
Abstract
Maintenance of normal core body temperature is vigorously defended by long conserved, neurovascular homeostatic mechanisms that assist in heat dissipation during prolonged, heat generating exercise or exposure to warm environments. Moreover, during febrile episodes, body temperature can be significantly elevated for at least several hours at a time. Thus, as blood cells circulate throughout the body, physiologically relevant variations in surrounding tissue temperature can occur; moreover, shifts in core temperature occur during daily circadian cycles. This study has addressed the fundamental question of whether the threshold of stimulation needed to activate lymphocytes is influenced by temperature increases associated with physiologically relevant increases in temperature. We report that the need for co-stimulation of CD4+ T cells via CD28 ligation for the production of IL-2 is significantly reduced when cells are exposed to fever-range temperature. Moreover, even in the presence of sufficient CD28 ligation, provision of extra heat further increases IL-2 production. Additional in vivo and in vitro data (using both thermal and chemical modulation of membrane fluidity) support the hypothesis that the mechanism by which temperature modulates co-stimulation is linked to increases in membrane fluidity and membrane macromolecular clustering in the plasma membrane. Thermally-regulated changes in plasma membrane organization in response to physiological increases in temperature may assist in the geographical control of lymphocyte activation, i.e., stimulating activation in lymph nodes rather than in cooler surface regions, and further, may temporarily and reversibly enable CD4+ T cells to become more quickly and easily activated during times of infection during fever.
Abbreviations
| APC | = | antigen-presenting cell |
| CD28 | = | cluster of differentiation 28 |
| CD3 | = | cluster of differentiation 3 |
| CD4 | = | cluster of differentiation 4 |
| CD8 | = | cluster of differentiation 8 |
| CTLA-4 | = | cytotoxic T-lymphocyte-associated protein 4 |
| CTxB | = | cholera toxin B subunit |
| Ct | = | cycle threshold |
| ELISA | = | enzyme-linked immunosorbant assay |
| EtOH | = | ethanol |
| FITC | = | fluoroisothiocyanate |
| GM1 | = | monosialotetrahexosylganglioside |
| IDEAS | = | imagestream data exploration and analysis software |
| IL-2 | = | interleukin 2 |
| LA | = | latrunculin A |
| MβCD | = | methyl-β-cyclodextrin |
| PD-1 | = | Programmed cell death-1 |
| PMA | = | phorbol 12-myristate 13-acetate |
| pMHC | = | peptide-major histocompatibility complexes |
| qRT-PCR | = | quantitative reverse transcription polymerase chain reaction |
| TCR | = | T cell receptor |
| TDI | = | time delay integration |
| TMA-DPH | = | trimethylammonium diphenylhexatriene |
| WBH | = | whole body hyperthermia. |
Introduction
The activation and subsequent proliferation of T lymphocytes is tightly controlled by ligation of separate receptors on the cell surface, including the antigen receptor and also co-stimulatory receptors, which are usually triggered by antigen presenting cells. These receptors, and others, collectively generate both positive and negative second signals that precisely regulate T cell function. Activation of T lymphocytes without the proper amount of co-stimulatory signals can lead to T cell deletion or anergy.Citation1-4 One of the best understood costimulatory receptors on T cells is CD28 and its function appears to be dependent on specific macromolecular organization of the plasma membrane.Citation5
From the earliest investigations which experimentally defined requirements for activation of lymphocytes,Citation6 investigators have utilized an in vitro culture temperature of precisely 37°C to mimic blood or body temperature. However, several observations suggest that temperature should be evaluated more completely as a variable which may modulate basic requirements for lymphocyte activation. For example, the core temperature of mice and humans normally undergoes a significant daily circadian flux (for mice the temperature shift is approximately 1.7°Celsius, ranging from 36.9 to 38.6°C).Citation7,8 Further, infection and inflammation can stimulate a 1–5 degree increase in core body temperature for hours at a time.Citation9-11 Thus, during fever, most lymphocytes will experience higher than normal temperatures for a sustained period of time prior to or during contact with antigen presenting cells which specifically engage T cell receptor (TCR) as well as CD28 receptors. There is intriguing evidence that sustained increases in temperature associated with fever result in significant survival benefits following infection in multiple vertebrate species,Citation12 including humans.Citation13-15 Therefore, thermal shifts which exist during the early stages of infection, when optimal co-stimulatory signals may not yet be generated, might help to improve, or speed, the host immune response. While previous research on the relationship between physiological temperature shifts and specific T cell receptors during activation is sparse, several studies using non-specific activators point strongly to the hypothesis that thermal signals may help to calibrate the requirements for activation. For example, very early studies on Con A-treated spleen cells incubated at fever-range temperature shows that their proliferation is increased compared to those maintained at 37°C,Citation10,16 while other studies show that the clonal expansion and proliferation of lymphocytes is enhanced.Citation17 More recently, Meinander et al. proposed that mild hyperthermia associated with fever could help to promote the elimination of excess T lymphocytes through promoting enhanced apoptosis.Citation18 In terms of antigen-specific effects of thermal stress, our lab has recently demonstrated that the activation and differentiation of antigen-specific CD8+ T cells into effector cells is enhanced by physiological range hyperthermia and accompanying this effect, we observed that mild heating of CD8 T cells resulted in the reversible clustering of GM1 CD-microdomains in the plasma membrane.Citation19,20 As a result of these data, it seems plausible that a physiologically-relevant temperature flux could affect the threshold of activation for T cells.
To test this hypothesis, we used several well-characterized CD4+ T cell systems, combined with their production of IL-2 as a functional read-out, since this is one of the most well characterized measures of activation. Using three different, well characterized cellular models for CD4+ T cell activation (cells isolated from human peripheral blood, Jurkat T cells grown in culture, and T cells isolated from CD28-deficient and Ova-specific transgenic mice), we obtained data which support the hypothesis that mild, fever-range heating significantly reduces the requirement for co-stimulation via CD28. Thus these new data suggest that fever, or mild hyperthermia could assist in generating a temporary state of heightened immune sensitivity during immune challenge, or during situations when optimal levels of co-stimulation for CD4+ T cell activity may not be immediately available.
Results
Mild heating augments IL-2 production by CD4+ T cells and reduces the requirement for CD28-mediated co-stimulation
Activation of T cells is initiated by the engagement of the TCR with antigen peptide-bound major histocompatibility complexes (pMHCs) on the surface of antigen presenting cells (APCs).Citation23 And although a weak T cell response can occur by strong, repeated TCR stimulation with high doses of antigen alone,Citation24 optimal T cell activation requires a co-stimulatory signal. To investigate the effect of mild heating on subsequent activation requirements for IL-2 production, freshly isolated human CD4+ cells were pre-incubated at 37 or 39.5°C for 6 hours and then stimulated at 37°C for 24 hours with serial dilutions of anti-CD3 antibodies (to mimic TCR-mediated signaling) and anti-CD28 antibodies (to mimic the co-stimulatory signal).Citation6 The results reveal that anti-CD28 ligation alone, in the absence of anti-CD3, is insufficient for IL-2 production at both temperatures (). But, even when a very small amount of anti-CD3 (0.02 μg/ml) is provided in the presence of anti-CD28, (concentrations which result in minimal amounts of IL-2 when cells are kept at 37°C) IL-2 production by the heat-pretreated cells is significantly increased () and this increase is also seen with higher concentrations of anti-CD3. Surprisingly, even when no co-stimulation via anti-CD28 was provided, heat pretreatment was still associated with some IL-2 production (), suggesting that mild heating is able to compensate, at least to some extent, for lack of CD28 mediated co-stimulation in human CD4+ T cells. Indeed, when we compared the amount of anti-CD28 needed to achieve comparable amounts of IL-2 production at 37°C vs. 39°C, we saw a significant difference. For example, at 37°C, 0.05 μg/mL of anti-CD28 was necessary to produce the same amount of IL-2 that was produced using only 0.001 μg/mL of anti-CD28 at 39.5°C even though 0.01 μg/mL of anti-CD3 was used in both cases (). Therefore, we conclude that treating CD4+ T cells with mild heat treatment lowers the requirement of CD28 mediated co-stimulation to produce IL-2.
Figure 1. Mild heating augments IL-2 production by CD4+ T cells and reduces the requirement for CD28-mediated co-stimulation. (A, B) Human peripheral blood (HPB) CD4+ T cells were pre-incubated at 37°C (gray bars) or 39.5°C (black bars) for up to 6 hours prior to activation with stimulating Abs. HPB CD4+ T cells were stimulated with anti-CD3 Ab in combination with increasing concentrations of anti-CD28 Ab (A), or anti-CD28 Ab in combination with increasing concentrations of anti-CD3 Ab (B). After 24 hrs at 37°C IL-2 production was measured by ELISA assay. (C) Jurkat T cells were pre-incubated at 37, and 39.5°C for 6 hours and stimulated using 3μg/ml of plate-bound anti-CD3 in combination with 0, 0.05, and 0.1μg/ml of soluble anti-CD28 for 24 hours at 37°C. Spent media was collected and the level of IL-2 cytokine in the supernatant was detected by an ELISA. (D) Jurkat T cells were pre-incubated at 37, and 39.5°C for 6 hours and stimulated using 2μg/ml of soluble anti-CD28 in combination with 0, 0.25, and 1μg/ml of plate-bound anti-CD28 for 24 hours at 37°C. Spent media was collected and the level of IL-2 cytokine in the supernatant was detected by an ELISA. (E) OVA-specific CD4+ T cells isolated by negative selection from the spleens of DO11.10 transgenic mice were incubated at 37 (gray bars) and 39.5°C (black bars) as above and stimulated with OVA pulsed A20 cells at 37°C for 24 hrs before the amount of IL-2 in the supernatant was determined by ELISA. *p < 0.05, when comparing the data at 37 vs. 39.5°C using an unpaired Student's t-test. The results are expressed as the mean ±s.d. and are representative of 3 or more independent experiments
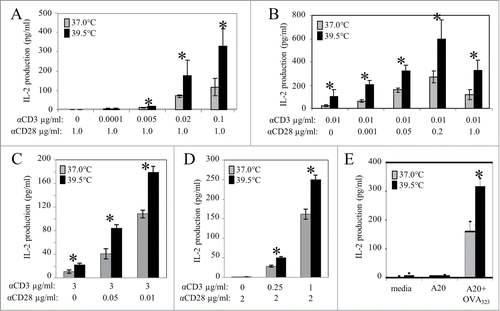
The CD4+ human T cell line Jurkat has long been used as a model system to define the requirements for T cell activation.Citation25 To determine whether this cell line responds to mild increases in temperature in a manner similar to that of freshly isolated human CD4+ T cells, Jurkat cells were stimulated with 3μg/ml of anti-CD3 and a range of anti-CD28. Results demonstrated that cells pre-exposed to 39.5°C require less anti-CD28 than cells pre-incubated at 37°C in order to achieve the same IL-2 output (). We also tested how mild hyperthermia affected IL-2 output following stimulation by serial dilutions of anti-CD3 along with a constant amount of anti-CD28. Once again, the results of this experiment revealed that pre-heating cells cannot compensate for a lack of TCR stimulation (anti-CD3) (). Therefore, just as with freshly isolated human blood CD4+ T cells, mild heating significantly increases IL-2 production by Jurkat T cells following stimulation with anti-CD3 alone (), but not when using anti-CD28 alone ().
Collectively, these data demonstrate that a mild, sustained increase in temperature can support and enhance IL-2 production by T cells stimulated with anti-CD3, even with very low, or even no co-stimulation by anti-CD28. In addition, these data emphasize the specificity of this effect since mild heating cannot replace the need for signaling through the TCR, nor does it result in any non-specific IL-2 release. However, while the use of antibodies specific for signaling receptors is useful for quantifying the contribution of temperature to activation requirements, it is also important to determine whether antigen-induced activation of CD4+ T cells can be affected by mild heating. To address this, we isolated CD4+ T cells from the spleens of DO11.10 transgenic mice whose T cell receptors are specific for OVA323-339 peptide (OVA;).Citation26 The cells were incubated at either 39.5 or 37°C for 6 hours before being stimulated with A20 B cells pulsed with OVA. The production of IL-2 was again monitored by an ELISA and the results clearly demonstrate that cytokine production is significantly greater when T cells are pre-heated. In contrast, OVA-specific CD4+ T cells stimulated with un-pulsed A20 cells produced no IL-2 when maintained at either temperature () again supporting the fact the mild heating does not induce IL-2 release in the absence of appropriate antigen receptor-induced triggering.
Mild heating enhances IL-2 mRNA production in CD4+ T cells
To determine whether transcription of IL-2 is affected by mild heating, we assessed the levels of IL-2 mRNA in Jurkat T cells following pre-incubation at either 37 or 39.5°C and subsequent activation by anti-CD3 ± anti-CD28. The fold changes in IL-2 mRNA production were monitored using quantitative real-time reverse transcriptase qRT-PCR ( and B). We found that IL-2 mRNA increased more than 7-fold in cells pre-incubated at 39.5°C starting 1 hour after stimulation with anti-CD3 alone and remained elevated until at least 12 hours following stimulation (). Mild heating also increased the level of IL-2 message in cells stimulated with both anti-CD3 and anti-CD28 starting 0.5 hour following stimulation, with a dramatic increase of greater than 100 fold beginning at 1 hour (). Mild heating not only increases early production of IL-2, but also prolongs transcription of this gene. These results along with data showing that mild heating alone does not result in any non-specific IL-2 release suggest that hyperthermia exerts its effect at the level of IL-2 gene activation rather than simply causing an increased release of pre-formed IL-2.
Figure 2. Mild heating enhances IL-2 and IL-2 mRNA production in Jurkat T cells. Following pre-incubation at 37°C or 39.5°C for 6 hours, Jurkat T cells were then stimulated with anti-CD3 Ab alone (0.1 µg/ml) (A) or anti-CD3 Ab in combination with anti-CD28 Ab ( both at 0.1 µg/ml) (B) at 37°C for different times as shown. The level of IL-2 mRNA was measured by quantitative real time RT-PCR (A and B) as described in supplemental methods. GAPDH was used as loading control for both experiments. Ct values were first normalized to GAPDH, then normalized to the Ct value of the 37°C group at the 0 time point. (C) CD4+ T cells isolated from human peripheral blood were incubated at 37 (gray bars) and 39.5°C (black bars) in the presence of cyclohexamide (striped bars) for 6 hours. Cyclohexamide was removed from the culture media and cells were stimulated using different stimuli at 37°C for 24 hours before IL-2 production was measured by ELISA. *p < 0.05, when comparing the data at 37 vs. 39.5°C using an unpaired Student's t-test. The results are expressed as the mean ± s.d. and are representative of 3 or more independent experiments.
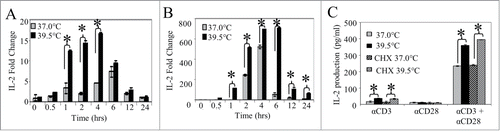
To determine whether new protein synthesis occurring during the heating period is necessary for thermally enhanced IL-2 production, human peripheral blood CD4+ T cells were treated with cyclohexamide to inhibit protein synthesis for the duration of heat treatment. Cyclohexamide was then removed in order to permit protein synthesis following activation at 37°C for 24 hour. Cells pre-treated with heat prior to activation again produced significantly more IL-2 than cells maintained at 37°C. Enhanced IL-2 was observed when both anti-CD3 and anti-CD28 were used for stimulation, and also when anti-CD3 was given alone (). Therefore, use of cycloheximide during the thermal treatment did not interfere with enhanced IL-2 production that occurred after stimulation with anti-CD3 ± anti-CD28. This evidence in combination with results that demonstrate there is no enhancement of CD3 and CD28 on heated and control cell surfaces (data not shown), indicates that new protein synthesis during the heating is not critical for enhanced IL-2 production in response to mild hyperthermia.
Mild heating enables IL-2 production by anti-CD3 stimulated CD4+ T cells isolated from CD28-deficient mice
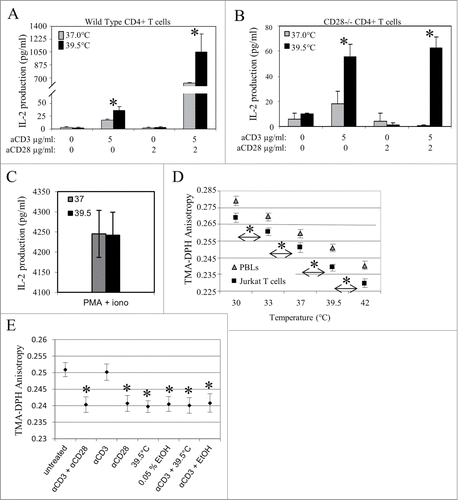
If mild heating can replace or reduce the need for CD28 co-stimulation, then it should be possible to see IL-2 production using anti-CD3 with heated CD4+ T cells obtained from a CD28-deficient mouse. For this purpose, we acquired CD28 knockout mice. Consistent with previous reports employing these mice,Citation24 the development their CD4+ T cells was normal, as we found that greater than 60% of splenocytes from both wild-type and CD28−/− mice were positive for both CD3 and CD4 (data not shown). In addition, we also verified the absence of CD28 on CD4+ T cells using immunofluorescent staining with anti-CD28 antibody (data not shown). We then investigated whether pre-treating these cells with mild hyperthermia (39.5°C) affected IL-2 production following stimulation by anti-CD3 ± anti-CD28. As expected, CD4+ T cells from wild-type C57BL/6 mice maintained at 37°C produced very low levels of IL-2 in response to CD3 stimulation alone, and high levels in response to a combination of anti-CD3 and anti-CD28 (). Furthermore, heat pre-treatment enhanced IL-2 production in each circumstance. Results from identical experiments conducted using CD4+ T cells from CD28-deficient mice also demonstrated the value of mild hyperthermia to the T cell response. Following pre-treatment with (39.5°C) and stimulation by anti-CD3 alone, CD4+ T cells from CD28-deficient mice, produced significantly higher levels of IL-2, compared to the amounts produced by cells maintained at 37°C throughout (). Since these cells do not express CD28, the amount of IL-2 measured was not further increased when cells were activated with the combination of anti-CD3 and anti-CD28, and stimulating these cells with anti-CD28 alone did not result in any IL-2 production at either incubation temperature. Moreover, mild heating did not increase IL-2 production when CD4+ CD28−/− T cells were stimulated with the mitogenic combination of PMA and ionomycin (), a treatment protocol which bypasses proximal TCR signaling events and, at the concentrations used, provides a maximal stimulus. Overall, these data suggest that a mild elevation in temperature can partially overcome the lack of CD28 receptors in cells isolated from the CD28-deficient mouse.
Figure 3. Mild heating enhances IL-2 production in CD4+ T cells isolated from CD28 deficient mice and is associated with an increase in membrane fluidity that is similar to the increase induced by various stimulants. CD4+ T cells from (A) wild type and (B) CD28−/− mice were incubated at 37°C (gray bars) and 39.5°C (black bars) for 6 hrs and stimulated with anti-CD3 Ab (5 µg/ml), anti-CD28 Ab (2 µg/ml) or the combination of both Abs at 37°C for 24 hrs before the amount of IL-2 in the supernatant was determined by ELISA. (C) Jurkat T cells were incubated at 37 (gray bars) and 39.5°C (black bars) for 6 hrs. Cells were stimulated with PMA (10ng/ml) and ionomycin (1 ng/ml) at 37°C for 24 hours and the number of IL-2 producing cells was determined by ELISA. (D) Effect of physiologically relevant temperature shifts on T cell membrane fluidity. T cells (freshly isolated human CD4+ T cells or Jurkat T cells) were resuspended in PBS and incubated at the indicated temperatures for 5 minutes. The plasma membrane fluidity of the cells was then monitored by measuring the fluorescence anisotropy of the membrane probe TMA-DPH, as described in the methods section. (E) A comparison of various treatments on T cell membrane fluidity. Jurkat T cells were stimulated with antibodies (3μg/ml anti-CD3 alone, 2μg/ml anti-CD28 alone, or 3μg/ml anti-CD3 + 2μg/ml anti-CD28) for 6 hours, briefly exposed to 39.5°C, briefly exposed to 0.05% EtOH, or simultaneously exposed to a combination of these treatments (as indicated). The cells were then resuspended in PBS, incubated at 37°C (unless otherwise indicated) for 5 minutes, and labeled with the fluorescent membrane probe, TMA-DPH. The plasma membrane fluidity of the cells was then monitored by measuring the TMA-DPH anisotropy, as described in the methods section. *, p < 0.05, when comparing the data at 37 vs. 39.5°C using an unpaired Student's t-test. Data presented is representative of at least 3 independent experiments.
Mild heating alters T cell plasma membrane fluidity in a manner comparable to co-stimulation
As mentioned above, previous publications have suggested that a primary signal in the cellular perception of a change in temperature is a change in the fluidity of plasma membrane lipids.Citation27-33 We investigated whether mild heat could affect the plasma membrane fluidity of T cells by measuring the steady-state fluorescence anisotropy of trimethylammonium diphenylhexatriene (TMA-DPH) in both Jurkat T cells and T lymphocytes freshly isolated from human donors. We found that increased temperatures correlate with significantly lower TMA-DPH anisotropy values, which represent a measure of the membrane order, and are in turn inversely proportional to plasma membrane fluidity (). Therefore, our data demonstrates that higher physiological temperatures correspond to increased plasma membrane fluidity in T cells. We then asked how the effects of mild heating on membrane order compared to that induced by a commonly used membrane fluidizer, ethanol (EtOH). For this purpose, the TMA-DPH anisotropy was measured as Jurkat T cells were exposed to different concentrations of EtOH. By doing so, we found that a concentration of 0.05% EtOH resulted in a fluidity increase comparable to that induced by mild heating (data not shown).
Calder et al. have previously shown that activation of freshly isolated T cells is accompanied by an increase in membrane fluidity.Citation34 Therefore, we compared thermally- and EtOH-induced changes in T cell membrane fluidity to that following stimulation through TCR and CD28 using Jurkat cells. To verify that Jurkat cells would undergo a change in membrane fluidity following activation similar to what was previously reported for freshly isolated lymphocytes,Citation34 we co- stimulated Jurkat T cells with anti-CD3 and anti-CD28 before measuring the TMA-DPH anisotropy of their plasma membranes. As expected, Jurkat T cells stimulated in this manner also displayed an increase in plasma membrane fluidity. () Next, we examined how the ligation of each receptor separately contributed to the observed increase in plasma membrane fluidity by stimulating Jurkat T cells with either anti-CD3 alone or anti-CD28 alone. We found that that stimulating with anti-CD28 alone led to a significant increase in plasma membrane fluidity, while stimulating with anti-CD3 alone had no effect (). Interestingly, when we directly compared the effects of 39.5°C or 0.05% EtOH on plasma membrane fluidity and those induced by anti-CD28 alone, or by anti-CD3/anti-CD28, we noted that the magnitude of the effects were comparable. (). This led us to investigate whether 39.5°C or 0.05% EtOH could increase the T cell plasma membrane fluidity of cells stimulated with anti-CD3 alone, to a level similar to that seen after stimulation with both anti-CD3 and anti-CD28. For this purpose, Jurkat T cells were stimulated with anti-CD3 alone before being exposed to 39.5°C or 0.05% EtOH. The data in reveals that exposing Jurkat T cells to 39.5°C results in the same membrane fluidity changes as does treatment with 0.05% EtOH and is similar to the increase in membrane fluidity similar to that induced by activation with both anti-CD3 and anti-CD28.
To further explore the similarity between the effects of alcohol and CD28 effects on membrane fluidity, we examined whether the increase in membrane fluidity seen upon treatment with mild heat was associated with the changes we observed in the requirements for CD28 ligation in CD4+ T cell activation. Briefly, Jurkat T cells were cultured at 37 or 39.5°C in the presence or absence of 0.05% EtOH. Following this incubation period, the cells were stimulated with plate-bound anti-CD3 for 24 hours. The supernatants were collected and IL-2 production was measured by an ELISA. Treating the cell pre-incubated at 37°C s with 0.05% EtOH enhanced IL-2 production to a level similar to what was observed after pre-incubation at 39.5°C (without EtOH). However, there was no EtOH-induced change in IL-2 production by cells pre-incubated at 39.5°C (). Therefore, we conclude that the increase in plasma membrane fluidity that is seen following exposure to 39.5°C or exposue to 0.05% EtOH is associated with the observed increase in cytokine production by CD4+ T cells upon stimulation.
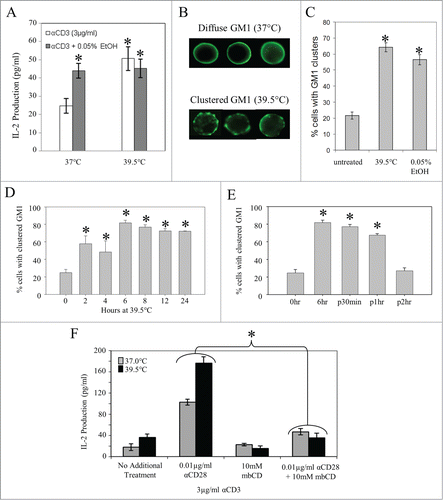
Figure 4. Mild membrane fluidizers promote lipid raft reorganization in vitro, and disruption of lipid rafts with MβCD eliminates thermally enhanced IL-2 production. (A) Effects of thermal and chemical fluidizers on IL-2 production in T cells stimulated through the TCR. Jurkat T cells were pre-incubated at 37, or 39.5°C in the presence or absence of 0.05% EtOH for 6 hours. The cells were then washed and stimulated with 3μg/ml of plate-bound anti-CD3 for 24 hours at 37°C. Spent media was collected and the level of IL-2 cytokine in the supernatant was detected by an ELISA. (B, C) Effects of thermal and chemical fluidizers on lipid raft organization in Jurkat T cells. Jurkat T cells were incubated at 37°C in the presence or absence of 0.05% EtOH, or at 39.5°C (no EtOH) for 6 hours. The cells were then fixed, and surface-labeled with Choleratoxin-B subunit to stain the lipid raft marker, GM1. Cells displaying GM1 clustering (B) were counted as positive cells, and the percentage of positive cells was determined and graphed (C). (D) Jurkat T cells were incubated at 39.5°C for different time periods, stained with FITC-CTxB, examined by fluorescence microscopy and the percentage of clustered GM1 cells was quantified. Asterisks denote a significant increase in GM1 clustering compared to untreated cells. (E) Cells were incubated at 39.5°C for 6 hrs and then returned to 37°C. At various time points cells were removed, stained with FITC-CTxB, examined by fluorescence microscopy and GM1 clustered cells were quantified. Asterisks denote a significant increase in GM1 clustering compared to untreated cells. (F) Jurkat T cells were incubated at 37 (gray bars) and 39.5°C (black bars) for 6 hrs with MβCD (10 mM) added during the last 30 min. Cells were stimulated with anti-CD3 Ab alone at 37°C for 24 hrs and the number of IL-2 producing cells was determined by ELISA. Data are representative of 3 independent experiments. *: p < 0.05, when comparing the data using an unpaired Student's t-test. The results are expressed as the mean ± s.d. from three independent experiments.
Thermally-induced increase in membrane fluidity is associated with altered T cell membrane organization
Since we established that the thermally-induced increase in IL-2 production does not require new protein synthesis during the heating phase, and is not accompanied by an increase in the surface expression of CD3 or CD28, we next examined whether mild heating affected the macromolecular organization at the plasma membrane. Our rationale was that sustained heating seems to be able to replace at least some of the function of co-stimulation through CD28, which is known to be mediated through cholesterol-dependant nanodomain reorganization at the cell surface.Citation35 In addition, Brameshuber et al. demonstrated that fever-range heat induced raft-destabilization/reorganization in living cells.Citation36 Could the co-stimulatory cues (e.g. a modest increase in fluidity) provided by mild heating induce nanodomain reorganization? The fact that even mild temperature shifts can significantly impact plasma membrane lipid organization Citation23,37,38 strongly suggests this possibility.
To explore this question, we investigated whether exposing Jurkat T cells to mild hyperthermia led to clustering of cholesterol dependent nano-domains. Jurkat T cells were cultured for 6 hours at various conditions: 37°C without EtOH, 37°C with 0.05% EtOH, or 39.5°C without EtOH. Micro-clusters of cholesterol dependent nano-domains were then visualized by staining with fluorescently labeled Choleratoxin B subunit (CTxB), which recognizes the ganglioside, GM1, a molecule known to be enriched in cholesterol dependent nano-domains. The percent of cells displaying GM1 clusters was assessed using epi-fluorescent microscopy. Representative images of diffuse and clustered GM1 staining patterns are displayed in . Approximately 20% of the ells cultured at 37°C without EtOH displayed GM1 clusters. This value increased to almost 60% when cells were exposed to 0.05% EtOH at 37°C and 65% when cells were cultured at 39.5°C without EtOH (). Therefore, we see that treatments associated with a modest increase in T cell membrane fluidity resulted in reorganization of signaling domains at the plasma membrane. We then investigated the kinetics of this thermally-induced rearrangement by exposing Jurkat T cells to 39.5°C and assessing GM1 distribution at various time points. Our results show that after 2 hours of heating, there is a significant increase in the percentage of Jurkat T cells with distinct GM1 clustering compared to control cells incubated at 37°C. This trend continues until the percentage of cells displaying clusters reaches its maximum value (80%) by 6-hours (). Extending the duration of hyperthermia beyond 6 hours did not further enhance the number of cells with GM1 clustering. We also demonstrated that this phenomenon is reversible; when cells were returned to 37°C following 6 hours of heating, the observed percentage of cells with GM1 clustering returned to baseline level 2 hours post-heating ().
Treatment with MβCD eliminates the effects of mild hyperthermia on T cell activation
To assess further the link between thermally-induced nanodomain clustering and enhanced IL-2 production, we asked whether disruption of cholesterol-dependant nanodomains could minimize or eliminate the effect of mild heating on IL-2 production. For this experiment, we used the drug methyl-β cyclodextrin (MβCD) which causes cholesterol to effuse from the membrane and is known to disrupt the formation of cholesterol-dependant nanodomains. Citation39,40 While overall cell survival was not affected by this treatment (data not shown), we observed a loss of thermal enhancement of IL-2 production when Jurkat T cells were treated with MβCD prior to activation with anti-CD3 (). However, MβCD treatment did not affect IL-2 production when using PMA and ionomycin to activate cells (data not shown). This is presumably due to the fact that these mitogens stimulate T cells downstream of the TCR/CD3-mediated signaling complex and do not depend upon nanodomain redistribution. These results further support the notion that mild heating affects CD4+ T cell activation through modulation of membrane domain organization.
Thermally induced effects on T cell plasma membrane fluidity are correlated with the reorganization of various cytoskeletal systems
Various cytoskeletal elements, including spectrin, ankyrin, and actin are known to act as barriers to lateral diffusion of lipids and proteins within the plasma membrane.Citation41-43 Furthermore, the formation of membrane domains, such as those which constitute the immune synapse following activation, is known to require cytoskeletal remodeling.Citation44,45 Therefore, we examined if mild heating induced similar changes in the subcellular distribution of various cytoskeletal proteins. For this purpose, Jurkat T cells were cultured under various conditions (listed on the x-axis in ) for 6 hours, and the distributions of spectrin and ankyrin were analyzed by immunofluorescent staining. Results show that the majority of untreated (37°C) Jurkat T cells display a diffuse staining pattern for spectrin and ankyrin, while the majority of cells stimulated with anti-CD3 and anti-CD28 contain aggregates of these molecules. Representative examples of diffuse and aggregated staining patterns are shown in . Exposure of the T cells to 39.5°C for 6 hours induced aggregate formation in almost 20% of the cells for each of the proteins. Likewise, culturing Jurkat cells at 37°C in the presence of 0.05% EtOH for 6 hours also resulted in the formation of aggregates in almost 20% of the cells (). Furthermore, when Jurkat T cells were stimulated by anti-CD28 alone, spectrin and ankyrin were rearranged to form aggregates in about 40% of Jurkat T cells, however, the response to stimulation by anti-CD3 alone was minimal (∼3% of cells). Thus, each treatment associated with a modest increase in membrane fluidity significantly altered the distribution of these cytoskeletal molecules, while the effect of anti-CD3 was minimal. Interestingly, if Jurkat T cells were pre-incubated at 39.5°C or at 37°C with 0.05% EtOH for 6 hours before being stimulated with anti-CD3 alone, the percentage of cells displaying cytoskeletal remodeling rose to over 30%, suggesting that a modest membrane fluidizer may act in a synergistic manner with TCR stimulaion to induce cytoskeletal redistribution. This provides additional evidence that thermally induced increases in plasma membrane fluidity may function in co-stimulation.
Figure 5. Thermally induced alterations in T cell plasma membrane fluidity are associated with cytoskeletal reorganization. (A, B) Jurkat T cells were stimulated with antibodies (anti-CD3 alone, anti-CD28 alone, or anti-CD3 + anti-CD28), exposed to mild hyperthermia (39.5°C), exposed to 0.05% EtOH, or simultaneously exposed to a combination of these treatments (as indicated) for 6 hours. The cells were then fixed and intracellularly stained for spectrin or ankyrin. Cells exhibiting cytoskeletal reorganization (A) were counted as positive cells and the percent of positive cells was assessed for each treatment was determined and graphed in (B). Asterisks denote a significant increase in cytoskeletal reorganization compared to untreated cells. (C) Jurkat T cells were incubated at 37, or 39.5°C for 7 hours. 1μM LA was added to the Jurkat T cell suspensions either before the incubation period, or for the final hour of the incubation period. The cells were then washed and stimulated with 3μg/ml plate-bound anti-CD3 with or without 0.1μg/ml soluble anti-CD28 (as indicated on the x-axis) for 24 hours at 37°C. Spent media was collected and the level of IL-2 cytokine in the supernatant was detected by an ELISA. *: p < 0.015 when comparing the data using an unpaired Student's t-test. The results are expressed as the mean ± s.d. from three independent experiments.
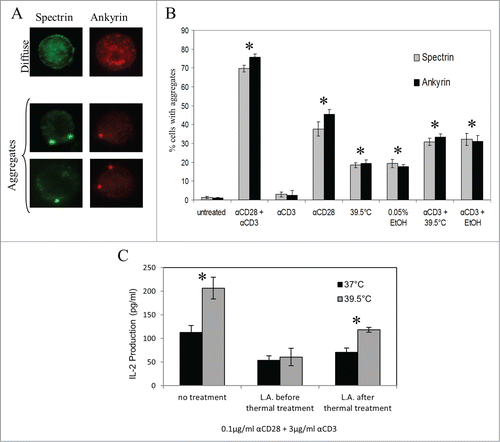
We also investigated whether actin polymerization was necessary to achieve the increase in cytokine production seen following exposure of T cells to mild heating. Here, we conducted an ELISA using a protocol similar to that used in , with one major change: we applied a potent inhibitor of actin polymerization, Latrunculin A (LA), either before or after the thermal incubation period. The data revealed that when actin polymerization is inhibited by LA prior to mild heat treatment, the enhanced IL-2 production is lost. However, if actin polymerization is inhibited after mild hyperthermia treatment, enhancement of IL-2 production is still apparent (). Therefore, the altered activation requirements and increased IL-2 production seen following pre-incubation at 39.5°C are dependent upon actin polymerization.
Exposing mice to mild hyperthermia results in membrane nanodomain clustering and cytoskeletal redistribution in lymphocytes
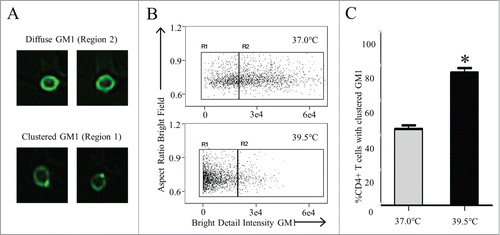
All of the experiments above were conducted in vitro. Therefore, we asked the critical question of whether these phenomena occurred in lymphocytes in vivo. This was accomplished by raising the core body temperature of mice into the fever-range by placing the mice in a warm air environment (as described in Materials and Methods and prior publications).Citation46 To determine the specific effects of heat on membrane nanodomain organization, CD4+ T cells purified from murine splenocytes of heated mice were stained with CTxB conjugated to FITC. An Imagestream flow cytometer, which provides a quantifiable means to assess morphological as well as phenotypical changes, was then employed to quantify the percentage of the CD4+ T cell population with a particular distribution of surface GM1. Representative images of cells collected by Imagestream with diffuse GM1 staining versus a clustered GM1 staining are shown in . By comparing the bright detail intensity feature to aspect ratio, we found that raising the core temperature of mice to a fever range for several hours resulted in significantly increased percentage of CD4+ T cells with aggregated nanodomains compared to mice maintained at 37°C (). These results are nearly identical to the results obtained in vitro using Imagestream flow cytometry on heated Jurkat T cells (data not shown).
Figure 6. Heat alone promotes lipid raft reorganization in vivo (A, B) C57BL/6 mice were heated at 39.5°C for 6 hours as described in Methods. Splenocytes were removed immediately following heating and stained with anti-CD4 Ab and FITC-CTxB. The diffuse GM1 and clustered GM1 on CD4+ T cells were examined by Imagestream flow cytometry with (A) representative images of diffuse GM1 and Clustered GM1 shown. (B) Aspect Ratio vs. Bright Detail Intensity features were used to gate on cells with clustered GM1 surface staining (C). The percentage of CD4+ T cells with clustered GM1 from mice maintained at 37°C (gray bar) and mice heated for 6 hrs at 39.5°C (black bar) was quantified.
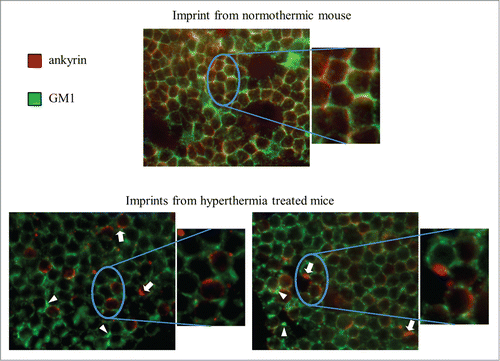
In addition to domain redistribution, we also investigated whether mild hyperthermia could alter plasma membrane and cytoskeletal organization in vivo. For this purpose, mice were again subjected to whole-body hyperthermia (WBH) for 6 hours, increasing their body temperature to 39.3±.1°C. The mice were then sacrificed and lymph node were removed and imprinted onto coverslips for in situ co-localization of the ganglioside, GM1, and the cytoskeletal protein, ankyrin. In lymph nodes isolated from control (37°C) BALB/c mice GM1 and ankyrin exhibited overlapping diffuse staining patterns around the cell surface, while lymph nodes isolated from mice exposed to WBH displayed subcellular aggregation of ankyrin (arrows), as well as clustering of GM1 on the cell surface (arrowheads) (). It should be noted that the imprint images are representative of a single focal plane and that each focal plane from imprints of mice treated with WBH displayed ankyrin aggregates and GM1 clusters in a subset of cells. Overall, these results demonstrate that elevation of the core temperature of mice can induce changes in plasma membrane and cytoskeletal organization of lymphocytes similar to those seen by mild in vitro heating and that these changes may be associated with more efficient cell signaling.Citation47
Figure 7. WBH induces redistribution of ankyrin and GM1 in vivo. Mice were incubated in a heated incubator (37.3°C) for 6 hours (control mice at room temperature − 23°C – in the dark). The mice were then sacrificed and their lymph nodes were removed and imprinted onto Alcian blue-coated coverslips for in situ immunofluorescent co-localization of GM1 (green) and ankyrin (red). Arrows denote representative ankyrin clusters while arrowheads denote representative GM1 clusters.
Discussion
We found that a mild, sustained increase in temperature not only enhances IL-2 production by CD4+ T cells receiving the canonical 2 activation signals, but it also enhances IL-2 production under conditions of insufficient co-stimulation of CD28 that would, if conducted at 37°C, result in greatly reduced IL-2 production. Even with no provision of anti-CD28, IL-2 production could still be measured at 39.5°C, but not at 37°C. Since co-stimulation via CD28 is considered necessary for full activation and IL-2 production at 37°C, these data suggest that mild hyperthermia may be able to mimic at least some of the responses elicited by CD28-mediated co-stimulation. However, the fact that mild heating could not replace the requirement for ligation of the antigen receptor () may represent a very important control mechanism ensuring that lymphocyte activation and release of potent cytokines does not occur during situations in which no antigen is present, e.g., during body warming exercise or normal circaidian shifts. Moreover, the fact that we did not begin to see significant enhancement of IL-2 production until several hours of heating further reduces the possibility that routine fluxes in body temperature could significantly affect activation requirements.
What are the possible molecular mechanisms by which ambient temperature influences T cell activation? Numerous studies show that optimal T cell activation involves reorganization of the plasma membrane, both in the order of membrane lipids and membrane cortex associated proteins, such as actin, spectrin, and vimentin.Citation48-50 Moreover, prior studies demonstrate that more severe hyperthermia (i.e., above 41°C) could also increase membrane fluidy (reviewed in Grimm et al.).Citation36,51 Here we provide evidence that reversible changes in fluidity occur immediately after exposing cells to mild hyperthermia and we were able to duplicate these effects using a known membrane fluidizer, 0.5% ethanol. We also demonstrated that co-stimulation through CD28 affects membrane fluidization in an identical manner, which is consistent with earlier publications that used various other cell types to show that signaling induced changes in plasma membrane fluidity correlate with that induced by temperature or solvents and vice-versa.Citation27-33 Collectively, these data suggest the intriguing possibility that heat can replace or mimic CD28 through its ability to alter membrane fluidity.
The observed effects of fever-range hyperthermia on T-cell stimulation persist over hours indicating that the changes in lipid fluidity likely modulate the macromolecular order in the membrane and membrane cortex. Under resting conditions, distinct macromolecular domains occur in the plasma membrane of T cells, but generally, these individual membrane domains are only a few nanometers in diameter and difficult to distinguish by microscopy.Citation52 However, when T cells are activated using TCR/CD28-receptors at 37°C, distinct membrane domains appear to cluster into larger aggregates that can be visualized using microscopic methods.Citation50,53 Furthermore, it has been previously noted that the geographical organization and specific composition of membrane microdomains may also be a critical target in the transduction of thermal signals to the cell.Citation54,55 We observed that CD4+ T lymphocytes exposed to a mild heating alone exhibit significant, yet reversible, clustering of plasma membrane macromolecular complexes (). Very importantly, this effect was also demonstrated to occur in vivo using CD4+ T cells isolated from animals receiving mild whole body hyperthermia (). Similarly, it has previously been shown that CD28 enhances the clustering of macromolecular signaling platforms in the plasma membrane, and that CD28s function is actually dependent on this membrane reorganization. In this way, perhaps mild hyperthermia and CD28 can each lower the T cell activation threshold. Whether mild heating acts as a co-stimulator itself, or through the enhancement of a signaling pathway downstream of CD28 signaling is an important question resulting from our observations. Our data shows that the greatest amount of IL-2 is produced when both heat and CD28 co-stimulation are used (), suggesting that there may be some non-overlapping or additive effects of heat and CD28 ligation in addition to observed similarities. Previous work by our group has revealed that treatment of natural killer cells and CD8 T cells with mild hyperthermia results in a similar clustering of membrane domains and activating receptors, which may assist with improved cytotoxicity of tumor cell targets.Citation19,20,56 Thus, there is a distinct possibility that several cell types use a similar strategy to respond to environmental temperature cues thatinfluence immune cell functions.
Further, earlier results from our group revealed a significant increase in the percentage of lymphocytes in spleens and lymph nodes with clustered cytoskeletal proteins (including vimentin, spectrin and actin) following systemic mild hyperthermia. Citation57,58 Here, using CD4+ T cells, we demonstrated that mild heating and ethanol (both of which increased membrane fluidity) caused an identical rearrangement of ankyrin and spectrin (). In addition to the spectrin and ankyrin cyoskeletal systems, actin polymerization has also been shown to play a prominent role in cholesterol dependent signal domain aggregation and synapse formation during T cell activation. Citation44,59-63 In turn, we looked at whether blockade of actin polymerization influenced the activating effects of mild heating and ethanol, and found that in both cases actin polymerization was necessary for enhancement of IL-2 production (). These data provide additional support for the hypothesis that thermal signals influence cytoskeletal reorganization in order to lower the threshold for T-cell activation.
While we have only focused here on the production of IL-2 as an important and well defined functional endpoint of ligation of CD3 and CD28, recent research shows that multiple independent signals are initiated upon CD28 ligation, including those involved in T cell metabolism, proliferation, survival, and tolerance. Moreover, there are several distinct members of the CD28 family, including those which can inhibit activation such as CTLA-4 and PD-1.Citation4 Determining whether mild heating differentially affects signaling pathways of these various CD28 family members will be necessary for achieving a more complete understanding of how the physiological microenvironment can impact lymphocyte function.
Finally, the possibility that a sustained increase in temperature could reduce the activation threshold of T lymphocytes may help to explain multiple studies that demonstrate a significant survival benefit associated with fever following infection in multiple vertebrates, including fish, amphibians, reptiles and mammalsCitation12 (e.g., including humans).Citation13 However, it is very important to point out that hyperthermia (as used here) differs in several important ways from an actual fever and that further study is needed to establish how activation thresholds are influenced during actual infections or tissue damage in which inflammatory signals may alter the signals that lymphocytes and other immune cells receive. Further study may lead to new strategies by which mild heating could be used therapeutically to improve pathologies, such as immunosenescence, that are linked to deficiencies in CD28 expression.Citation64
Methods
Cells, reagents and antibodies
Jurkat T cells (Clone E6–1) and A20 B cell line (ATCC) were maintained in RPMI1640 with 2 mM L-glutamine, 1.5 g/L sodium bicarbonate, 4.5 g/L glucose, 10 mM HEPES, and 10 mM sodium pyruvate, 10% FBS at 37°C with 5% CO2. Human CD4+ T cells were purified from peripheral blood of healthy donors and murine CD4+ T cells were purified from the spleens of CD28−/− mice, and DO11.10 transgenic mice (Jackson Labs) by negative selection using Macs isolation kit following manufacturer's instructions (Miltenyi Biotech). All procedures using mice were done with IACUC approval. MβCD (Sigma Aldrich) and Latrunculin A (Molecular Probes) were used as described. FITC-conjugated CTxB (Sigma Aldrich), anti-CD3 (Zymed), anti-CD28, anti-spectrin αII, and anti-ankyrin G (Santa Cruz Biotechnology) Alexa Fluor 568 and 488 - F(ab')2 goat anti-mouse IgG (Molecular Probes), FITC-conjugated anti-CD3, PE-conjugated anti-CD4 (BD PharMingen), PE-conjugated anti-CD28 and anti-CD86 (eBioscience) were used for immunofluorescent staining and flow cytometry.
In vitro heating protocol
Various human and murine CD4+ T cells were incubated at 37°C or 39.5°C by placing culture flasks in incubators maintained at the appropriate temperature with 5% CO2. Incubator temperatures were calibrated and accuracy routinely maintained by use of a digital thermometer (resolution = 0.1°C; accuracy ± 0.3°C). Specific incubation times are described in detail in supplemental materials and in figure legends.
In vivo heating protocol
Whole body hyperthermia (WBH) was administered to mice to raise core body temperature to 39.5°C. Mice were injected i.p. with 1.0mL sterile, non-pyrogenic 0.9% saline before WBH to prevent dehydration. Mice were then placed in preheated cages without access to water (to circumvent the possibility of water-induced cooling) and transferred to an environmental chamber (Memmert model BE500, East Troy, WI), the temperature of which is adjusted to maintain the animals average core body temperature at 39.5 – 40°C for 6 hours. Body temperatures were monitored using a microchip transponder (BMDS, Seaford, DE) implanted subcutaneously into the dorsal thoracic area one week prior to WBH.
IL-2 ELISA assay
For pre-activation thermal treatments, Jurkat T cells, CD4+ T cells isolated from HPB, or CD4+ T cells isolated from CD28−/− mice were incubated at 37°C or 39.5°C for times indicated up to 6 hours then activated by plate-bound anti-CD3 Ab with or without soluble anti-CD28 Ab for 24 hrs at 37°C. Supernatant was collected and IL-2 cytokine was measured by ELISA as described above. For cyclohexamide treatment, cyclohexamide (10 µM) was added to the culture media for the duration of the 6 hour pre-activation thermal treatment. Cyclohexamide was removed by washing 3 times with pre-warmed media before cells were stimulated and IL-2 cytokine was measured by ELISA. CD4+ T cells (5 × 105) from the spleens of DO11.10 transgenic mice were incubated as above and activated by (5 × 105) A20 cells which had been pulsed for 1 hour at 37°C with 2 µg/ml OVA323 peptide. After 24 hours at 37°C IL-2 cytokine was measured in the supernatant as above. For Latrunculin A (LA) treatment, LA was added to the culture media before and during the 6-hour pre-activation thermal treatment or for the final hour of the thermal incubation period. LA was then removed by washing the cells 3 times with pre-warmed media before cells were stimulated and IL-2 cytokine was measured by ELISA.
RNA isolation and RT-PCR/qRT-PCR assays
After heat treatment, Jurkat T cells were stimulated with soluble anti-CD3 Ab (0.1 µg/ml) alone or in combination with soluble anti-CD28 Ab (0.1 µg/ml) for various time periods. Total RNA was isolated with an RNA isolation kit according to the manufacturer's instructions (Qiagen). Reverse transcriptase polymerase chain reaction (RT-PCR) was performed as follows: 1 µg RNA was incubated with 500 ng poly(dT) and a mixture of dNTPs at 65°C for 10 min. Samples were then mixed with 40 U RNaseOUT, 200 U Superscript II reverse transcriptase, 0.02 mmol DTT (Invitrogen). First-strand cDNA synthesis reactions were incubated at 42°C for 50 min and inactivated at 70°C for 10 min. Samples were stored at −80°C until use. PCR was carried out in 1× PCR buffer, 75 µmol MgCl2, dNTP mix, 10 pmol of each specific primer, 1 U Taq polymerase (Invitrogen), 5% DMSO and 2 µl cDNA with the following specifications: 35 cycles of denaturation at 94°C for 5 min, annealing at 60°C for 1 min, and elongation at 72°C for 1 min. The primers (Integrated DNA Technologies Inc.) used were IL-2 sense: 5′-ATG TAC AGG ATG CAA CTC TCT T-3′; IL-2 anti-sense: 5′-GTC AGT GTT GAG ATG ATG CTT TGA C-3′; GAPDH sense: 5′-CCA CCC ATG GCA AAT TCC ATG GCA-3′; GAPDH anti-sense: 5′-TCT AGA CGG CAG GTC AGG TCC ACC-3′. PCR products were separated in a 2% agarose gel in 40 mM Tris, pH 8.1 containing 0.5 µg/ml ethidium bromide and visualized using the BioDoc-It™ system (UVP Inc.).
Quantitative RT-PCR (qPCR) using Fast Start Universal SYBR Green Master Mix (Invitrogen) was used to quantify IL-2 mRNA expression, as well as expression of the housekeeping gene GAPDH. Primer sequences were as follows: IL-2 sense: 5′-GAA TGG AAT TAA TAA TTA CAA GAA TCC C-3′ and IL-2 antisense: 5′-GAC ACT GAA GAT GTT TCA GTT CTG T-3′; GAPDH sense: 5′-GTT CGA CAG TCA GCC GCA TC-3′ and GAPDH antisense: 5′-GGA ATT TGC CAT GGG TGG A-3; (Molecular Probes). Each qPCR reaction was carried out in a total volume of 25 µl containing 12.5 µl Fast Start Universal SYBR Green Master Mix (Invitrogen), and the gene-specific primer set. One µg cDNA was added to each reaction well. qRT-PCR was performed in duplicate using a 7900HT qPCR thermocycler (Applied Biosystems). The thermal profile consisted of 1 cycle of 90°C for 10 minutes followed by 40 cycles of 95°C for 15 seconds and 60°C for 60 seconds and a dissociation step of 95°C for 15 seconds and 60°C for 15 seconds. This was followed by a final hold at 25°C. 7900 HT optics was programmed to collect the fluorescence signal from the 60°C plateau. Threshold cycle values were determined from the exponential phase of amplification using SDS2.3 software (Applied Biosystems).
Fluorescence Anisotropy Measurements
T cell membrane fluidity was monitored by measuring the steady-state fluorescence anisotropy of TMA-DPH (Molecular Probes) following previous recommendations. Citation21,22
Jurkat T cells were suspended at a concentration of 1·105cells/ml of PBS. A cuvette containing 3ml of cell suspension was incubated at the desired temperature. After the cell suspension reached thermal equilibrium, 5 µl of 1·10−3 M TMA-DPH was added to the cuvette. The measurements were performed using a PTI spectrofluorometer equipped with emission and excitation polarizers, as well as a thermally regulated sample chamber. The TMA-DPH anisotropy was calculated from its polarization value obtained with the excitation and emission wavelengths of 360 and 430, respectively.
When conducting these measurements in stimulated T cells, the cells were incubated in the presence of antibodies for 6 hours prior to the measurements. However, temperature and EtOH concentration were altered immediately before the measurements.
Intercellular Immunofluorescent Staining
After treating the cells as indicated for 6 hours, they were adhered to Alcian blue coated coverslips for 10 min, fixed in 2% formalin for 15 min. For internal staining, cells were fixed, permeabilized with 0.2% Triton-X 100 for 5–10 min and blocked with 4% BSA for 1 hr. Primary antibodies were applied for 1 hour, followed by 30 minutes with the secondary antibodies. For GM1 staining, adhered cells were incubated with 5µg/ml FITC-CTxB for 30 min at room temperature and mounted onto slides with mounting media containing DAPI (Vector laboratories). Coverslips were then mounted onto slides with mounting media containing DAPI (Vector laboratories). The slides were examined using a Zeiss Axiosktop2 microscope, and the images were acquired using the manufacture's software.
ImageStream data acquisition and analysis
Splenocytes were isolated from C57BL/6 mice immediately following WBH, fixed with 2% PFA, and stained with FITC-CTxB. Cell images were acquired using an ImageStream flow cytometer (Amnis, Seattle, WA) by excitation with a 488-nm laser, and a time delay integration (TDI) CCD camera. Ten,000 images were analyzed using ImageStream Data Exploration and Analysis Software (IDEAS). Spectral compensation was digitally performed on a pixel-by-pixel basis prior to data analysis. In-focus cells were evaluated after gating on live, single cells based on an aspect ratio near 1 and a low area of the bright field. Cells were gated by aspect ratio and area of the bright field, gated on CD4+ T cells, and bright detail intensity of FITC-CTxB staining was used to measure lipid raft aggregation.
Lymph Node Imprints
Freshly isolated lymph nodes from BALB/c mice were removed and imprinted on coverslips. Coverslips were fixed, permeabilized and immunofluorescently stained for GM1 and ankyrin as described above. The slides were examined using a Zeiss Axiosktop2 microscope, and the images were acquired using the manufacture's software.
Statistical analysis
Results are expressed as mean and standard deviation. Student's two-tailed t test was used for comparing experimental groups, unless otherwise stated, with p value < 0.05 considered significant.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were reported.
Acknowledgments
The authors thank Drs. Scott Abrams, Michelle Appenheimer, Bonnie Hylander, Michael Nemeth, and Kathleen Kokolus for their helpful comments on the data and manuscript and Cheryl H. Rozanski for her assistance in breeding the CD28−/− mice. The authors also thank Drs. Sharon Evans and John Subjeck for many helpful discussions regarding this research.
Funding
This research was supported by research grants from the National Cancer Institute PO1 CA094045 and RO1 CA071599, and in part, by the NCI Cancer Center Support Grant to the Roswell Park Cancer Institute CA016056.
References
- Schwartz RH. Costimulation of T lymphocytes: the role of CD28, CTLA-4, and B7/BB1 in interleukin-2 production and immunotherapy. Cell 1992; 71: 1065-8; PMID:1335362; http://dx.doi.org/10.1016/S0092-8674(05)80055-8
- June CH, Bluestone JA, Nadler LM, Thompson CB. The B7 and CD28 receptor families. Immunol Today 1994; 15: 321-31; PMID:7522010
- Dustin ML, Cooper JA. The immunological synapse and the actin cytoskeleton: molecular hardware for T cell signaling. Nat Immunol 2000; 1: 23-9; PMID:10881170
- Riley JL, June CH. The CD28 family: a T-cell rheostat for therapeutic control of T-cell activation. Blood 2005; 105: 13-21; PMID:15353480; http://dx.doi.org/10.1182/blood-2004-04-1596
- Tavano R, Gri G, Molon B, Marinari B, Rudd CE, Tuosto L, Viola A. CD28 and lipid rafts coordinate recruitment of Lck to the immunological synapse of human T lymphocytes. J Immunol 2004; 173: 5392-7; PMID:15494485
- Schwartz RH. T cell anergy. Annu Rev Immunol 2003; 21: 305-34; PMID:12471050
- LeMay LG, Otterness IG, Vander AJ, Kluger MJ. In vivo evidence that the rise in plasma IL 6 following injection of a fever-inducing dose of LPS is mediated by IL 1 beta. Cytokine 1990; 2: 199-204; PMID:2104223; http://dx.doi.org/10.1016/1043-4666(90)90016-M
- Refinetti R, Menaker M. The circadian rhythm of body temperature. Physiol Behav 1992; 51: 613-37; PMID:1523238
- Hardy J, Bard P. Chapter 56. In: VB Mountcastle, editors. Medical Physiology, 1992; pp. 1305-42
- Hanson DF. Fever and the immune response. The effects of physiological temperatures on primary murine splenic T-cell responses in vitro. J Immunol 1993; 151: 436-48; PMID:8326136
- Guyton AC. Chapter 73. In: Elsevier, editors. Textbook of Medical Physiology, 2006
- Kluger MJ. Fever. Pediatrics 1980; 66: 720-4; PMID:7432877
- Plaisance KI, Kudaravalli S, Wasserman SS, Levine MM, Mackowiak PA. Effect of antipyretic therapy on the duration of illness in experimental influenza A, Shigella sonnei, and Rickettsia rickettsii infections. Pharmacotherapy 2000; 20: 1417-22; PMID:11130213
- Kluger MJ. Fever: role of pyrogens and cryogens. Physiol Rev 1991; 71: 93-127; PMID:1986393
- Hasday JD, Fairchild KD, Shanholtz C. The role of fever in the infected host. Microbes and infection / Institut Pasteur 2000; 2: 1891-904; PMID:11165933
- Smith JB, Knowlton RP, Agarwal SS. Human lymphocyte responses are enhanced by culture at 40 degrees C. J Immunol 1978; 121: 691-4; PMID:150449
- Roberts NJ. Impact of temperature elevation on immunologic defenses. Rev Infect Dis 1991; 13: 462-72; PMID:1866550; http://dx.doi.org/10.1093/clinids/13.3.462
- Meinander A, Söderström TS, Kaunisto A, Poukkula M, Sistonen L, Eriksson JE. (2007) Fever-like hyperthermia controls T lymphocyte persistence by inducing degradation of cellular FLIPshort. J Immunol 2007; 178: 3944-53; PMID:17339495; http://dx.doi.org/10.4049/jimmunol.178.6.3944
- Mace TA, Zhong L, Kilpatrick C, Zynda E, Lee CT, Capitano M, Minderman H, Repasky EA. Differentiation of CD8+ T cells into effector cells is enhanced by physiological range hyperthermia. J Leukoc Biol 2011; 90: 951-62; PMID:21873456; http://dx.doi.org/10.1189/jlb.0511229
- Mace TA, Zhong L, Kokolus KM, Repasky EA. Effector CD8+ T cell IFN-γ production and cytotoxicity are enhanced by mild hyperthermia. Int J Hyperthermia 2012; 28: 9-18; PMID:22235780; http://dx.doi.org/10.3109/02656736.2011.616182
- Kuhry J-G, Duportail G, Bronner C, Laustriat G. Plasma membrane fluidity measurements on whole living cells by fluorescence anisotropy of trimethylammoniumdiphenylhexatriene. Biochim Biophys Acta 1985; 845: 60-7; PMID:3978130; http://dx.doi.org/10.1016/0167-4889(85)90055-2
- Ferretti G, Offidani AM, Simonetti O, Valentino M, Curatola G, Bossi G. Changes in Membrane Properties of Erythrocytes and Polymorphonuclear Cells in Psoriasis. Biochem Med Metab B 1989; 41: 132-8; http://dx.doi.org/10.1016/0885-4505(89)90018-2
- Lin J, Miller MJ, Shaw AS. The c-SMAC: Sorting it all out (or in). J Cell Biol 2005; 170: 177-82; PMID:16009722; http://dx.doi.org/10.1083/jcb.200503032
- Shahinian A, Pfeiffer K, Lee KP, Kundig TM, Kishihara K, Wakeham A, Kawai K, Ohashi PS, Thompson CB, Mak TW. Differential Costimulatory Requirements in CD28-Deficient Mice. Science 1993; 261: 609-12; PMID:7688139; http://dx.doi.org/10.1126/science.7688139
- Abraham RT, Weiss A. Jurkat T cells and development of the T-cell receptor signalling paradigm. Nat Rev Immunol 2004; 4: 301-8; PMID:15057788; http://dx.doi.org/10.1038/nri1330
- Murphy KM, Heimberger AB, Loh DY. Induction by antigen of intrathymic apoptosis of CD4+CD8+TCRlo thymocytes in vivo. Science 1990; 250: 1720-3; PMID:2125367; http://dx.doi.org/10.1126/science.2125367
- Vigh L, Los DA, Horvath I, Murata N. The primary signal in the biological perception of temperature: Pd-catalyzaed hydrogenation of membrane lipids stimulated the expression of the desA gene in Synechocystis PCC6803. Proc Natl Acad Sci USA 1993; 90: 9090-4; PMID:8415659; http://dx.doi.org/10.1073/pnas.90.19.9090
- Carratu L, Franceschelli S, Pardini CL, Kobayashi GS, Horvath I, Vigh L, Maresca B. Membrane lipid perturbation modifies the setpoint of the temperature of heat shock response in yeast. Proc Natl Acad Sci USA 1996; 93: 3870-5; PMID:8632982; http://dx.doi.org/10.1073/pnas.93.9.3870
- Ibolya Horváth, Attila Glatz, Viktória Varvasovszki, Zsolt Török, Tibor Páli, Gábor Balogh, Eszter Kovács, Levente Nádasdi, Sándor Benkö, Ferenc Joó, et al. Membrane physical state controls the signaling mecahismof the heat shock response in synechocysistics pcc6803: identification of hsp17 as a fluidity gene. Proc Natl Acad Sci USA 1998; 95: 3513-8; PMID:9520397; http://dx.doi.org/10.1073/pnas.95.7.3513
- Balogh G, Horvath I, Nagy E, Hoyk Z, Benko S, Bensaude O, Vígh L. The hyperfluidization of mammalian cell membranes acts as a signal to initiate the heat shock protein response. FEBS Journal 2005; 272: 6077-86; PMID:16302971; http://dx.doi.org/10.1111/j.1742-4658.2005.04999.x
- Shigapova N, Torok Z, Balogh G, Goloubinoff P, Vigh L, Horváth I. Membrane fluidization triggers membrane remodeling which affects the thermotolerance in escherichia coli. Biochem Biophys Res Commun 2005; 328: 1216-23; PMID:15708006; http://dx.doi.org/10.1016/j.bbrc.2005.01.081
- Nagy E, Balogi Z, Gombos I, Akerfelt M, Björkbom A, Balogh G, Török Z, Maslyanko A, Fiszer-Kierzkowska A, Lisowska K, et al. Hyperfluidization-coupled membrane microdomain reorganization is linked to activation of the heat shock response in a murine melanoma cell line. Proc Natl Acad Sci USA 2007; 104: 7945-50; PMID:17470815; http://dx.doi.org/10.1073/pnas.0702557104
- Balogh G, Péter M, Liebisch G, Horváth I, Török Z, Nagy E, Maslyanko A, Benko S, Schmitz G, Harwood JL, et al. Lipidomics reveals membrane lipid remodelling and release of potential lipid mediators during early stress responses in a murine melanoma cell line. Biochim Biophys Acta 2010; 1801: 1036-47; PMID:20430110; http://dx.doi.org/10.1016/j.bbalip.2010.04.011
- Calder PC, Yaqoob D, Harvey DJ, Watts A, Newsholme EA. Incorporation of fatty acid by concanavalin A-stimulated lymphocytes and the effect on fatty acid composition and membrane fluidity. Biochem J 1994; 300: 509-18; PMID:8002957
- Kovacs B, Parry RV, Ma Z, Fan E, Shivers DK, Freiberg BA, Thomas AK, Rutherford R, Rumbley CA, Riley JL, et al. Ligation of CD28 by its natural ligand CD86 in the absence of TCR stimulation induces lipid raft polarization in human CD4 T cells. J Immunol 2005; 175: 7848-54; PMID:16339520; http://dx.doi.org/10.4049/jimmunol.175.12.7848
- Brameshuber M, Weghuber J, Ruprecht V, Gombos I, Horváth I, Vigh L, Eckerstorfer P, Kiss E, Stockinger H, Schütz GJ. Imaging of mobile long-lived nanoplatforms in the live cell plasma membrane. J Biol Chem. 2010; 285: 41765-71; PMID:20966075; http://dx.doi.org/10.1074/jbc.M110.182121
- Garcia-Saez AJ, Chiantia S, Schwille P. Effect of line tension on the lateral organization of lipid membranes. J Biol Chem 2007; 282: 33537-44; PMID:17848582; http://dx.doi.org/10.1074/jbc.M706162200
- Kuzmin PI, Akimov SA, Chizmadzhev YA, Zimmerberg J, Cohen FS. Line tension and interaction energies of membrane rafts calculated from lipid splay and tilt. Biophys J 2005; 88: 1120-33; PMID:15542550; http://dx.doi.org/10.1529/biophysj.104.048223
- Xavier R, Brennan T, Li Q, McCormack C, Seed B. Membrane compartmentation is required for efficient T cell activation. Immunity 1998; 8: 723-32.; PMID:9655486; http://dx.doi.org/10.1016/S1074-7613(00)80577-4
- Kabouridis PS, Janzen J, Magee AL, Ley SC. Cholesterol depletion disrupts lipid rafts and modulates the activity of multiple signaling pathways in T lymphocytes. Eur J Immunol 2000; 30: 954-63; PMID:10741414; http://dx.doi.org/10.1002/1521-4141(200003)30:3%3c954::AID-IMMU954%3e3.0.CO;2-Y
- Bennett V, Healy J. Membrane domains based on ankyrin and spectrin associated with cell–cell interactions. Cold Spring Harb Perspect Biol 2009; 1: 1-17; http://dx.doi.org/10.1101/cshperspect.a003012
- Fujiwara T, Ritchie K, Murakoshi H, Jacobson K, Kusumi A. Phospholipids undergo hop diffusion in compartmentalized cell membrane. J Cell Biol 2002; 157: 1071-81; PMID:12058021; http://dx.doi.org/10.1083/jcb.200202050
- Saxton MJ. The spectrin network as a barrier to lateral diffusion in erythrocytes. Biophys J 1989; 55: 21-8; PMID:2930822; http://dx.doi.org/10.1016/S0006-3495(89)82776-6
- Pradhan D, Morrow JS. The spectrin-ankyrin skeleton controls CD45 surface display and interleukin-2 production. Immunity 2002; 17: 303-15; PMID:12354383; http://dx.doi.org/10.1016/S1074-7613(02)00396-5
- Fuller CL, Braciale VL, Samelson LE. All roads lead to actin: the intimate relationship between TCR signaling and the cytoskeleton. Immunol Rev 2003; 191: 220-36; PMID:12614363; http://dx.doi.org/10.1034/j.1600-065X.2003.00004.x
- Pritchard MT, Ostberg JR, Evans SS, Burd R, Kraybill W, Bull JM, Repasky EA. Protocols for simulating the thermal component of fever: preclinical and clinical experience. Methods 2004; 32: 54-62; PMID:14624878; http://dx.doi.org/10.1016/S1046-2023(03)00187-7
- Wang WC, Goldman LM, Schleider DM, Appenheimer MM, Subjeck JR, Repasky EA, Evans SS. Fever-range hyperthermia enhances L-selectin-dependent adhesion of lymphocytes to vascular endothelium. J Immunol 1998; 160: 961-9; PMID:9551935
- Janes P W, Ley SC, Magee AI, Kabouridis PS. The role of lipid rafts in T cell antigen receptor (TCR) signalling. Semin Immunol 2000; 12: 23-34; PMID:10723795
- He HT, Lellouch A, Marguet D. Lipid rafts and the initiation of T cell receptor signaling. Semin Immunol 2005; 17: 23-33; PMID:15582486
- Janes PW, Ley SC, Magee AI. Aggregation of lipid rafts accompanies signaling via the T cell antigen receptor. J Cell Biol 1999; 147: 447-61; PMID:10525547
- Grimm MJ, Zynda ER, Repasky EA. Temperature Matters: Cellular Targets of Hyperthermia in Cancer Biology and Immunology. In Prokaryotic and Eurkaryotic Heat Shock Proteins in Infections Disease. Dordrecht: Springer, 2010; 4:267-306.
- Schade AE, Levine AD. Lipid raft heterogeneity in human peripheral blood T lymphoblasts: a mechanism for regulating the initiation of TCR signal transduction. J Immunol 2002; 168: 2233-9; PMID:11859110
- Tuosto L, Parolini I, Schröder S, Sargiacomo M, Lanzavecchia A, Viola A. Organization of plasma membrane functional rafts upon T cell activation. Eur J Immunol 2001; 31: 345-9; PMID:11180097
- Lillemeier B, Mörtelmaier M, Forstner M, Huppa J, Groves J, Davis M. TCR and Lat are expressed on separate protein islands on T cell membranes and concatenate during activation Nature Immunology (2009); 11 (1), 90-96; PMID:20010844; http://dx.doi.org/10.1038/ni.1832
- Millan J, Qaidi M, Alonso MA. Segregation of co-stimulatory components into specific T cell surface lipid rafts. Eur J Immunol 2001; 31: 467-73; PMID:11180111
- Ostberg JR, Dayanc BE, Yuan M, Oflazoglu E, Repasky EA. Enhancement of natural killer (NK) cell cytotoxicity by fever-range thermal stress is dependent on NKG2D function and is associated with plasma membrane NKG2D clustering and increased expression of MICA on target cells. J Leukoc Biol 2007; 82: 1322-31; PMID:17711975
- Wang XY, Ostberg JR, Repasky EA. Effect of fever-like whole-body hyperthermia on lymphocyte spectrin distribution, protein kinase C activity, and uropod formation. J Immunol 1999; 162: 3378-87; PMID:10092792
- Hughes CS, Repasky EA, Bankert RB, Johnson RJ, Subjeck JR. Effects of hyperthermia on spectrin expression patterns of murine lymphocytes. Radiat Res 1987; 112: 116-23; PMID:3659292
- Valitutti S, Dessing K, Aktories K, Gallati H, Lanzavecchia A. Sustained signaling leading to T cell activation results from prolonged T cell receptor occupancy: role of T cell actin cytoskeleton. J Exp Med 1995; 181: 577-84; PMID:7836913
- Rozdzial MM, Malissen B, and Finkel TH. Tyrosin-phosphorylated T cell receptor ζ chain associates with the actin cytoskeleton upon activation of mature T lymphocytes. Immunity 1995; 3: 623-33; PMID:7584152; http://dx.doi.org/10.1016/1074-7613(95)90133-7
- Rodgers W, Zavzavadjian J. Glycolipid-enriched membrane domains are assembled into membrane patches by associating with the actin cytoskeleton. Exp Cell Res 2001; 267: 173-83; PMID:11426936
- Villalba M, Bi K, Rodriguez F, Tanaka Y, Shoenberger S, Altman A. Vav1/Rac-dependent actin cytoskeleton reorganization is required for lipid raft clustering in T cells. J Cell Biol 2001; 155: 331-8; PMID:11684704
- Kumari S, Curado S, Mayya V, Dustin ML. T cell antigen receptor activation and actin cytoskeleton remodeling. Biochim Biophys Acta. 2014; 1838: 546-56; PMID:23680625; http://dx.doi.org/10.1016/j.bbamem.2013.05.004
- Topp MS, Riddell SR, Akatsuka Y, Jensen MC, Blattman JN, Greenberg PD. Restoration of CD28 expression in CD28- CD8+ memory effector T cells reconstitutes antigen-induced IL-2 production. J Exp Med 2003; 198: 947-55; PMID:12963692; http://dx.doi.org/10.1084/jem.20021288