
Genotoxic agents that induce bulky or helix-distorting (‘disruptive’) nucleotide lesions can provoke a number of responses, ranging from the induction of checkpoints, senescence or apoptosis, to nucleotide substitutions and recombinogenic double-strand DNA (dsDNA) breaks.Citation1 An integral picture of how these divergent responses to DNA damage are orchestrated is lacking, possibly owing to the fact that, usually, non-physiologically high DNA damage loads are used to study checkpoint signaling and dsDNA breaks, whereas low loads are used to investigate the induction of mutations.
Checkpoint induction requires the presence of patches of single-stranded DNA (ssDNA), covered with the ssDNA binding protein RPA to recruit the damage signaling kinase ATR that, in turn, phosphorylates the checkpoint kinase CHK1.Citation1,2 In replicating cells, ssDNA patches may originate from the arrest of the replicative polymerases by disruptive lesions, followed by downstream repriming, or by convergence of replication from an adjacent replicon. These post-replicative ssDNA gaps have been proposed to be dominant inducers of checkpoint responsesCitation3 although evidence for this currently is lacking. Post-replicative, lesion-containing, ssDNA patches can be filled by so-called DNA damage tolerance (DDT) pathways that include lesion bypass by specialized DNA translesion synthesis (TLS) polymerases. DDT pathways allow the completion of replication of damaged genomes, which precludes checkpoint induction and prevents the breakage of post-replicative gaps to recombinogenic dsDNA breaks. On the flip side, TLS polymerases have the propensity to ‘misincorporate’ opposite damaged, including non-instructive, nucleotides, which makes TLS the source of genotoxin-induced nucleotide substitutions.Citation2
Checkpoint, apoptotic and mutagenic responses induced by low doses of genotoxic agents are modulated not only by TLS and by nucleotide excision repair (NER) that removes most disruptive nucleotide lesions prior to replication. Also DNA mismatch repair (MMR) proteins appear to play a role in these responses, following exposure to low doses of genotoxic agents.Citation4 This elusive function of MMR proteins is distinct from canonical MMR that repairs misincorporations of the processive DNA polymerases at undamaged templates, but also from the minor MMR pathway that acts at slightly damaged, mismatched, bases.
To elucidate the mechanistic basis of the involvement of the MSH2/MSH6 mismatch binding heterodimer in pleiotropic responses to disruptive nucleotide lesions we studied mouse embryonic stem (ES) cells with targeted disruptions in major players in NER (the Xpa gene), error-prone TLS (the Rev1 gene) or MMR (Msh2 or Msh6).Citation5 These cell lines were treated with low doses of UVC light, that induces cyclobutane pyrimidine dimers and (6–4)pyrimidine-pyrimidone photolesions [(6–4)PP]. Only the strongly disruptive, genotoxic, clastogenic and mutagenic (6–4)PP are substrates for NER in mouse cells.
While disruption of Msh2/Msh6 in wild type ES cells resulted in a strong increase of the mutagenicity of UVC, this increase was even more pronounced when Msh2/Msh6 was disrupted in a NER-deficient background.Citation5 Conversely, its disruption in cells carrying a (partial) inactivation of Rev1 led to an only mild increase in the mutagenicity of UVC. In other words: Msh2/Msh6 acts independently of NER to suppress the mutagenicity of (6–4)PP, but in an epistatic fashion with error-prone TLS. Most UVC-induced mutations that were prevented by Msh2/Msh6 in Xpa-deficient or wild type cells resided at pyrimidine dimers, the signature of TLS errors at photolesions. Indeed, TLS at a site-specific (6–4)PP within a transfected replicating plasmid was most mutagenic in Msh2/Msh6-deficient cells. In UVC-exposed ES cells, Msh2/Msh6 appeared to mediate the induction of persistent patches of ssDNA with embedded (6–4)PP lesions [called ss(6–4)PP], and these patches were associated with checkpoint induction. The transfer of persistent ss(6–4)PP through mitosis to the next cell cycle resulted in their collapse to dsDNA breaks that were associated with the induction of delayed apoptosis. Combined, our results strongly suggest that a novel, Msh2/Msh6-dependent excision repair pathway, called post-TLS repair, excises errors by TLS at disruptive nucleotide lesions, while inducing rapid checkpoint signaling, and delayed apoptosis (). Of note, since MSH2/MSH6 can directly interact with ATRCitation6 it formally cannot be excluded that checkpoint induction depends on this interaction, rather than on the ssDNA gaps generated by post-TLS repair per se. Remarkably, also TLS polymerase η binds to MSH2/MSH6.Citation7 Possibly, this interaction facilitates gap filling following Msh2/Msh6-dependent excision of the TLS error.
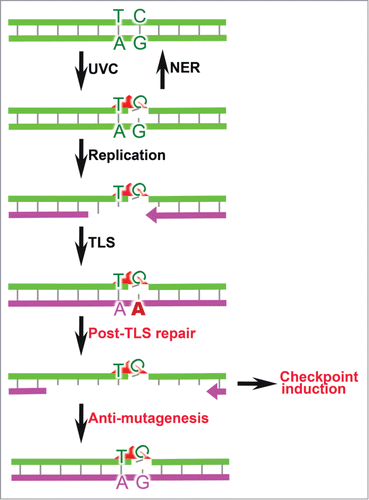
Figure 1. Responses to (6–4)PP. Most UVC-induced (6–4)PP (red triangle) will be repaired by NER. Unrepaired (6–4)PP are skipped by processive replication, necessitating to invoke error-prone TLS. Inadvertent TLS errors are excised by post-TLS repair. The resulting ss(6–4)PP patches induce checkpoint signaling and, during the subsequent cell cycle, can collapse (not shown). Error-free filling of the gaps induced by post-TLS repair mitigates the mutagenicity of UVC.
The discovery of post-TLS repair raises a host of questions of fundamental and clinical nature. How can MSH2/MSH6 specifically bind and initiate repair of incorrect nucleotides incorporated by TLS opposite lesions that do not engage in basepairing? Are the downstream actors in post-TLS repair the same as those involved in canonical MMR? What provides the strand discrimination signal for post-TLS repair in the absence of a proximal replication fork, which during canonical MMR might provide strand directionality? What is the relevance of DNA damage signaling by post-TLS repair for in vivo responses to genotoxic agents? The spontaneous mutator phenotype resulting from a defect in canonical MMR is involved in rapid cancer development in the familial cancer predisposition Lynch syndrome.Citation4 However, aberrant mutagenic, checkpoint and apoptotic responses to dietary or endogenous genotoxic agents in the intestine of Lynch syndrome patients may explain the inclination of cancer toward the colon. We anticipate that these, and many more, questions will be addressed in the years to come.
References
- Ciccia A, Elledge, SJ. Mol Cell 2010; 40:179-204; PMID:20965415; http://dx.doi.org/10.1016/j.molcel.2010.09.019
- Chang DJ, Cimprich KA. Nat Chem Biol 2009; 5:82-90; PMID:19148176; http://dx.doi.org/10.1038/nchembio.139
- Novarina D, et al. DNA Repair 2011; 10:751-9; PMID:21602108; http://dx.doi.org/10.1016/j.dnarep.2011.04.030
- Jiricny J. Cold Spring Harb Perspect Biol 2013; 5:a012633; PMID:23545421; http://dx.doi.org/10.1101/cshperspect.a012633
- Tsaalbi-Shtylik A, et al. J Cell Biol 2015; 209:33-46; http://dx.doi.org/10.1083/jcb.201408017
- Liu Y, et al. J Biol Chem 2010; 285:5974-82; PMID:20029092; http://dx.doi.org/10.1074/jbc.M109.076109
- Wilson TM, et al. J Exp Med 2005; 201:637-45; PMID:15710654; http://dx.doi.org/10.1084/jem.20042066