
Abstract
In response to replication stress ATR signaling through CHK1 controls the intra-S checkpoint and is required for the maintenance of genomic integrity. Homologous recombination (HR) comprises a series of interrelated pathways that function in the repair of DNA double strand breaks and interstrand crosslinks. In addition, HR, with its key player RAD51, provides critical support for the recovery of stalled forks during replication. High levels of RAD51 are regularly found in various cancers, yet little is known about the effect of the increased RAD51 expression on intra-S checkpoint signaling. Here, we describe a role for RAD51 in driving genomic instability caused by impaired replication and intra-S mediated CHK1 signaling by studying an inducible RAD51 overexpression model as well as 10 breast cancer cell lines. We demonstrate that an excess of RAD51 decreases I-Sce-I mediated HR despite formation of more RAD51 foci. Cells with high RAD51 levels display reduced elongation rates and excessive dormant origin firing during undisturbed growth and after damage, likely caused by impaired CHK1 activation. In consequence, the inability of cells with a surplus of RAD51 to properly repair complex DNA damage and to resolve replication stress leads to higher genomic instability and thus drives tumorigenesis.
Abbreviations
| ATR | = | Ataxia Telangiectasia and Rad3 related |
| CHK1 | = | checkpoint serine/threonine kinase |
| DDR | = | DNA damage response |
| DSB | = | DNA double strand break |
| dsDNA | = | double-stranded DNA |
| GFP | = | green fluorescent protein |
| HR | = | homologous recombination |
| ICLs | = | interstrand cross-links |
| MMC | = | mitomycin C; Pon A, ponasterone A |
| RPA | = | relication protein A |
| SSB | = | single strand break |
| ssDNA | = | single-stranded DNA |
Introduction
One of the major cellular processes to prevent genomic instability is the repair of DNA double strand breaks (DSBs) mediated by non-homologous end joining (NHEJ) or homologous recombination (HR). HR is preferentially active during S and G2 phase and there is increasing evidence that HR is primarily responsible for the repair of DSBs during DNA replication to restart stalled and recover broken replication forks.Citation1-3 Because cells are most vulnerable to genomic instability during replication and segregation of chromosomes at mitosis,Citation4 RAD51 as the key player in HR has a fundamental role in controlling genomic stability.Citation5,6 The potential sources of spontaneous genomic instability during S phase are discontinuities at the replication fork, caused primarily by stalling of replication forks, which is provoked for instance by nucleotide depletion, base damage, single strand breaks or cross links. In this regard HR is one of several interrelated pathways that function in the repair of “one-sided” DSBs and interstrand crosslinks (ICLs).Citation7 The diverse functions of HR and its regulation are reflected in the need for complex protein networks. A core of HR protein comprising RPA, RAD51, RAD52, RAD54, RAD51B, C, D, XRCC2 and XRCC3 perform their function together with additional factors, such as proteins like BRCA1, BRCA2, TP53 and the Fanconi anemia proteins A-P. If inactivated by mutation, all of them promote a genomic instable phenotype reflected by an increase of spontaneous DNA damage and resulting in a higher susceptibility to cancers. Although the regulation of HR is not completely understood, a precisely adjusted amount of RAD51 seems to be necessary for cellular maintenance. This can be concluded from (i) the lethality when RAD51 is knocked out,Citation8,9 (ii) the alteration of radiosensitivity by a lowered or higher expression,Citation10,11 and (iii) the elevated expression regularly observed in human tumor cell lines derived from several origins.Citation12 Supporting the idea that a well-balanced HR is necessary to avoid genomic instability, we and others could show a negative effect of RAD51 overexpression on prognosis for tumor patients after combined radio/chemotherapy, as well as after surgery alone.Citation13-15 This clearly demonstrates that prognosis is not determined by increased repair capacity of the irradiation-induced DNA damage after RAD51 overexpression. In line with this, overexpression of RAD51 does not necessarily improve repair capacity in cellular HR models. All scenarios affecting HR were observed, such as a reduction, no change or an increase in HR events in cells with RAD51 overexpression.Citation16–20 Similarly, a reduction in HR was also observable in cells with BRCA2 overexpression.Citation21 However, overexpression of RAD51 restores HR function, if repair proteins like BRCA1, BRCA2 or FANCD2 involved in HR are lost,Citation22-26 with the consequence that genomic stability could also be restored to some extent.Citation3,25,27-30
In response to stalled replication forks or replication stress ATR signaling through CHK1 is the major pathway and the key regulator of the intra-S checkpoint.Citation31-33 CHK1-mediated DNA damage response is required for the maintenance of genomic integrity and it is generally agreed that, upon replication stress, long stretches of single-stranded DNA (ssDNA) arise, which are quickly covered by replication protein A (RPA). RPA-coated ssDNA then recruits the ATR–ATR-interacting protein (ATRIP) complex followed by TopBP1 and the 9–1–1 complex, thus activating the ATR signaling pathway and its downstream phosphorylation targets including CHK1 kinase.Citation34-36 ATR-mediated CHK1 phosphorylation at Ser317 and Ser345 stimulates CHK1 kinase and releases it from chromatin to facilitate the intra-S checkpoint response.Citation37,38 One major function of the intra-S phase checkpoint is the regulation of DNA replication initiation. As a consequence of CHK1 activation, neighboring, dormant origins of replication are activated. At the same time, distant origins are restricted from firing.Citation39 In this way, the cell prioritizes on completion of DNA replication that has already been started. Even in the absence of DNA damage, ATR and CHK1 regulate replication timing and normal S-phase progression, as ATR or Chk1 inhibition, down-regulation as well as inactivation of the CHEK1 gene all cause a slow-down of fork progression and extensive origin firing.Citation3,40-42
CHK1 is also important for the activation of HR, as the phosphorylation of RAD51 or BRCA2 by CHK1 plays a critical role in the recruitment of RAD51 to sites of DNA damage.Citation43,44 Conversely, we have recently been able to show that haploinsufficiency in the PALB2 gene, another RAD51 DNA loading factor is sufficient to diminish the ATR/CHK1 response,Citation45 resulting in pronounced replication defects such as increased origin firing. Thus, HR and intra-S checkpoint are apparently tightly intertwined.
Therefore, we hypothesized that an oversupply of RAD51 should not only affect HR repair in general, but also influence DNA replication and the activation of the ATR-CHK1 signaling pathway. To further elucidate the consequences of high RAD51 expression, we examined HR, DNA replication, intra-S checkpoint and CHK1 kinase activation in an isogenic cellular system with high RAD51 expression as well as in 10 breast cancer cell lines with RAD51 expression ranging from low to high. Here we report that an excess of RAD51 decreases I-Sce-I mediated HR, perturbs DNA replication, and incompletely activates intra-S-phase checkpoint signaling. In consequence, cells display excessive dormant origin firing as well as reduced recruitment of other repair factors, resulting in genomic instability. Our data thus suggest that only an optimized RAD51 activity allows efficient activation of ATR-CHK1 intra-S checkpoint signaling in response to replication stress.
Results
High levels of RAD51 increases genomic instability
We and others have shown that high level expression of RAD51 is an independent negative prognostic marker for cancer patients, which is not attributable to a resistance to tumor therapy.Citation13,14 Despite its role in maintaining genomic integrity, it has been proposed that an aberrant increase in RAD51 expression found in tumor cells contributes to genomic instability.Citation20,25 The same appears to be true for the downregulation of RAD51,Citation46-49 and we therefore hypothesized that only a balanced RAD51 dose ensures error-free HR. To determine the consequences of high RAD51 levels on genomic stability, HR, DNA replication, the intra-S-phase signal cascade and cellular resistance were analyzed in a model system with inducible RAD51 expression and in 10 breast cancer cell lines.
An isogenetic U2OS cell model with ponasterone-inducible RAD51 expression shows a normal level of RAD51 under non-induced conditions and after induction by Pon A an approximately 3-fold ectopic RAD51 expression (). This is comparable to breast cancer cell lines with high RAD51 expression (), without any further increase seen at higher concentrations of Pon A (Fig. S1A). The observed higher expression of RAD51 does not only result in an up-regulation of total RAD51 but also in an increase of RAD51 in the nuclear as well as the chromatin-bound fraction (Fig. S1A). Also ATR expression and CHK1 seem to be affected in the inducible system, showing a slight increase in ATR and a slight decrease in CHK1 in cells with high RAD51 levels (Fig. S1C, D).
Figure 1. High levels of RAD51 increase genomic instability. Protein expression patterns in (A) U2OS cells transfected with a hormone-inducible RAD51 expression vector and (B) breast cancer cell lines. Induction of high RAD51 in U2OS cells was performed by addition of 1 µM Pon A for 24 h. (top) Protein was assessed by RAD51 immunoblotting with β-actin as loading control. (bottom) Quantification of RAD51 expression relative to levels of β-actin. Mean values of at least 3 independent protein extracts are shown. (C) RAD51 foci (left) and γH2AX-foci (right) in untreated U2OS cells with RAD51 overexpression. Exponentially growing cells were fixed 24 h after induction of RAD51 overexpression, immuno-stained for RAD51 or γH2AX, counterstained with DAPI and foci formation was quantified. Columns depict the mean number of RAD51 foci per cell and bars represent the standard error of the mean of at least 3 experiments. Statistical analysis was performed using Student's t-test. (D) Chromatid-type (left) and chromosome-type (right) aberrations in untreated U2OS cells with RAD51 overexpression. Representative examples for both types of chromosome aberrations are shown in the middle. Exponentially growing cells 24 h after induction of RAD51 overexpression were treated with 0.2 µM colcemid for 4 h and collected at metaphase. Giemsa stained chromatid-type or chromosome-type aberrations were scored and expressed as G2- or G1-aberrations per cell. Columns depict data of at least 3 experiments and statistical analysis was performed using Student's t-test.
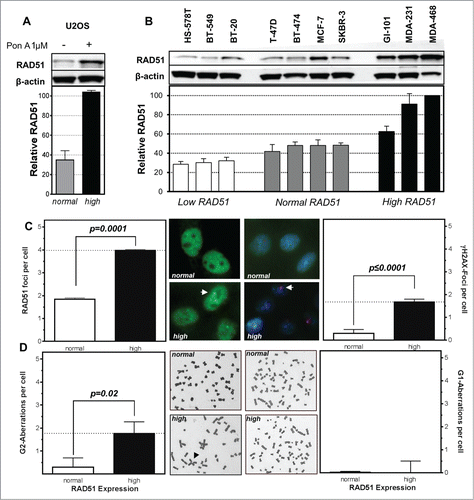
The breast cancer cell lines showed a broad variation in the expression of RAD51 ranging from low (<35% expression compared to the highest expressing cell line MDA-468) and normal (40–50%) to high (>60%) (). Mutations in BRCA1 or BRCA2 seem not to be responsible for the variation in the expression pattern in the 10 breast cancer cell lines as these do not harbor any common BRCA1 or BRCA2 mutations (Table S1). Expression of other relevant HR-related proteins, such as BRCA2, FANCD2 and CHK1 also varied, but according to their own pattern. Only ATR seemed to be more highly expressed in cell lines with moderate or high RAD51 expression (Fig. S1E).
Functionality of RAD51 was tested by immunofluorescence microscopic detection of RAD51 foci. USO2 cells with high RAD51 clearly exhibited RAD51 foci ( left) indicating functional filament formation on ssDNA in the untreated situation. This went along with an increase of spontaneous double-strand breaks (DSB), supported by a 2-fold increase in RAD51 foci per cell (p = 0.0001) and a 6 fold higher number of γH2AX foci per cell (P ≤ 0.0001; right). This view was further strengthened by analysis of U2OS cells with high levels of RAD51 in metaphase, where a 6-fold increase in the number chromatid-type aberrations (p = 0.02) ( left) was observed. In contrast, there was no increase in the number of chromosome-type aberrations ( right). Both observations indicate that high RAD51 levels caused increased genomic instability during replication or at later time points in the cell cycle but not in early S or G1 phase.
High levels of RAD51 reduces HR capacity and sensitizes cancer cells to MMC
To test functionality of the RAD51 protein, HR was quantified using a GFP reporter assayCitation50 where a DSB, introduced by the I-SceI endonuclease and repaired via HR, leads to GFP-positive cells. In representative flow cytometry profiles, HR repair events represented a population shown in green, distinguishable from all other repair events shown in red ().
Figure 2. High levels of RAD51 decrease HR-frequency and sensitize cells to MMC. (A) Representative examples for I-SceI-induced recombination frequencies in U2OS cells. Cells were transfected with the I-Sce-I linearized pGC substrate (bottom) and monitored as the number of GFP positive cells by FACS analysis. Transfection without linearized plasmids served as control (top). (B) Relative HR-frequency in U2OS cells with normal and high RAD51 expression after stable (left) or transient (right) transfection of the GFP-reporter. For both experiments a high level of RAD51 was induced for 24 h before transfection and HR-frequencies were calculated 24 h later. Columns depict the percentage of recombination events and bars represent the standard deviation of at least 3 experiments. Statistical analyses were performed using Student's t-test. (C) Relative HR-frequency in breast cancer cell lines with low, normal and high RAD51 expression. Cells were transiently transfected and HR-frequencies were calculated 24 h after transfection. Symbols depict the percentage of recombination events and bars represent the standard deviation of at least 3 experiments. Statistical analyses were performed using Student's t-test. (D) Influence of RAD51 protein expression in different breast cancer cell lines on HR frequency. Data were taken from and fitted by linear regression analysis. (E) Cellular sensitivity to MMC in U2OS and (F) breast cancer cell lines. Exponentially growing cells were treated with increasing doses up to 60 nM MMC for 24h (U2OS) or with 1.5 µM (breast cancer cell lines) for 1 h and cellular sensitivity was monitored by colony formation. Data of at least 3 independent experiments were fitted by linear regression analysis (A) or statistical analysis was performed using Student's t-test (B).
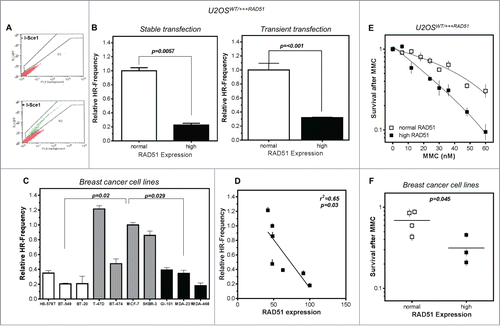
A 4-fold and a 3-fold decrease in the HR frequency after stable integration as well as transient transfection of the GFP-reporter was observed in U2OS cells with high RAD51 expression compared to normal expression, with 0.23 ± 0.04 and 0.32 ± 0.007 compared to 1.00 ± 0.075 and 1.00 ± 0.09 (p = 0.0057; P < 0.001), respectively ( left and right). In breast cancer cell lines with low as well as high RAD51 expression, a reduction in the HR frequency was observed compared to cells with intermediate RAD51 expression (p = 0.02; p = 0.029) (). For cell lines with intermediate and high RAD51 expression a clear correlation of HR frequency and RAD51 expression was observed (r2 = 0.65; p = 0.03) (). This reduction in HR in cells with high RAD51 expression was not influenced by differences in cell cycle distribution between the cell lines (Fig. S2A) and strongly suggests that an excess of RAD51 increases genomic instability by suppression of HR. Based on this observation we defined the 4 cell lines with an intermediate RAD51 expression as the cell lines with normal RAD51 expression.
The consequences of loss of RAD51 on HR frequency are extensively described in the literature.Citation11 Therefore we focused further experiments on long-term effects of high RAD51 expression upon genotoxic stress. Cell survival was examined following exposure to MMC. U2OS cells induced to over-express RAD51 showed a higher sensitivity to MMC, accompanied by an increase of cells in the G2 phase relative to non-induced cells with normal RAD51 expression (; Fig. S2B). Breast cancer cell lines with differences in RAD51 expression behaved similarly, showing the same trend in sensitization in cells with high compared to normal RAD51 expression, with an up to 2-fold increase in cellular sensitivity (p = 0.045; and r2 = 0.48; p = 0.08; Fig. S2C).
At replication forks ICL repair takes place by overlapping functions of the HR and the FA pathway.Citation51 To exclude that the dramatic increase in sensitivity against MMC in cells with high RAD51 expression could be due to a failure in the activation of the FA pathway, FANCD2 monoubiquitination as a marker for the activated FA pathway was measured and expressed as L/S ratio (activated/non-activated) of FANCD2.Citation52 Cells with high RAD51 expression were able to activate FANCD2, but to a much lower extent compared to cells with normal RAD51 expression. This was apparent in the inducible U2OS model (Fig. S2D) and the breast cancer cell line panel (). On average, MMC treatment increased activation of FANCD2 about 15-fold in U2OS cells with normal RAD51 expression, but only less than 2-fold in U2OS cells with high RAD51 expression, respectively. This reduced activation of FANCD2 was, however, not observable for breast cancer cell lines with high RAD51 expression. Thus, the increased ICL sensitivity of cells with high levels of RAD51 could not fully be attributed to impaired FA pathway signaling at replication forks.
Perturbance of DNA replication in cells with high levels of RAD51
RAD51 has been reported to participate in the reactivation of stalled replication forks.Citation3,29,30 Therefore, the DNA fiber assay was used to explore the effect of high RAD51 levels on replication processes. The labeling protocol and representative examples of replication tracts are shown in ). The mean replication fork speed in U2OS cells with normal RAD51 expression was 0.82 ± 0.01 kb/min (). After induction of high RAD51 expression by addition of 1 µM Pon A, replication tracts were substantially shortened to 0.69 ± 0.01 kb/min (P < 0.0001; ). In addition, 11 independent U2OS cell clones that varied in RAD51 levels upon induction of ectopic RAD51 expression ranging from normal to high (, upper chart) were analyzed. Replication fork speed declined with increasing level of RAD51 with an r2 = 0.78 for regression analysis of tract length against logarithm of RAD51 expression (p = 0.003), indicating that already a small excess in RAD51 expression perturbs normal replication. This observation is also supported by the analysis of fork symmetry (, white arrows), showing a decrease of IdU/CldU ratio from 1.0 to 0.88 (Fig. S2F), indicative of a statistically significant increase of asymmetric replication tracts in cells with high RAD51 expression compared to controls (p = 0.02; ).
Figure 3. Perturbance of DNA replication processes in U2OS cells with high levels of RAD51. (A) Schematic of single DNA fiber analysis. Red tracts CldU; green tracts IdU. (B) Examples of various types of tracts like symmetric and asymmetric replication tracts in cells with normal (top) or high (bottom) RAD51 expression. (C) Replication fork speed in untreated U2OS cells with normal and high RAD51 expression. Cells were sequentially pulse-labeled with CldU followed by IdU and replication fork speed (kb/min) was detected by immunofluorescence with specific antibodies. Statistical analysis was performed using Student's t-test. (D) Percentage of asymmetrical replication tracts in cells with normal and high RAD51 expression. IdU/CldU length ratios from at least 1,400 replication tracts of at least 3 independent experiments were calculated by dividing IdU/CldU tracts and statistical analysis was performed using Student's t-test (right). (E) Influence of RAD51 protein expression in different U2OS strains on replication fork speed. Induction of high RAD51 expression was performed by addition of 1 µM Pon A for 24 hrs. After removal RAD51 expression was assessed over 120 days post Pon A treatment by Western analysis in total cell lysates from exponentially growing cells; β-actin was used as a loading control (top). Lane 11 represents RAD51 expression 24 hrs after induction. In parallel, replication fork speed was detected by immunofluorescence with specific antibodies at the same time points and plotted against relative RAD51 expression.. Data were fitted by linear regression analysis. (F) Examples and (G) relative frequency of 1st pulse origin firing in untreated U2OS cells with normal and high RAD51 expression. Cells were sequentially pulse-labeled with CldU followed by IdU, origins detected by immunofluorescence with specific antibodies and the percentage of 1st origins relative to all replication structures was calculated. Statistical analysis of at least 3 different experiments was performed using Student's t-test. (H) Quantification of ori-to-ori distances in U2OS cells with normal and high RAD51 expression. Length between adjacent origins was measured and expressed as ori-to-ori distances in kb. Data were collected from 3 different experiments and statistical analysis was performed using Student's t-test.
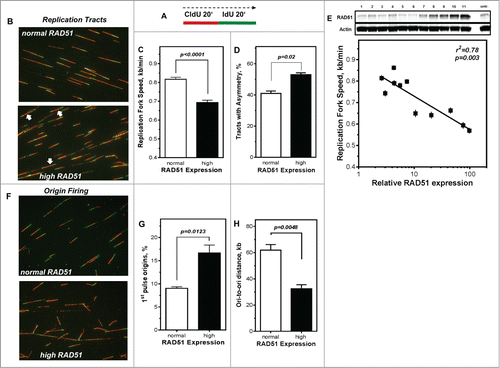
To exclude the possibility that tract length shortening resulted from degradation of the newly synthesized strand at stalled replication forks that has been observed for other HR-mutants,Citation25 DNA fiber analysis was utilized to monitor replication perturbation by marking newly synthesized DNA strands just before and after replication stress provoked by the exposure to HU (Fig. S2G). The mean replication fork speed seemed to be maintained in the presence of HU at high levels of RAD51, showing only a small, comparable shortening of tract length for both RAD51 expression levels. Thus, high levels of RAD51 perturbed progression of normal DNA replication but did not interfere with the protection of newly synthesized DNA strands at stalled replication forks.
Replication fork speed often correlates inversely with origin firing.Citation53,54 This was also observable for cells with high RAD51 expression, showing nearly a doubling in the frequency of 1st pulse origins, (17% ± 1.7 compared to 9% ± 1.7 in U2OS cells with normal RAD51 expression, p = 0.0123; ). Also, the mean inter-origin distance is significantly reduced in U2OS cells with high levels of RAD51, showing 33 kb ± 5.3, compared to 62 kb ± 7.3 in cells with normal RAD51 expression (p = 0.0048; ).
Taken together, fiber analysis, in particular the reduced replication fork speed rates together with the strong increase in origin usage, indicates that an increase in RAD51 levels aggravates spontaneous fork stalling and leads to an increased use of dormant origins.
Activation of CHK1 is diminished in cells with high RAD51 and results in reduced fork speed and increased origin firing after DNA damage
We next tested whether changes in intra-S-phase signaling were responsible for the genomic instability, perturbance of DNA replication and higher sensitivity to MMC observed in cells with high RAD51 levels. To this end, we monitored CHK1 expression and activation as well as the impact of various types of DNA damage on the regulation of DNA replication. A small change in CHK1 protein expression was visible in cells with high compared to normal RAD51 expression ( and Fig. S1E). After challenge of non-induced U2OS cell with 1.5 µM MMC, phosphorylation of CHK1 on Ser345 (pCHK1) increased steadily for 24 h ( left). In response to 200 μM hydrogen peroxide, a rapid but transient CHK1 phosphorylation reached its maximum already 15 min after treatment (, right). In contrast, U2OS cells induced to express high RAD51 levels showed only an attenuated phosphorylation of CHK1, corresponding to half of the phosphorylation of non-induced U2OS cells at the same time points (). Also breast cancer cell lines with high RAD51 expression showed a trend to become less phosphorylated at CHK1Ser345 as well as at the autophosporylation site CHK1S296 (right) after MMC treatment (p = 0.27; p = 0.06 left and right), which was nevertheless not statistically significant. Because phosphorylation of CHK1 showed these remarkable differences it was of interest to test whether these could be manifested in the regulation of DNA replication after different types of DNA damage. Exponentially growing U2OS cells were sequentially pulse-labeled with CldU and IdU and DNA damaging agents were added during the second labeling period. Cells were treated with MMS to induce base damage, a low concentration of hydrogen peroxide to induce mainly single strand breaks, MMC to induce interstrand cross-links, a high concentration of hydrogen peroxide to induce DSBs and topotecan to induce one-ended DSBs at replication forks (). Data shown were normalized to untreated controls. All treatments led to a reduction of the replication fork speed in cells with normal RAD51 expression. The smallest effect was observed after MMC treatment (0.93 ± 0.02 kb/min), the strongest after agents inducing DSBs (0.55 ± 0.02 kb/min and 0.54 ± 0.01 for one- and 2-ended DSBs, respectively), base damage and SSB being in between (0.78 ± 0.01 kb/min after MMS and 0.77 ± 0.01 kb/min after low hydrogen peroxide). Remarkably, cells with high RAD51 expression responded much more sensitively, with a significantly stronger reduction in replication fork speed after all damages induced (0.69 ± 0.11 kb/min (P < 0.0001) after MMS, 0.66 ± 0.01 kb/min (P < 0.0001) after low hydrogen peroxide, 0.66 ± 0.13 kb/min (P < 0.0001) after MMC, 0.48 ± 0.08 kb/min (P < 0.0001) after high hydrogen peroxide and 0.44 ± 0.08 kb/min (P < 0.0001) after topotecan) This clearly showed that cells with high RAD51 expression stalled or slowed down DNA replication more severely upon DNA damage.
Figure 4. Activation of CHK1 is diminished in cells with high levels of RAD51, resulting in reduced fork speed and increased dormant origin activation after DNA damage. (A) Activation of CHK1 after DNA damage. Exponentially growing U2OS cells with normal or high RAD51 expression were exposed to 1.5 µM MMC or 200 µM hydrogen peroxide for 1 h, total protein was extracted at the indicated time points, CHK1 and pCHK1 (Ser345) proteins were visualized by immunoblotting. pCHK1 was densitometrically quantified relative to levels of total CHK1 protein and β-actin served as a loading control. (B) Activation of CHK1 after DNA damage in breast cancer cell lines. Exponentially growing cells were exposed to 1.5 µM MMC for 1 h, total protein was extracted at time points indicated, CHK1 and pCHK1 (Ser345) (left) or pCHK1 (S296) (right) proteins were detected by immunoblotting. pCHK1 was quantified relative to levels of total CHK1 protein signals and β-actin served as a loading control. Statistical analysis of at least 3 different protein extracts was performed using Student's t-test. (C) Effect of DNA damage on replication fork speed, (D) 2nd pulse origin firing (left) and 2nd pulse origin firing adjacent to stalled replication forks (right). Cells were sequentially pulse-labeled, DNA damage was induced during the IdU label and fiber structures were detected by immunoflourescence. Damage was induced by treatment with 500 µM MMS, 50 µM or 200 µM hydrogen peroxide, 1.5 µM MMC or 1 µM topotecan. Replication fork speed was expressed as kb/min and origin firing was calculated relative to all replication structures analyzed. Error bars represent the standard error of the mean (SEM), *** corresponds to P < 0.001.
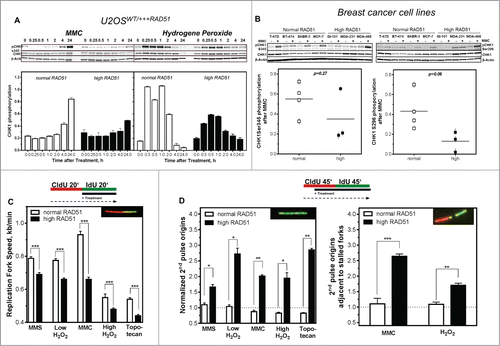
Besides the inhibition of elongation, the CHK1-mediated DNA replication checkpoint is also able to inhibit distant replication factory activation.Citation39 Therefore, we analyzed origin firing during the later IdU pulse, termed 2nd pulse origins, i.e. after DNA damage treatment (). After treatment, U2OS cells with normal RAD51 expression showed only little change in origin firing after base damage and SSB induction (1.01 ± 0.06 and 1.04 ± 0.05, respectively). As expected a strong reduction in origin firing was observed after MMC and the treatments causing DSBs (0.8 ± 0.05, 0.89 ± 0.02 and 0.83 ± 0.03 for MMC, high hydrogen peroxide and topotecan, respectively), reflecting the proper CHK1-mediated intra-S-phase DNA damage response signaling. In contrast, the U2OS cells with high RAD51 expression failed to decrease origin firing after all treatments (1.7 ± 0.08 after MMS, 2.7 ± 0.2 after low hydrogen peroxide, 2.01 ± 0.01 after MMC, 1.95 ± 0.2 after high hydrogen peroxide and 1.96 ± 0.06 after topotecan, (, left), supporting the hypothesis that an excess of RAD51 interferes with intra-S checkpoint signaling and thus prevents cells from blocking origin activation after damage induction. The fact that 2nd pulse origin firing is increased rather than diminished, suggests that high levels of RAD51 cause an extensive activation of dormant origins triggered by excessive replication fork stalling or the inability to reactivate stalled forks after DNA damage. Analysis of 2nd pulse origins in the vicinity of stalled replication forks supported this hypothesis, showing a strong increase after treatment with MMC and low hydrogen peroxide in U2OS cells with high RAD51 expression and, as expected, no effect in U2OS cells with moderate RAD51 expression (, right).
Inhibition of CHK1 does not alter cellular sensitivity in cells with high RAD51 levels
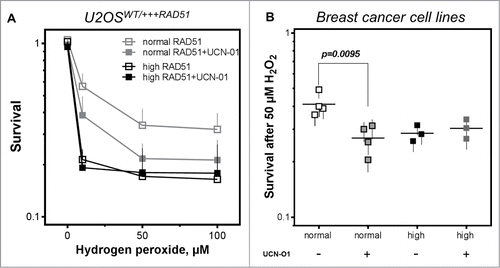
To directly test the hypothesis, that the failure to properly activate CHK1, followed by an altered DNA damage response, is responsible for the higher cellular sensitivity in cells with high RAD51 expression, cellular sensitivity was investigated in the presence of the CHK1 inhibitor UCN-O1. As expected, UCN-O1 treatment sensitized U2OS cells with normal RAD51 expression in the presence of hydrogen peroxide whereas it did not alter the already high sensitivity of cells with high RAD51 expression (, left). This observation is also visible in breast cancer cell lines, showing an increase in sensitivity after UCN-O1 treatment in cells with normal RAD51 expression and no further sensitization in cells with high RAD51 in the presence of CHk1 inhibition (, right) strongly supporting the idea, that an excess of RAD51 impairs the intra-S-phase DNA damage response mediated by CHK1.
Figure 5. Inhibition of CHK1 does not alter cellular sensitivity in cells with high RAD51 levels. (A) Sensitivity to hydrogen peroxide in U2OS cells with high RAD51 levels. Exponential growing cells were treated for 1 h with increasing hydrogen peroxide concentrations in the presence or absence of a 2 h pre-incubation with 100 nM UCN-O1 24 h after induction of high RAD51 expression and sensitivity was analyzed using the colony formation assay. Mean ± SEM of 3 independent experiments is shown. (B) Sensitivity to hydrogen peroxide after inhibition of CHK1 in breast cancer cell lines. Exponential growing cells were treated with 50 µM hydrogen peroxide in the presence or absence of a 2 h pre-incubation with100 nM UCN-O1 and sensitivity was monitored by colony formation. Mean ± SEM of at least 3 independent experiments is shown and statistical analysis was performed using Student's t-test.
Lower number of RPA foci in cells with high levels of RAD51
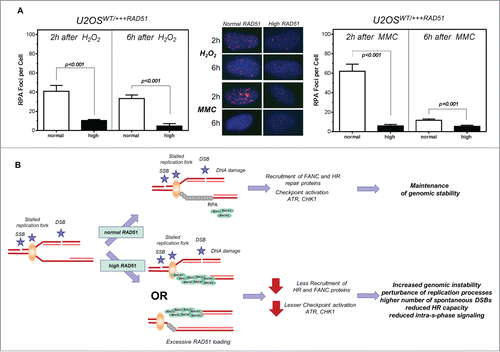
RAD51 and RPA compete and collaborate at replication forksCitation55 and RPA is required for the recruitment of ATR and the ATR-mediated CHK1 activation at sites of DNA damage.Citation34 Hence, RPA foci formation was monitored after treatment with hydrogen peroxide (, left) or MMC (, right) in U2OS cells with normal and high levels of RAD51. A 4–6 -fold decrease in the formation of RPA foci was observed after treatment with hydrogen peroxide or MMC in U2OS cells with high RAD51 expression compared to normal expression, with only 10.4 ± 1.13 and 4.69 ± 2.53 RPA foci per cell after 2 h and 6 h compared to 40.9 ± 6.1 and 33.38 ± 3.37 after hydrogen peroxide and 11.63 ± 0.7 and 5.39 ± 0.65 after 2 h and 6 h compared to 62.0 ± 7.3 and 5.89 ± 1.47 (P < 0.001; P < 0.001, respectively).
Figure 6. Lower number of RPA foci in cells with high levels of RAD51. Model of increased genomic instability driven by high levels of RAD51. (A) RPA foci formation after treatment with hydrogen peroxide (left) or MMC (right) in U2OS cells with high RAD51 levels. Exponential growing cells were treated for 1 h with 200 µM hydrogen peroxide or 1.5 µM MMC and foci formation was monitored 2 h and 6 h after treatment. Columns depict the mean number of RPA foci per cell and error bars represent the standard error of the mean of at least 3 experiments. Statistical analysis was performed using Student's t-test. (B) Model. A replication fork can stall for various reasons, including one-ended and 2- ended DSBs, resulting in single stranded DNA stretches, which are quickly protected by RPA. This leads to the activation of the ATR-mediated signal cascade and the recruitment of DNA repair proteins. Importantly, in the presence of an oversupply of RAD51 protein, RAD51 may be recruited prematurely. Therefore, the S-phase checkpoint signal cascade is disturbed; stalled forks are not reactivated appropriately, resulting in excessive dormant origin loading followed by higher genomic instability.
Discussion
In this study, we demonstrate that an excess of RAD51 has a role in driving genomic instability caused by impaired replication and intra-S-phase CHK1-mediated signaling. We show for a panel of breast cancer cell lines that an excess of RAD51 decreases I-Sce-I mediated HR, reduces elongation rates and generates excessive firing of dormant origins, caused by a failure to properly activate CHK1.
Surprisingly, a high level of RAD51 expression results in a significantly lower frequency of I-Sce-I induced HR events compared to cells with low RAD51 expression (). This is in line with some previous studies,Citation16,17 whereas others found no changeCitation20 or even an increase in HRCitation18,19 after RAD51 overexpression. Suppression of HR events was also observed after overexpression of other HR proteins such as BRCA2.Citation21 Therefore it seems that increasing the level of factors involved in HR is not entirely sufficient to increase repair capacity of the whole HR machinery. It is more likely that constituents of this multi-protein complex have to be in a delicate equilibrium to maintain the balance between HR, DNA repair and replication. In this equilibrium RAD51 plays the most important role, as it is ultimately delivered to the DNA by the HR machinery and catalyzes the critical step during HR, the strand pairing and exchange. This explains why HR in BRCA1 and BRCA2-depleted cells is restorable by the over-expression of RAD51,Citation23 but not by RAD52 or RAD54.Citation24 Notably, the initial step of HR, the binding to single stranded DNA and formation of a nucleofilament appear not to be compromised by high RAD51 expression, as RAD51 foci were readily formed (). This might also explain why the initial step of HR, the binding to single stranded DNA to form a nucleofilament was not prevented by high RAD51 expression, visible by proper RAD51 foci formation.Citation12 Thus, initiation is not inhibited by the surplus of RAD51, but rather the later steps are compromised, leading to the observed reduction of I-Sce-I induced HR. This view is supported by the observation that human RAD54 promotes the dissociation of RAD51 from dsDNA and that translocase depletion leads to the accumulation of RAD51 on chromosomes, forming complexes that are not associated with markers of DNA damage.Citation56 The most prominent phenotype driven by high RAD51 expression is increased genomic instabilityCitation10,16,57 that we also detected as an increase of chromatid-type aberrations occurring in DNA, which has already been replicated (). It is accompanied by elevated spontaneous gamma-H2AX fociCitation12,18,57 (, right). Thus, our observations suggest that cells with high RAD51 expression are defective in the repair of replication-associated DNA damage. This notion gets strong support from the DNA fiber analysis. Not only did cells with increased RAD51 levels suffer from reduced elongation rates, they also displayed increased spontaneous initiation (). The elevated fork asymmetry and the reduced ori-to-ori distances strongly suggest that these cells experience more frequent or extended fork arrest leading to an increase in dormant origin firing. This view is further strengthened by the large number of origins fired after various DNA damages (). In fact, after MMC and low hydrogen peroxide, dormant origin firing became directly visible as 2nd pulse origins in the immediate vicinity of a 1st pulse tract that likely represented fork stalling immediately after addition of MMC (). Proper DNA duplication is highly dependent on intact HR and any disturbances in this pathway give rise to genomic instability.Citation1,58,59 BRCA2, XRCC2 and RAD51 overexpressing cells have been previously found to exhibit reduced replication fork progression,Citation60 and increased origin firing has been observed in breast cancer patients with reduced expression of PALB2.Citation45 Nevertheless, in all these cases forks remain stable, whereas cells deficient in FANCD2 and BRCA2 experience degradation of nascent DNA at the fork.Citation26,27
At physiological levels, RAD51 is loaded onto ssDNA at stalled replication forks in order to protect nascent DNA from degradation and to promote fork reassembly.Citation3,26,29,30,61 We suggest that at supraphysiological levels of RAD51 there is an excessive loading of this protein at stalled or disrupted forks (). This probably prevents reactivation of the stalled forks, so that the cell has to resort to dormant origin firing. On the other hand, the premature replacement of RPA () by RAD51 on ssDNA at stalled forks is also expected to interfere with replication checkpoint signaling mediated by ATR that is dependent on RPA.Citation35 This scenario is consistent with the observed reduction of CHK1 activation (). CHK1 represents the major effector kinase activated by ATR. Its substrates include p53 and DNA repair proteins such as BRCA2 and FANCECitation43,62,63 which promote fork stability by controlling replication initiation and elongation.Citation64,65 Furthermore, recruitment of RAD51 to sites of replication-dependent DNA damage has been reported to require phosphorylation of RAD51 and BRCA2 by CHK1Citation64,66 and loss of CHK1 function impedes the FA-mediated DNA damage response.Citation65 In particular, ATR/CHK1-mediated phosphorylation of FANCI and FANCD2 is essential for efficient monoubiquitination of these proteins. It is probably due to this negative feedback loop that we did not observe increased FANCD2 monoubiquitination in MMC-treated cells (Fig. S2B, C) despite strongly increased fork stalling (data not shown). Therefore it seems likely that the observed endogenous reduction in CHK1 activation is responsible for the sensitization to MMC ()Citation67 and cisplatinCitation23 in cells with high RAD51 expression.
Based on our model (), the consequence of high RAD51 levels during DNA replication is excessive dormant origin firing that would lead to DSBs at disrupted forks and/or accumulation of un-replicated DNA at the end of S phase. This is consistent with the elevated genomic instability observed in cells and cancers with high RAD51 expression. At the same time, it explains why BCR/Abl expressing cells with high RAD51 are sensitive to MMC, but acquire resistance after simultaneous overexpression of BLM,Citation67 as BLM is important for the resolution of late replication intermediates.Citation68
In summary, our results indicate that genomic instability in cells with increased RAD51 levels and the bad prognosis in cancers with high RAD51 may not be due to a defect in HR per se, but rather owing to the interference with DNA replication, especially the reactivation of stalled or disrupted forks and the underlying checkpoint signaling.
Materials and Methods
Cell lines, chemicals and drugs
Experiments were carried out with the cell lines U2OS stably transfected of an ecdysone-inducible expression system (Invitrogen) carrying RAD51 cloned into vector pIND and vector pVgRXR.Citation69 Cells were grown in DMEM with 10% fetal calf serum (FCS, Sigma-Aldrich) supplemented with neomycin and zeocin to maintain transfection and experiments were carried out 24 h after addition of 1 µM Pon A.
The breast cancer cell lines BT-20, HS-578T, GI-101, MCF-7, MDA-MB-231, MDA-MB-468 and SKBR-3 were cultivated in DMEM and BT-474, BT-549 and T-47D in RPMI media supplemented with 10% FCS, 2 mM glutamine, 100 U/mL penicillin und 100 μg/mL streptomycin at 37°C at 10 % CO2 or 5% CO2 respectively.
Homologous targeting assay
HR frequency was measured by stable or transient transfection of I-Sce-I linearized form of the pGC plasmid.Citation50 Briefly, 1 µg of I-Sce-I linearized plasmid was transfected into cells using FuGENE (Roche) in a 1:3 µg/µl ratio according to the manufacturer's instruction. After 24 h cells were harvested and the fraction GFP positive cells, representing cells, that have successful repaired by HR, was determined by flow cytometry.
Western blot and immune staining
Total Protein was extracted from exponentially growing cells and 40 µg/ml were resolved by SDS-PAGE using a 4–15% gradient gel (Bio-Rad Laboratories). Fractionated protein was prepared with the Subcellular protein fractionation kit for cultured cells (Thermo Fisher Scientific) according to manufactures instructions. After transfer to a Nitrocellulose membrane (Licor) or PVDF (Chemiluminescence), proteins were detected by Anti-ATR [2B5] (1:1.000, abcam), BRCA2 [2A9] (10 µg/ml, Smith, Grosse and Pospiech, to be published elsewhere), FANCD2 [FI17] (1:2.000, Santa Cruz), anti-CHK1 [2G1D5] (Cell Signaling, 1:750), anti-CHK1-pSer345 [133D3] (Cell Signaling, 1:750) RAD51 [14B4] (1:2.000, GeneTex) or anti-β-actin [AC-15] (Sigma, 1:20.000) and IRDYE 680 conjugated anti-mouse IgG (Licor, 1:7.500 or 15.000) or IRDYE 800 conjugated anti-rabbit IgG (Licor 1:7.500 or 15.000) or for chemiluminescence detection ECLTM Anti-mouse IgG (1:2000, GE Healthcare) and ECLTM Anti-rabbit IgG (1:2000, GE Healthcare) were used.
For immunofluorescence staining, cells were seeded on culture slides and incubated with 1 µM Pon A, or mock-treated. Cells were fixed with 2% formaldehyde in PBS for 15 min, permeabilized on ice for 5 min using 0.2% Triton X-100 in PBS and blocked with 0.15% (w/v) glycine and 3% (w/v) BSA in PBS overnight at 4°C followed by 30 min at 37°C. Foci were detected using anti-RAD51 (1:2.000), or Anti-phospho-histone H2A.X [JBW301] (Millipore, 1:100) followed by Fluoresceinisothiocyanate-linked anti-rabbit IgG (Amersham) or Alexa Fluor 594 goat anti-mouse IgG (invitrogen) and mounted by using mounting medium with 4,6-diamidino-2-phenlyindole (Vector Laboratories). Fluorescence images were captured using a Zeiss Axioplan 2 epifluorescence microscope equipped with a charge-coupled device camera and Optimas software. For quantitative analysis, foci were counted by fluorescence microscopy using a 1,000-fold magnification. 100 cells per dose per slide and experiment were evaluated blindly.
DNA fiber assay
Exponentially growing cells were pulse-labeled with 25 μM CldU (Sigma) and 250 μM IdU (Sigma) for the times specified. Where indicated, the cells were exposed to methyl-methanesulfonate (MMS), hydrogen peroxide, mitomycin C (MMC), topotecan or hydroxyurea. Labeled cells were harvested and Fiber spreads were prepared from 0.5 × 106 cells/ml. Slides were incubated in 2.5 M HCl for 90 min and then washed several times in PBS, followed by incubation in blocking buffer (2% BSA in PBS with 0.1% Tween) for 1 h. Acid-treated fiber spreads were stained with monoclonal rat anti-BrdU (Oxford Biotechnologies, 1:1000) antibody to detect CldU, followed by monoclonal mouse anti-BrdU (Becton Dickinson, 1:15000) to detect IdU. Secondary antibodies were goat anti-rat AlexaFluor555 and goat anti-mouse AlexaFluor488 (both Invitrogen, 1:500). Primary antibodies were diluted in blocking buffer, incubated for 1 h with rat anti-BrdU antibody and over-night with mouse anti-BrdU antibody followed by extensive washes in PBS. Secondary antibodies were applied for 1.5 h, and slides were mounted in Immuno-Fluor mounting medium (MP Biomedicals). Fiber tracts were examined using fluorescence microscopy as described above. Pictures were taken from randomly selected fields with untangled fibers and analyzed using the ImageJ software package.Citation45 For structure analyses, the frequencies of the different classes of fiber tracks were classified; red-green (ongoing replication tracts), red (stalled forks/termination), green-red-red-green (1st order origin) and green (2nd order origin). For fork speed analyses, replication fork speeds of CldU and IdU were measured and micrometer values were converted into kilobases. A conversion factor for the length of a labeled track of 1 μm = 2.59 kb was used.Citation45 A minimum of 100 individual fibers was analyzed for each experiment and the means of at least 3 independent experiments are presented.
Metaphase spread analysis
For metaphase spreads exponential growing cells were treated with colcemid (0.02 µg/ml) overnight, incubated with 0.0075 M KCl, fixed with methanol/acetic acid (3:1), dropped onto microscope slides, stained with 5% giemsa and mounted with entellan before imaging with a Zeiss Axioplan 2 microscope. 100 metaphases per experiment were counted.
Cell cycle analysis
Cell cycle analysis of exponential growing cells was performed by flow cytometry (FACS). Cells were fixed, washed with PBS, incubated with propidium iodide (10 µg/ml) RNase (5 µg/ml, Serva) and data were analyzed by flow cytometry using the ModFitLT software on a FAC-Scan (Becton Dickinson) from a cell population under exclusion of debris.
Clonogenic survival
For survival assays 250 cells were seeded in a 6-well plate 6 h before treatment and cells were cultured for 14 days. Cells were fixed and stained with 1% crystal violet (Sigma-Aldrich, St. Louis, MO). Colonies with more than 50 cells were counted and normalized to untreated samples. Each survival curve represents the mean of at least 3 independent experiments.
Statistical analysis
Statistical analysis, curve fitting and graphs were performed by means of the computer program Prism 6.02 (GraphPad Software). Data are given as mean (SE) of 3–5 replicate experiments. Unless stated otherwise, significance was tested by Student's t-test.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
1055996_supplemental_files.pptx
Download MS Power Point (2.6 MB)Acknowledgment
The authors thank Alexandra Zielinski for excellent technical assistance.
Funding
This work was supported by Deutsche Krebshilfe [70-1932-Di2]; Roggenbuck-Foundation and BMU [3610S30016].
Related Research Data
References
- Arnaudeau C, Lundin C, Helleday T. DNA double-strand breaks associated with replication forks are predominantly repaired by homologous recombination involving an exchange mechanism in mammalian cells. J Mol Biol 2001; 307:1235-45; PMID:11292338; http://dx.doi.org/10.1006/jmbi.2001.4564
- Llorente B, Smith CE, Symington LS. Break-induced replication: what is it and what is it for? Cell Cycle 2008; 7:859-64; PMID:18414031; http://dx.doi.org/10.4161/cc.7.7.5613
- Petermann E, Orta ML, Issaeva N, Schultz N, Helleday T. Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol Cell 2010; 37:492-502; PMID:20188668; http://dx.doi.org/10.1016/j.molcel.2010.01.021
- Burrell RA, McGranahan N, Bartek J, Swanton C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature 2013; 501:338-45; PMID:24048066; http://dx.doi.org/10.1038/nature12625
- Orr SJ, Gaymes T, Ladon D, Chronis C, Czepulkowski B, Wang R, Mufti GJ, Marcotte EM, Thomas NS. Reducing MCM levels in human primary T cells during the G(0)–>G(1) transition causes genomic instability during the first cell cycle. Oncogene 2010; 29:3803-14; PMID:20440261; http://dx.doi.org/10.1038/onc.2010.138
- Aze A, Zhou JC, Costa A, Costanzo V. DNA replication and homologous recombination factors: acting together to maintain genome stability. Chromosoma 2013; 122:401-13; PMID:23584157; http://dx.doi.org/10.1007/s00412-013-0411-3
- Helleday T, Lo J, van Gent DC, Engelward BP. DNA double-strand break repair: from mechanistic understanding to cancer treatment. DNA Repair 2007; 6:923-35; PMID:17363343; http://dx.doi.org/10.1016/j.dnarep.2007.02.006
- Lim DS, Hasty P. A mutation in mouse rad51 results in an early embryonic lethal that is suppressed by a mutation in p53. Mol Cell Biol 1996; 16:7133-43; PMID:8943369
- Sonoda E, Sasaki MS, Buerstedde JM, Bezzubova O, Shinohara A, Ogawa H, Takata M, Yamaguchi-Iwai Y, Takeda S. Rad51-deficient vertebrate cells accumulate chromosomal breaks prior to cell death. EMBO 1998; 17:598-608; http://dx.doi.org/10.1093/emboj/17.2.598
- Vispe S, Cazaux C, Lesca C, Defais M. Overexpression of Rad51 protein stimulates homologous recombination and increases resistance of mammalian cells to ionizing radiation. Nucleic Acids Res 1998; 26:2859-64; PMID:9611228; http://dx.doi.org/10.1093/nar/26.12.2859
- Koch K, Wrona A, Dikomey E, Borgmann K. Impact of homologous recombination on individual cellular radiosensitivity. Radiother Oncol 2009; 90:265-72; PMID:18804300; http://dx.doi.org/10.1016/j.radonc.2008.07.028
- Raderschall E, Stout K, Freier S, Suckow V, Schweiger S, Haaf T. Elevated levels of Rad51 recombination protein in tumor cells. Cancer Res 2002; 62:219-25; PMID:11782381
- Li Y, Yu H, Luo RZ, Zhang Y, Zhang MF, Wang X, Jia WH. Elevated expression of Rad51 is correlated with decreased survival in resectable esophageal squamous cell carcinoma. J Surg Oncol 2011; 104:617-22; PMID:21744352; http://dx.doi.org/10.1002/jso.22018
- Tennstedt P, Fresow R, Simon R, Marx A, Terracciano L, Petersen C, Sauter G, Dikomey E, Borgmann K. RAD51 overexpression is a negative prognostic marker for colorectal adenocarcinoma. Int J Cancer 2013; 132:2118-26; PMID:23065657; http://dx.doi.org/10.1002/ijc.27907
- Le Scodan R, Cizeron-Clairac G, Fourme E, Meseure D, Vacher S, Spyratos F, de la Lande B, Cvitkovic F, Lidereau R, Bieche I. DNA repair gene expression and risk of locoregional relapse in breast cancer patients. Int J Radiat Oncol Phys 2010; 78:328-36; http://dx.doi.org/10.1016/j.ijrobp.2009.07.1735
- Kim PM, Allen C, Wagener BM, Shen Z, Nickoloff JA. Overexpression of human RAD51 and RAD52 reduces double-strand break-induced homologous recombination in mammalian cells. Nucleic Acids Res 2001; 29:4352-60; PMID:11691922; http://dx.doi.org/10.1093/nar/29.21.4352
- Paffett KS, Clikeman JA, Palmer S, Nickoloff JA. Overexpression of Rad51 inhibits double-strand break-induced homologous recombination but does not affect gene conversion tract lengths. DNA Repair 2005; 4:687-98; PMID:15878310; http://dx.doi.org/10.1016/j.dnarep.2005.03.003
- Yanez RJ, Porter AC. Differential effects of Rad52p overexpression on gene targeting and extrachromosomal homologous recombination in a human cell line. Nucleic Acids Res 2002; 30:740-8; PMID:11809887; http://dx.doi.org/10.1093/nar/30.3.740
- Lambert S, Lopez BS. Characterization of mammalian RAD51 double strand break repair using non-lethal dominant-negative forms. EMBO 2000; 19:3090-9; http://dx.doi.org/10.1093/emboj/19.12.3090
- Richardson C, Stark JM, Ommundsen M, Jasin M. Rad51 overexpression promotes alternative double-strand break repair pathways and genome instability. Oncogene 2004; 23:546-53; PMID:14724582; http://dx.doi.org/10.1038/sj.onc.1207098
- Magwood AC, Mundia MM, Baker MD. High levels of wild-type BRCA2 suppress homologous recombination. J Mol Biol 2012; 421:38-53; PMID:22579622; http://dx.doi.org/10.1016/j.jmb.2012.05.007
- Takata M, Sasaki MS, Tachiiri S, Fukushima T, Sonoda E, Schild D, Thompson LH, Takeda S. Chromosome instability and defective recombinational repair in knockout mutants of the five Rad51 paralogs. Mol Cell Biol 2001; 21:2858-66; PMID:11283264; http://dx.doi.org/10.1128/MCB.21.8.2858-2866.2001
- Martin RW, Orelli BJ, Yamazoe M, Minn AJ, Takeda S, Bishop DK. RAD51 up-regulation bypasses BRCA1 function and is a common feature of BRCA1-deficient breast tumors. Cancer Res 2007; 67:9658-65; PMID:17942895; http://dx.doi.org/10.1158/0008-5472.CAN-07-0290
- Lee SA, Roques C, Magwood AC, Masson JY, Baker MD. Recovery of deficient homologous recombination in Brca2-depleted mouse cells by wild-type Rad51 expression. DNA Repair 2009; 8:170-81; PMID:18992372; http://dx.doi.org/10.1016/j.dnarep.2008.10.002
- Schild D, Wiese C. Overexpression of RAD51 suppresses recombination defects: a possible mechanism to reverse genomic instability. Nucleic Acids Res 2010; 38:1061-70; PMID:19942681; http://dx.doi.org/10.1093/nar/gkp1063
- Schlacher K, Christ N, Siaud N, Egashira A, Wu H, Jasin M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 2011; 145:529-42; PMID:21565612; http://dx.doi.org/10.1016/j.cell.2011.03.041
- Schlacher K, Wu H, Jasin M. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 2012; 22:106-16; PMID:22789542; http://dx.doi.org/10.1016/j.ccr.2012.05.015
- Yata K, Bleuyard JY, Nakato R, Ralf C, Katou Y, Schwab RA, Niedzwiedz W, Shirahige K, Esashi F. BRCA2 coordinates the activities of cell-cycle kinases to promote genome stability. Cell Rep 2014; 7:1547-59; PMID:24835992; http://dx.doi.org/10.1016/j.celrep.2014.04.023
- Hashimoto Y, Puddu F, Costanzo V. RAD51- and MRE11-dependent reassembly of uncoupled CMG helicase complex at collapsed replication forks. Nat Struct Mol Biol 2012; 19:17-24; http://dx.doi.org/10.1038/nsmb.2177
- Hashimoto Y, Ray Chaudhuri A, Lopes M, Costanzo V. Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nat Struc Mol Biol 2010; 17:1305-11; http://dx.doi.org/10.1038/nsmb.1927
- Min W, Bruhn C, Grigaravicius P, Zhou ZW, Li F, Kruger A, Siddeek B, Greulich KO, Popp O, Meisezahl C, et al. Poly(ADP-ribose) binding to Chk1 at stalled replication forks is required for S-phase checkpoint activation. Nat Comm 2013; 4:2993
- Cimprich KA, Cortez D. ATR: an essential regulator of genome integrity. Nat Rev 2008; 9:616-27; http://dx.doi.org/10.1038/nrm2450
- Feijoo C, Hall-Jackson C, Wu R, Jenkins D, Leitch J, Gilbert DM, Smythe C. Activation of mammalian Chk1 during DNA replication arrest: a role for Chk1 in the intra-S phase checkpoint monitoring replication origin firing. J Cell Biol 2001; 154:913-23; PMID:11535615; http://dx.doi.org/10.1083/jcb.200104099
- Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 2003; 300:1542-8; http://dx.doi.org/10.1126/science.1083430
- Nam EA, Cortez D. ATR signalling: more than meeting at the fork. Biochem 2011; 436:527-36; http://dx.doi.org/10.1042/BJ20102162
- Sokka M, Parkkinen S, Pospiech H, Syvaoja JE. Function of TopBP1 in genome stability. SubCell Biochem 2010; 50:119-41; PMID:20012580
- Smits VA, Reaper PM, Jackson SP. Rapid PIKK-dependent release of Chk1 from chromatin promotes the DNA-damage checkpoint response. Curr Biol 2006; 16:150-9; http://dx.doi.org/10.1016/j.cub.2005.11.066
- Zhao H, Piwnica-Worms H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol Cell Biol 2001; 21:4129-39; PMID:11390642; http://dx.doi.org/10.1128/MCB.21.13.4129-4139.2001
- Ge XQ, Blow JJ. Chk1 inhibits replication factory activation but allows dormant origin firing in existing factories. J Cell Biol 2010; 191:1285-97; PMID:21173116; http://dx.doi.org/10.1083/jcb.201007074
- Syljuasen RG, Sorensen CS, Hansen LT, Fugger K, Lundin C, Johansson F, Helleday T, Sehested M, Lukas J, Bartek J. Inhibition of human Chk1 causes increased initiation of DNA replication, phosphorylation of ATR targets, and DNA breakage. Mol Cell Biol 2005; 25:3553-62; PMID:15831461; http://dx.doi.org/10.1128/MCB.25.9.3553-3562.2005
- Petermann E, Maya-Mendoza A, Zachos G, Gillespie DA, Jackson DA, Caldecott KW. Chk1 requirement for high global rates of replication fork progression during normal vertebrate S phase. Mol Cell Biol 2006; 26:3319-26; PMID:16581803; http://dx.doi.org/10.1128/MCB.26.8.3319-3326.2006
- Maya-Mendoza A, Petermann E, Gillespie DA, Caldecott KW, Jackson DA. Chk1 regulates the density of active replication origins during the vertebrate S phase. EMBO 2007; 26:2719-31; http://dx.doi.org/10.1038/sj.emboj.7601714
- Sorensen CS, Hansen LT, Dziegielewski J, Syljuasen RG, Lundin C, Bartek J, Helleday T. The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nat Cell Biol 2005; 7:195-201; PMID:15665856; http://dx.doi.org/10.1038/ncb1212
- Bahassi EM, Ovesen JL, Riesenberg AL, Bernstein WZ, Hasty PE, Stambrook PJ. The checkpoint kinases Chk1 and Chk2 regulate the functional associations between hBRCA2 and Rad51 in response to DNA damage. Oncogene 2008; 27:3977-85; PMID:18317453; http://dx.doi.org/10.1038/onc.2008.17
- Nikkila J, Parplys AC, Pylkas K, Bose M, Huo Y, Borgmann K, Rapakko K, Nieminen P, Xia B, Pospiech H, et al. Heterozygous mutations in PALB2 cause DNA replication and damage response defects. Nat Comm 2013; 4:2578; http://dx.doi.org/10.1038/ncomms3578
- Bindra RS, Schaffer PJ, Meng A, Woo J, Maseide K, Roth ME, Lizardi P, Hedley DW, Bristow RG, Glazer PM. Down-regulation of Rad51 and decreased homologous recombination in hypoxic cancer cells. Mol Cell Biol 2004; 24:8504-18; PMID:15367671; http://dx.doi.org/10.1128/MCB.24.19.8504-8518.2004
- Mukherjee A, Karmakar P. Attenuation of PTEN perturbs genomic stability via activation of Akt and down-regulation of Rad51 in human embryonic kidney cells. Mol Carcinogen 2013; 52:611-8; http://dx.doi.org/10.1002/mc.21903
- Wang Y, Huang JW, Calses P, Kemp CJ, Taniguchi T. MiR-96 downregulates REV1 and RAD51 to promote cellular sensitivity to cisplatin and PARP inhibition. Cancer Res 2012; 72:4037-46; PMID:22761336; http://dx.doi.org/10.1158/0008-5472.CAN-12-0103
- Gasparini P, Lovat F, Fassan M, Casadei L, Cascione L, Jacob NK, Carasi S, Palmieri D, Costinean S, Shapiro CL, et al. Protective role of miR-155 in breast cancer through RAD51 targeting impairs homologous recombination after irradiation. P Natl Acad Sci USA 2014; 111:4536-41; http://dx.doi.org/10.1073/pnas.1402604111
- Mansour WY, Schumacher S, Rosskopf R, Rhein T, Schmidt-Petersen F, Gatzemeier F, Haag F, Borgmann K, Willers H, Dahm-Daphi J. Hierarchy of nonhomologous end-joining, single-strand annealing and gene conversion at site-directed DNA double-strand breaks. Nucleic Res 2008; 36:4088-98; http://dx.doi.org/10.1093/nar/gkn347
- Deans AJ, West SC. DNA interstrand crosslink repair and cancer. Nat Rev Cancer 2011; 11:467-80; PMID:21701511; http://dx.doi.org/10.1038/nrc3088
- Garcia-Higuera I, Taniguchi T, Ganesan S, Meyn MS, Timmers C, Hejna J, Grompe M, D'Andrea AD. Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol Cell 2001; 7:249-62; PMID:11239454; http://dx.doi.org/10.1016/S1097-2765(01)00173-3
- Zhong Y, Nellimoottil T, Peace JM, Knott SR, Villwock SK, Yee JM, Jancuska JM, Rege S, Tecklenburg M, Sclafani RA, et al. The level of origin firing inversely affects the rate of replication fork progression. J Cell Biol 2013; 201:373-83; PMID:23629964; http://dx.doi.org/10.1083/jcb.201208060
- Conti C, Seiler JA, Pommier Y. The mammalian DNA replication elongation checkpoint: implication of Chk1 and relationship with origin firing as determined by single DNA molecule and single cell analyses. Cell Cycle 2007; 6:2760-7; PMID:17986860; http://dx.doi.org/10.4161/cc.6.22.4932
- Krejci L, Altmannova V, Spirek M, Zhao X. Homologous recombination and its regulation. Nucleic acids Res 2012; 40:5795-818; PMID:22467216; http://dx.doi.org/10.1093/nar/gks270
- Mason JM, Dusad K, Wright WD, Grubb J, Budke B, Heyer WD, Connell PP, Weichselbaum RR, Bishop DK. RAD54 family translocases counter genotoxic effects of RAD51 in human tumor cells. Nucleic acids Res 2015; 43:3180-96; PMID:25765654; http://dx.doi.org/10.1093/nar/gkv175
- Lundin C, Schultz N, Arnaudeau C, Mohindra A, Hansen LT, Helleday T. RAD51 is involved in repair of damage associated with DNA replication in mammalian cells. J Mol Biol 2003; 328:521-35; PMID:12706714; http://dx.doi.org/10.1016/S0022-2836(03)00313-9
- Budzowska M, Kanaar R. Mechanisms of dealing with DNA damage-induced replication problems. Cell Biochem Biophys 2009; 53:17-31; PMID:19034694; http://dx.doi.org/10.1007/s12013-008-9039-y
- Petermann E, Helleday T. Pathways of mammalian replication fork restart. Nat Rev 2010; 11:683-7; http://dx.doi.org/10.1038/nrm2974
- Daboussi F, Thacker J, Lopez BS. Genetic interactions between RAD51 and its paralogues for centrosome fragmentation and ploidy control, independently of the sensitivity to genotoxic stresses. Oncogene 2005; 24:3691-6; PMID:15782136; http://dx.doi.org/10.1038/sj.onc.1208438
- Pefani DE, O'Neill E. Safeguarding Genome stability: RASSF1A tumour suppressor regulates BRCA2 at stalled forks. Cell Cycle 2015:0; [Epub ahead of print]; PMID:25927241
- Wang X, Kennedy RD, Ray K, Stuckert P, Ellenberger T, D'Andrea AD. Chk1-mediated phosphorylation of FANCE is required for the Fanconi anemia/BRCA pathway. Mol Cell Biol 2007; 27:3098-108; PMID:17296736; http://dx.doi.org/10.1128/MCB.02357-06
- Lossaint G, Besnard E, Fisher D, Piette J, Dulic V. Chk1 is dispensable for G2 arrest in response to sustained DNA damage when the ATM/p53/p21 pathway is functional. Oncogene 2011; 30:4261-74; PMID:21532626; http://dx.doi.org/10.1038/onc.2011.135
- Collis SJ, Ciccia A, Deans AJ, Horejsi Z, Martin JS, Maslen SL, Skehel JM, Elledge SJ, West SC, Boulton SJ. FANCM and FAAP24 function in ATR-mediated checkpoint signaling independently of the Fanconi anemia core complex. Mol Cell 2008; 32:313-24; PMID:18995830; http://dx.doi.org/10.1016/j.molcel.2008.10.014
- Guervilly JH, Mace-Aime G, Rosselli F. Loss of CHK1 function impedes DNA damage-induced FANCD2 monoubiquitination but normalizes the abnormal G2 arrest in Fanconi anemia. Hum Mol Genet 2008; 17:679-89; PMID:18029388; http://dx.doi.org/10.1093/hmg/ddm340
- Hansen LT, Lundin C, Helleday T, Poulsen HS, Sorensen CS, Petersen LN, Spang-Thomsen M. DNA repair rate and etoposide (VP16) resistance of tumor cell subpopulations derived from a single human small cell lung cancer. Lung Cancer 2003; 40:157-64; PMID:12711116; http://dx.doi.org/10.1016/S0169-5002(03)00026-6
- Slupianek A, Schmutte C, Tombline G, Nieborowska-Skorska M, Hoser G, Nowicki MO, Pierce AJ, Fishel R, Skorski T. BCR/ABL regulates mammalian RecA homologs, resulting in drug resistance. Mol Cell 2001; 8:795-806; PMID:11684015; http://dx.doi.org/10.1016/S1097-2765(01)00357-4
- Chu WK, Hickson ID. RecQ helicases: multifunctional genome caretakers. Nat Rev Cancer 2009; 9:644-54; PMID:19657341; http://dx.doi.org/10.1038/nrc2682
- Maacke H, Jost K, Opitz S, Miska S, Yuan Y, Hasselbach L, Luttges J, Kalthoff H, Sturzbecher HW. DNA repair and recombination factor Rad51 is over-expressed in human pancreatic adenocarcinoma. Oncogene 2000; 19:2791-5; PMID:10851081; http://dx.doi.org/10.1038/sj.onc.1203578