mobility shift assay (EMSA) using purified p53
Double stranded oligonucleotides (Sigma) corresponding to the p53 binding site on the SREBP-1c promoter (sequences given in ) were prepared by annealing equimolar concentration of sense and antisense oligonucleotides. These oligonucleotides were labeled with γ-32P and incubated separately with purified p53 protein (2 µg) in a buffer containing 12% glycerol, 2 mM EDTA, 1 mM DTT, 8 mM Tris–HCl (pH 8), incubated at 37°C for 30 minutes. For competition assays, unlabelled double stranded oligonucleotides were added in ratios of 1:1 and 1:10 to the reaction mixture. Samples were separated on a non reducing 5% polyacyrlamide gel. The gel was dried and autoradiographed on a phoshorimager (BioRad, CA, USA).
Chromatin immunoprecipitation assay (ChIP)
ChIP assays were performed to validate the binding of p53 at the predicted site by the method as described previously.Citation9 HepG2 cells were transfected with the scramble or CRT siRNA for 48 h and cross linked for 15 min with 1% formaldehyde. Cells were lysed and sonicated for shearing the chromatin to an average size of 500 bp. Twenty five percent of lysate was used for input chromatin isolation and rest of the lysate was used for chromatin immunoprecipitation with anti-p53 antibody or mouse IgG antibody using protein-A sepharose beads. Phenol- chloroform and ethanol precipitation were used for DNA isolation. p53 enriched DNA was quantified with real time PCR using SREBP-1c promoter specific primers for Site1 ()
Animals and primary hepatocytes
Experiments were performed in accordance with the Institutional Animal Ethics Committee. Twelve week old male normal (C57BL/Ks-leprdb/+) and diabetic (C57BL/Ks-leprdb/leprdb) mice (n = 3 ) were obtained from the Animal House Facility of the CSIR-Central Drug Research Institute, Lucknow, India. Liver tissues were dissected, washed and stored at −80°C until further use. Tissues were homogenized in RIPA lysis buffer and 30 μg protein of each tissue were probed for CRT, p53 and SREBP-1c levels by Western Blot analyses as described above. Vinculin was used as the loading control. For primary hepatocytes, C57 BL/6J mice (8–10 wk old, weighing 18–20 g) were obtained from the National Institute of Nutrition (Hyderabad, India) and acclimatized for 1 week under standard laboratory conditions (25 ± 2°C, 55% humidity) before starting the experiments. Mouse hepatocytes were isolated after liver perfusion with 20 ml liver perfusion medium (GIBCO Laboratory, NY) and 20 ml liver digest medium (GIBCO Laboratory, NY). Hepatocytes were washed thrice with hepatocyte wash medium (GIBCO Laboratory, NY) and 3 × 105 cells were seeded on corning cellbind surface 6-well plates in DMEM medium (Sigma Chemical Co., St. Louis, MO, USA) supplemented with 10% Fetal bovine serum (GIBCO Laboratory, NY) and 1% antibiotic–antimycotic (100 units/ml penicillin, 0.1 mg/ml streptomycin and 0.25 µg/ml amphotericinB). After 24 h the media were changed and cells were transfected with scramble siRNA or p53 siRNA or CRT siRNA and stained with Bodipy (Invitrogen) as described above to assess the lipid accumulation.
Densitometry Analysis
Densitometric analysis was performed with Alpha DigiDoc 1201 software (Alpha Innotech Corporation, CA, USA). Each band was marked with the same size rectangle box and intensities were analyzed by the program after subtraction of the background intensity.
Statistical Analysis
Experiments were done in triplicate and statistical significance was calculated by the student's t-test. A value of at least p < 0.05 was considered as statistically significant.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
1046654_supplemental_files.zip
Download Zip (28.1 KB)Funding
This work was supported by funding from the Council of Scientific and Industrial Research (CSIR), New Delhi, India (EXP0009). SV, PT and KK acknowledge ICMR, CSIR and UGC, New Delhi, India for their fellowships.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
Related Research Data
References
- Gelebart P, Opas M, Michalak M. Calreticulin, a Ca2+-binding chaperone of the endoplasmic reticulum. Int J Biochem Cell Biol 2005; 37:260-6; PMID:15474971; http://dx.doi.org/10.1016/j.biocel.2004.02.030
- Trombetta ES, Parodi AJ. Quality control and protein folding in the secretory pathway. Annu Rev Cell Dev Biol 2003; 19:649-76; PMID:14570585; http://dx.doi.org/10.1146/annurev.cellbio.19.110701.153949
- Pandey AK, Munjal N, Datta M. Gene expression profiling and network analysis reveals lipid and steroid metabolism to be the most favored by TNFalpha in HepG2 cells. PLoSOne 2010; 5:e9063-; PMID:20140224; http://dx.doi.org/10.1371/journal.pone.0009063
- Hotamisligil GS, Arner P, Caro JF, Atkinson RL, Spiegelman BM. Increased adipose tissue expression of tumor necrosis factor-α in human obesity and insulin resistance. J Clin Invest 1995; 95:2409-15; PMID:7738205; http://dx.doi.org/10.1172/JCI117936
- Kido Y, Burks DJ, Withers D, Bruning JC, Kahn CR, White MF, Accili D. Tissue-specific insulin resistance in mice with mutations in the insulin receptor, IRS-1, and IRS-2. J Clin Invest 2000; 105:199-205; PMID:10642598; http://dx.doi.org/10.1172/JCI7917
- Tamemoto H, Kadowaki T, Tobe K, Yagi T, Sakura H, Hayakawa T, Terauchi Y, Ueki K, Kaburagi Y, Satoh S, et al. Insulin resistance and growth retardation in mice lacking insulin receptor substrate-1. Nature 1994; 372:182-6; PMID:7969452; http://dx.doi.org/10.1038/372182a0
- Withers DJ, Gutierrez JS, Towery H, Burks DJ, Ren JM, Previs S, Zhang Y, Bernal D, Pons S, Shulman GI, et al. Disruption of IRS-2 causes type 2 diabetes in mice. Nature 1998; 391:900-4; PMID:9495343; http://dx.doi.org/10.1038/36116
- Ramos RR, Swanson AJ, Bass J. Calreticulin and Hsp90 stabilize the human insulin receptor and promote its mobility in the endoplasmic reticulum. Proc Natl Acad Sci USA 2007; 104:10470-5
- Vig S, Pandey AK, Verma G, Datta M. C/EBPalpha mediates the transcriptional suppression of human calreticulin gene expression by TNFalpha. Int J Biochem Cell Biol 2012; 44:113-22; PMID:22024156; http://dx.doi.org/10.1016/j.biocel.2011.10.004
- Marchesini G, Brizi M, Bianchi G, Tomassetti S, Bugianesi E, Lenzi M, McCullough AJ, Natale S, Forlani G, Melchionda N. Nonalcoholic fatty liver disease: a feature of the metabolic syndrome. Diabetes 2001; 50:1844-50; PMID:11473047; http://dx.doi.org/10.2337/diabetes.50.8.1844
- Bacon BR, Farahvash MJ, Janney CG, Neuschwander-Tetri BA. Nonalcoholic steatohepatitis: an expanded clinical entity. Gastroenterology 1994; 107:1103-9; PMID:7523217
- Lee RG. Nonalcoholic steatohepatitis: a study of 49 patients. HumPathol 1989; 20:594-8; PMID:2656500
- Powell EE, Cooksley WG, Hanson R, Searle J, Halliday JW, Powell LW. The natural history of nonalcoholic steatohepatitis: a follow-up study of forty-two patients for up to 21 years. Hepatology 1990; 11:74-80; PMID:2295475; http://dx.doi.org/10.1002/hep.1840110114
- Sanyal AJ, Campbell-Sargent C, Mirshahi F, Rizzo WB, Contos MJ, Sterling RK, Luketic VA, Shiffman ML, Clore JN. Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology 2001; 120:1183-92; PMID:11266382; http://dx.doi.org/10.1053/gast.2001.23256
- Hernandez-Toro J, Prieto C, De las RJ. APID2NET: unified interactome graphic analyzer. Bioinformatics 2007; 23:2495-7; PMID:17644818; http://dx.doi.org/10.1093/bioinformatics/btm373
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 2003; 13:2498-504; PMID:14597658; http://dx.doi.org/10.1101/gr.1239303
- Wu CH, Apweiler R, Bairoch A, Natale DA, Barker WC, Boeckmann B, Ferro S, Gasteiger E, Huang H, Lopez R, et al. The Universal Protein Resource (UniProt): an expanding universe of protein information. Nucleic Acids Res 2006; 34:D187-D91; PMID:16381842; http://dx.doi.org/10.1093/nar/gkj161
- Barsky A, Gardy JL, Hancock RE, Munzner T. Cerebral: a Cytoscape plugin for layout of and interaction with biological networks using subcellular localization annotation. Bioinformatics 2007; 23:1040-2; PMID:17309895; http://dx.doi.org/10.1093/bioinformatics/btm057
- Bader GD, Hogue CW. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics 2003; 4:2; PMID:12525261; http://dx.doi.org/10.1186/1471-2105-4-2
- Qiu Y, Michalak M. Transcriptional control of the calreticulin gene in health and disease. Int J Biochem Cell Biol 2009; 41:531-8; PMID:18765291; http://dx.doi.org/10.1016/j.biocel.2008.06.020
- Michalak M, Groenendyk J, Szabo E, Gold LI, Opas M. Calreticulin, a multi-process calcium-buffering chaperone of the endoplasmic reticulum. Biochem J 2009; 417:651-66; PMID:19133842; http://dx.doi.org/10.1042/BJ20081847
- Hisaoka M, Matsuyama A, Nakamoto M. Aberrant calreticulin expression is involved in the dedifferentiation of dedifferentiated liposarcoma. Am J Pathol 2012; 180:2076-83; PMID:22429966; http://dx.doi.org/10.1016/j.ajpath.2012.01.042
- Wang X, Zhao X, Gao X, Mei Y, Wu M. A new role of p53 in regulating lipid metabolism. J Mol Cell Biol 2013; 5:147-50; PMID:23258697; http://dx.doi.org/10.1093/jmcb/mjs064
- Cheung EC, Vousden KH. The role of p53 in glucose metabolism. Curr Opin Cell Biol 2010; 22:186-91; PMID:20061129; http://dx.doi.org/10.1016/j.ceb.2009.12.006
- Ide T, Brown-Endres L, Chu K, Ongusaha PP, Ohtsuka T, El-Deiry WS, Aaronson SA, Lee SW. GAMT, a p53-inducible modulator of apoptosis, is critical for the adaptive response to nutrient stress. Mol Cell 2009; 36:379-92; PMID:19917247; http://dx.doi.org/10.1016/j.molcel.2009.09.031
- Assaily W, Rubinger DA, Wheaton K, Lin Y, Ma W, Xuan W, Brown-Endres L, Tsuchihara K, Mak TW, Benchimol S. ROS-mediated p53 induction of Lpin1 regulates fatty acid oxidation in response to nutritional stress. Mol Cell 2011; 44:491-501; PMID:22055193; http://dx.doi.org/10.1016/j.molcel.2011.08.038
- Finck BN, Gropler MC, Chen Z, Leone TC, Croce MA, Harris TE, Lawrence JC, Jr., Kelly DP. Lipin 1 is an inducible amplifier of the hepatic PGC-1alpha/PPARalpha regulatory pathway. Cell metabolism 2006; 4:199-210; PMID:16950137; http://dx.doi.org/10.1016/j.cmet.2006.08.005
- Jiang P, Du W, Wang X, Mancuso A, Gao X, Wu M, Yang X. p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat Cell Biol 2011; 13:310-6; PMID:21336310; http://dx.doi.org/10.1038/ncb2172
- Mesaeli N, Phillipson C. Impaired p53 expression, function, and nuclear localization in calreticulin-deficient cells. Mol Biol Cell 2004; 15:1862-70; PMID:14767071; http://dx.doi.org/10.1091/mbc.E03-04-0251
- Roder K, Zhang L, Schweizer M. SREBP-1c mediates the retinoid-dependent increase in fatty acid synthase promoter activity in HepG2. FEBS Lett 2007; 581:2715-20; PMID:17531980; http://dx.doi.org/10.1016/j.febslet.2007.05.022
- Repa JJ, Liang G, Ou J, Bashmakov Y, Lobaccaro JM, Shimomura I, Shan B, Brown MS, Goldstein JL, Mangelsdorf DJ. Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRalpha and LXRbeta. Genes Dev 2000; 14:2819-30; PMID:11090130; http://dx.doi.org/10.1101/gad.844900
- Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest 2002; 109:1125-31; PMID:11994399; http://dx.doi.org/10.1172/JCI0215593
- memiya-Kudo M, Shimano H, Yoshikawa T, Yahagi N, Hasty AH, Okazaki H, Tamura Y, Shionoiri F, Iizuka Y, Ohashi K, et al. Promoter analysis of the mouse sterol regulatory element-binding protein-1c gene. J Biol Chem 2000; 275:31078-85; PMID:10918064; http://dx.doi.org/10.1074/jbc.M005353200
- Yahagi N, Shimano H, Matsuzaka T, Najima Y, Sekiya M, Nakagawa Y, Ide T, Tomita S, Okazaki H, Tamura Y, et al. p53 Activation in adipocytes of obese mice. J Biol Chem 2003; 278:25395-400; PMID:12734185; http://dx.doi.org/10.1074/jbc.M302364200
- Javitt NB. Hep G2 cells as a resource for metabolic studies: lipoprotein, cholesterol, and bile acids. FASEB J. 1990; 4:161-8; PMID:2153592
- Mavri-Damelin D, Eaton S, Damelin LH, Rees M, Hodgson HJ, Selden C. Ornithine transcarbamylase and arginase I deficiency are responsible for diminished urea cycle function in the human hepatoblastoma cell line HepG2. Int J BIochem Cell Biol 2007; 39:555-64; PMID:17098461; http://dx.doi.org/10.1016/j.biocel.2006.10.007
- Du P, Kibbe WA, Lin SM. lumi: a pipeline for processing Illumina microarray. Bioinformatics 2008; 24:1547-8; PMID:18467348; http://dx.doi.org/10.1093/bioinformatics/btn224
- Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 2008; 3:1101-8; PMID:18546601; http://dx.doi.org/10.1038/nprot.2008.73
- Ricchi M, Odoardi MR, Carulli L, Anzivino C, Ballestri S, Pinetti A, Fantoni LI, Marra F, Bertolotti M, Banni S, et al. Differential effect of oleic and palmitic acid on lipid accumulation and apoptosis in cultured hepatocytes. J Gastroen Hepatol 2009; 24:830-40; PMID:19207680; http://dx.doi.org/10.1111/j.1440-1746.2008.05733.x
- Uhlen M, Oksvold P, Fagerberg L, Lundberg E, Jonasson K, Forsberg M, Zwahlen M, Kampf C, Wester K, Hober S, et al. Towards a knowledge-based Human Protein Atlas. Nat Biotechnol 2010; 28:1248-50; PMID:21139605; http://dx.doi.org/10.1038/nbt1210-1248
- Mishra A, Vij M, Kumar D, Taneja V, Mondal AK, Bothra A, Rao V, Ganguli M, Taneja B. Integration host factor of Mycobacterium tuberculosis, mIHF, compacts DNA by a bending mechanism. PLoSOne 2013; 8:e69985; PMID:23922883; http://dx.doi.org/10.1371/journal.pone.0069985