
Genomic DNA is continuously damaged by external and endogenous factors, resulting in different DNA lesions. Once DNA damage is detected, a multifaceted signaling network is activated that halts the cell cycle, initiates repair and, in some instances induces apoptotic cell death. The rapid accumulation of signaling and repair factors in the proximity of broken DNA ends is crucial in cellular DNA damage response (DDR) to DNA double-strand breaks (DSB). Such a type of lesion is mainly repaired by either homologous recombination (HR) or non-homologous end joining (NHEJ) pathways. The proper choice of the repair pathway is critical for genomic stability and cancer. Tumor cells have limited ability to maintain genomic stability owing to defective DNA damage checkpoints/response. Antitumor strategies are exploiting this vulnerability of cancer cells to spare normal tissues from toxicity.Citation1 Indeed, the recent past years have seen the emergence of new antitumor therapeutic opportunities taking advantage of knowledge on defects of components of DNA repair pathways. Genetic studies in model organisms have suggested the potential of profiling drug ability to selectively kill cells exhibiting a specific molecular context, suggesting the feasibility of strategies that have been already implemented to treat human cancer.Citation2 For instance, cells carrying mutations in genes involved in HR such as BRCA1 or BRCA2 have been shown to be sensitive to inhibitors of poly-ADP-ribose polymerase (PARP)Citation3 that are used as monotherapy in patients with mutated BRCA cancers, representing a successful example of drugs targeting vulnerable features of tumor cells.
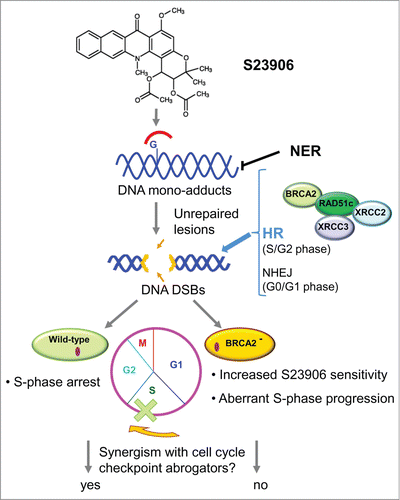
In this issue of Cell Cycle, Rocca et al.Citation4 report that BRCA2 is needed for both DNA repair and cell cycle arrest in mammalian cells exposed to the antitumor agent S23906, an acronycine derivative forming monofunctional DNA adducts. A unique property of S23906 lies in its ability to destabilize the duplex structure in the proximity of the DNA adducts leading to the formation of bulky lesions quickly processed through Nucleotide Excision Repair (NER). When left unrepaired, S23906 adducts are, at the contact of the replication fork, converted into toxic DSB that are processed through HR thanks to the action of BRCA2, but not through NHEJ repair.Citation4 Of note, the higher sensitivity to S23906 of BRCA2-deficient cells in comparison to wild-type cells was associated with the lack of S-phase arrest and of synergistic interaction with known cell cycle checkpoint abrogators (UCN-01 and AZD77662).Citation4 Thus, the increased sensitivity of BRCA2-deficient cells to S23906 was linked to both an aberrant S-phase progression and a defective HR repair. The findings of Rocca and colleagues suggest that tumors with defective DNA damage response, specifically with deficiencies in NER and HR (e.g., lacking functional BRCA2), may be particularly sensitive to S23906 and its related derivatives, thereby suggesting a molecular signature to be used for patient selection in clinical trials. Such a finding extends the number of vulnerable features that may be considered in the path toward personalized treatment of cancer and underlines the need to precisely characterize the molecular mechanism of action of anticancer drugs. However, some caveats should be considered. In fact, although multiple molecular pathways are implicated in the repair of specific DNA damage lesions induced by antitumor agents which inhibit DNA functions, and certain enzymes such as PARP-1, Polθ or TDP-1 appear to play a peculiar role in DNA repair, not all of them can reveal synthetic lethal relationships. Interestingly, besides PARP, Polθ has been recently suggested to represent a promising therapeutic target.Citation5 Conversely, inhibition of TDP-1, a unique enzyme which acts to repair DNA encumbered with topoisomerase I (Top1) protein adducts may result in principle in improvement of the antitumor activity of Top1 poisons.Citation6 However, the redundancy of the pathways implicated in repair of Top1-mediated damage appears to limit the efficacy of this approach. In fact, TDP1-mediated repair of Top I DNA damage seems to occur through pathways also implicating BRCA1 and XRCC1, in keeping with studies showing that TDP1 per se only facilitates single-strand break (SSB) repair.Citation7
As our understanding of the mechanism of DDR increases, the potential ways to manipulate this pathway for the development of novel therapeutics will emerge.Citation1 The availability of biomarkers to detect patients with particular checkpoint or DDR defects may allow. stratifying patients for personalized cancer treatments.Citation1
Figure 1. A schematic representation of the mechanism of S23906-induced bulky DNA adducts processing in mammalian cells. In the absence of functional NER, adducts are converted into DSBs processed through HR thanks to BRCA2 action. The S23906-sensitivity is increased in cells in BRCA2 deficient (BRCA2−) cells. DSBs, double strand breaks; HR, homologous recombination; NER, nucleotide excision repair; NHEJ, non-homologous end joining.
References
- Bolderson E, et al. Clin Cancer Res 2009; 15:6314–20; PMID: 19808869; http://dx.doi.org/10.1158/1078-0432.CCR-09-0096
- Perego P, et al. Pharmacol Rev 2000; 52:477–92; PMID: 11121507
- Farmer H, et al. Nature 2005; 434:917–21; PMID: 15829967; http://dx.doi.org/nature03445.
- Rocca CJ, et al. Cell Cycle 2015.
- Mateos-Gomez PA, et al. Nature 2015; 518:254–7; PMID: 25642960; http://dx.doi.org/10.1038/nature14157
- Perego P, et al. Biochem Pharmacol 2012; 83:27–36; PMID: 21978643; http://dx.doi.org/10.1016/j.bcp.2011.09.021
- Katyal S, et al. EMBO J 2007; 26:4720–31; PMID: 17914460