
Abstract
DNA damage, binding of drugs to DNA or a shortage of nucleotides can decrease the rate or completely halt the progress of replication forks. Although the global rate of replication decreases, mammalian cells can respond to replication stress by activating new replication origins. We demonstrate that a moderate level of stress induced by inhibitors of topoisomerase I, commencing in early, mid or late S-phase, induces activation of new sites of replication located within or in the immediate vicinity of the original replication factories; only in early S some of these new sites are also activated at a distance greater than 300 nm. Under high stress levels very few new replication sites are activated; such sites are located within the original replication regions. There is a large variation in cellular response to stress – while in some cells the number of replication sites increases even threefold, it decreases almost twofold in other cells. Replication stress results in a loss of PCNA from replication factories and a twofold increase in nuclear volume. These observations suggest that activation of new replication origins from the pool of dormant origins within replication cluster under conditions of mild stress is generally restricted to the original replication clusters (factories) active at a time of stress initiation, while activation of distant origins and new replication factories is suppressed.
Abbreviations
| Cpt | = | camptothecin |
| Tpt | = | topotecan |
| EdU | = | 5-ethynyl-2′-deoxyuridine |
| BrdU | = | 5-bromo-2′-deoxyuridine |
| PCNA | = | proliferating cell nuclear antigen |
| EGFP | = | Enhanced Green Fluorescent Protein |
| γH2AX | = | histone H2AX phosphorylated on Ser139. |
Introduction
Mammalian cells replicate their genome by initiating incorporation of nucleotides in over 30,000 distinct replication origins.Citation1 Under a fluorescence microscope regions of active replication appear as numerous foci, arranged in patterns characteristic for early, mid or late S-phase. Each of these foci is thought to consist of several clusters of active replication forks. The number of distinct replication regions active at any given time is estimated to reach 1100Citation2 to 1400 in early S-phase,Citation3 as demonstrated by fluorescence confocal and high resolution microscopy supplemented by appropriate image analysis and deconvolution. One replication focus is thought to contain several active replication forks,Citation2,4 and the time required to complete replication within this chromatin region is approximately 45 minutes.Citation2
In early and mid S-phase replication origins are activated in euchromatin. Heterochromatin is replicated in mid and late S-phase. The length of S-phase is under strict control and replication of all DNA in a mammalian cell is usually completed within approximately 8 hours. However, under conditions of stress, the active replication forks may stall and the global rate of nucleotide incorporation can decrease. This may occur when the pool of precursors is depleted, DNA damage is inflicted or factors that interfere with replication, like intercalating drugs, bind to DNA.
It is important to recognize that any change in global replication rates measured as the amount of a precursor incorporated per cell within a given time can be brought about by a change in the number of active replication forks, as well as by a change in the rate of nucleotide incorporation. Moreover, it has been suggested that activation of replicating forks in one sub-region of the nucleus may be accompanied by inhibition of replication in other regions.Citation5,6 Thus, in order to understand cellular response to factors that interfere with replication, the effects of replication stress need to be investigated locally, within chromatin sub-domains, replication factories and individual replication forks, and considered in terms of the number of active replication sites, as well as local, rather than just global rates of nucleotide incorporation. The existing knowledge about activation of replication origins under conditions of stress is derived primarily from studies of DNA fibers pulled out of nuclei.Citation7-12 We note, however, that these studies do not provide information about distances between the originally active and newly activated replication regions in 3D space of individual nuclei. They also provide no information about replication rates in individual replication factories, nor about the spectrum of reactions of individual cells within a population to stress. This gap can be filled by advanced quantitative 3D microscopy. The work reported here is focused on understanding the local sub-nuclear response to replication stress, in the context of the whole nucleus, caused by the topoisomerase I inhibitor topotecan, a known inducer of replication-related double strand breaks.Citation13,14
Two principal, seemingly opposing mechanisms of response to replication stress were described previously. One of them entails activation of new replication origins, while the other is based on a global halt of replication or a decrease in the number of active replication sites.
Activation of new replication origins was postulated and demonstrated nearly 40 years ago.Citation7,15 Taylor concluded that under standard conditions cells activate only 1 out of 15 to 20 potentially available replication origins. Since the rate of nucleotide incorporation was lower under conditions of stress, apparently the cells attempted to compensate by recruiting more replication origins. These origins would not have been used, had the stress not been induced. Anglana et al.Citation16 arrived at a similar conclusion that, under standard conditions, when progression of replication forks is not impeded and their progress is rapid, only a specific subset of replication origins is activated. However, in conditions of replication stress induced by a shortage of precursors or DNA damage, the speed of fork progress diminishes, and additional replication origins are activated;Citation7,16 reviewed in ref.Citation5
In contrast to these studies, stalling replication forks due to DNA damage or exhaustion of the pool of available precursors and a global halt of replication were reported by several groups.Citation8,17,19 A study by Petermann et al.Citation20 demonstrated that after brief replication blocks induced by hydroxyurea (HU, 2 mM, 1 to 2 hour blocks) most replication forks resume progression, but after long HU blocks (24 hours) their reactivation does not occur. The reduced number of restarting forks after long HU blocks was accompanied by an increased frequency of firing of new origins. These data suggest that stalled replication forks retain the ability to restart for some time but subsequently become inactivated in a process that coincides with accumulation of DNA damage and double strand break (DSB) formation.
The abovementioned observationsCitation7,19,20 suggest that 2 types of response to replication stress arising from a depletion of DNA precursors can occur: activation of previously dormant replication origins when the replication stress is brief, or a global… previously dormant replication origins when the replication stress is brief, or a global halt to replication and inhibition of activation of new origins after a longer period of precursor shortage. It is still unclear if only one of these 2 principal mechanisms is activated under certain stress conditions or if both are activated simultaneously in different regions of the nucleus.
In this report we describe sub-nuclear localization of newly activated replication foci with respect to the regions of the stalled replication forks, and replication inhibition under the conditions of topotecan-induced replication stress, using quantitative 3-dimensional confocal microscopy and live cell imaging.
Results
Replication under optimal conditions
In order to provide a basis for investigating decreased replication rates and activation of new replication foci under conditions of drug-induced stress, we first imaged replication patterns of A549 cells under optimal growth conditions. At least 3 distinct replication patterns were visually distinguished by imaging PCNA (, Suppl. Movie SM1) or sites of incorporation of DNA precursors (). These replication patterns were similar to the ones reported previously in A549 and other cell lines Citation14,21,22 The typical replication patterns were more easily recognized in images of a central slice than in maximum intensity projections of the whole nucleus (, compare the left and the right column).
Figure 1. Replication patterns observed in A549 cells under standard conditions, and the numbers of replication foci in the absence and presence of replication stress induced by camptothecin. (A) changes of the pattern of replication foci visualized in live cells by detecting PCNA bound to DNA. Images are maximum intensity projections of the whole nucleus, recorded at 20 minutes time intervals. See also Suppl. Movie SM1. (B) central slices of the 3D images of nuclei with incorporated precursor EdU labeled by ‘click’ reaction (left column) show typical replication patterns characteristic for early, mid and late S-phase; the right column – maximum intensity projections. (C) a schematic representation of the experiment where replication foci were imaged in untreated or in camptothecin-treated cells, by detecting the incorporated EdU (a – untreated cells; b – cells exposed to 0.2 μM Cpt for 60 minutes; EdU was present prior and during exposure to the drug). Note, that the number of foci labeled here embraces the foci active before and during the stress. (D) the numbers of EdU-incorporation foci in untreated and Cpt-treated cells, in early, mid, and late S-phase.
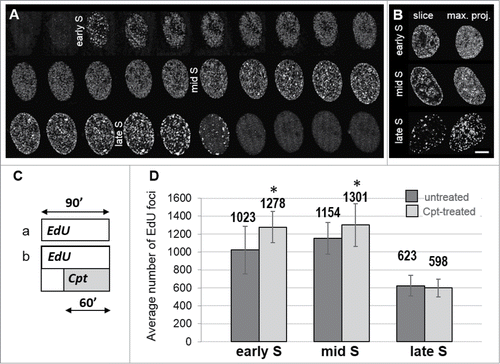
In accordance with previous reports,Citation2,22 we define early S-phase sub-stage as the period when a large number of small replication foci are detected; these foci are relatively evenly distributed throughout chromatin. Early S lasted for approximately 4.5 h. In mid S-phase replication was active predominantly in foci located under the nuclear envelope and in perinucleolar regions, while less replication foci were seen in other regions of the nucleus. Mid S lasted for approximately 3 h. Late S is characterized by replication in a relatively low number of large heterochromatin regions. This phase lasted approximately 1 hour (). Not only the distribution, but also the number of recognizable replication foci changed during S-phase. Using our imaging and data processing approach we typically identified approximately 1,000 active foci at any time during the first 5 hours of S-phase, more than 1,150 foci in mid S, and some 600 foci in late S phase (, left columns).
The number of replication foci under stress conditions
Camptothecin and its more stable derivative topotecan (Tpt) are known inhibitors of type I topoisomerase. These drugs stall the movement of replication forks and induce DSBs.Citation23 When cells were challenged with Cpt for 1 hour, despite the global inhibition of replication, the average number of active replication foci increased significantly (). Notably, the increase was highest in early S-phase (25%) and still significant in mid S-phase (13%). In late S-phase, the number of replication foci in Cpt-treated and untreated cells was similar.
Since stalling active replication forks by Cpt apparently resulted in activation of new ones and an increase in the total number of replication foci, the question arose about the actual number of stalled and newly activated foci and about the localization of the newly activated replication regions with respect to the replication foci that had been active under physiological conditions immediately prior to the exposure to topoisomerase I inhibitor. This issue was investigated using 2 pulses of DNA precursors and is described below.
Patterns of newly activated replication foci under conditions of stress – 2 pulse experiments
In order to enable specific detection of replication foci active at time of the first contact with Tpt, and new replication origins, activated under stress, cells were exposed to 2 pulses of different DNA precursors, EdU and BrdU. First a pulse (EdU) was applied for 15 minutes before induction of replication stress by Tpt. The second pulse (BrdU) was given at the end of the exposure to the drug (15 minutes before removal of Tpt and cell fixation; the total time of exposure to Tpt was 0.5 h, 2 h or 4 h). It is important to consider that under similar experimental conditions, and using the same concentrations of both precursors, BrdU is incorporated with much higher efficiency than EdU. Therefore, using our protocol and this sequence of precursor pulses, replication regions that were active before replication stress were labeled with EdU, while regions activated during the last 15 minutes of exposure to Tpt generated exclusively BrdU signal (, ). Analysis of distances between the replication foci activated under stress and the nearest original replication foci (that were active under physiological conditions) is given in .
Figure 2. Images of a distribution of replication foci active prior (EdU, green) and at the end (BrdU, red) of exposure to topotecan lasting for 0.5 h (B) or 4 h (D), in early, mid and late S-phase (A, C, untreated cells). 3D images were deconvolved and normalized; maximum intensity projections of 10 central confocal planes are shown.
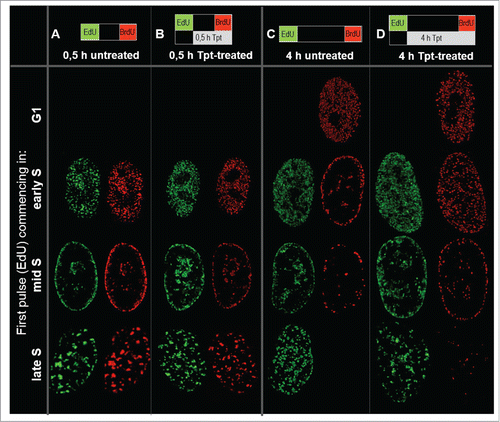
Figure 3. Two color images of DNA replication prior (EdU, green) and at the end (BrdU, red) of Tpt exposure (0.5 h, commencing in early or late S-phase (A, B), or 4 h, commencing in early or mid S (C, D)), demonstrating close proximity of the replication sites activated under stress in relation to the original factories. Maximum intensity projections of 10 central confocal sections are shown; note, that when the newly activated foci overlap with the original ones, they appear yellow (green plus red). Scale bars: nuclei 5 μm, insets 500 nm.
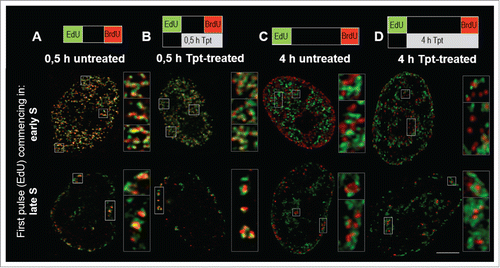
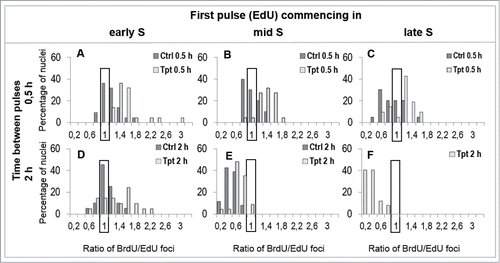
Figure 4. Histograms of distances between replication foci active 0.5, 2 or 4 h apart (EdU – the first pulse, BrdU – the second pulse), in the 3 subphases of replication in cells under optimal growth conditions (A–G), and in cells under stress conditions (i.e., – EdU prior to stress, BrdU – under stress) (H–P).
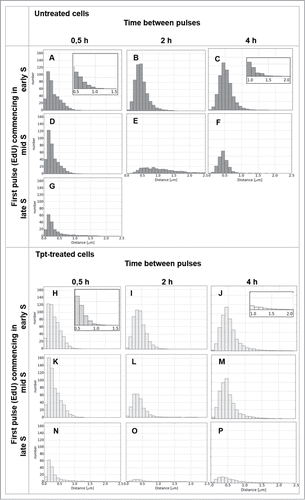
Figure 5. The spectrum of reactions of cells to Tpt-induced stress (0.5 or 2 h). The percentage of cells, in which the numbers of replication foci increased or decreased under the conditions of stress, are shown. For instance, in early S all cells exposed to Tpt increased the percentage of replication sites within 0.5h by a factor of 1.2 up to 3.0, in comparison with untreated cells (A), however in late S-phase some cells increased the number of active replication sites by a factor of 1.6, but other cells decreased it by a factor of 0.6 (C).
Images in demonstrate that during a 0.5 h exposure to Tpt (in early, mid and late S) replication continued and the pattern of replication foci did not change significantly and appeared very similar to the pattern seen without the drug exposure (). However, a 4 h exposure to Tpt resulted in a halt to the usual succession of patterns characteristic for early, mid and late S-phase (). While the untreated cells progressed through S-phase and displayed typical changes of replication patterns, the treated cells preserved the pattern characteristic for the phase in which the exposure to Tpt commenced. Although replication still continued, the treated cells incorporated less DNA than the untreated cells, as demonstrated by the relative intensities of signals in the presence vs. absence of Tpt (see below). Similar observations, namely an inhibition of replication and a delay in progression through the cell cycle were reported in HT29 human colon adenocarcinoma under the influence of camptothecin at 2.5 μM.Citation24 Interestingly, we noticed that the size of nuclei in the treated cells was significantly larger than in untreated cells (). The lack of change in the replication pattern, accompanied by a lower rate of DNA synthesis, suggests that replication continued mostly in the regions where it had been active at the time when the drug was introduced, and that there was no activation of replication in distant regions. In order to verify this statement we compared the patterns of DNA replication prior and at the end of Tpt exposure, and analyzed the distances between the regions of incorporation of the precursors of the first and the second pulse, using the analytical approach which was described recently.Citation14 These analyses are described in the following section.
Analysis of distances between new and original replication foci
In untreated cells, when the 2 pulses were separated by 30 min., in early S as well as mid S, replication was generally continued in the same regions or in the immediate vicinity (closer than 300 nm) of the original replication area ( – images, – quantitative analysis). Only in the case of late S cells a significant number of replication sites were activated at larger distances from the original mid S sites (). When the time interval between the 2 precursor pulses was longer (2 h and 4 h) active replication was found in many new regions, separated from the original sites by more than 300 nm (images in , quantitative analysis in and ). This is consistent with typical reported replication patterns in untreated cells.Citation14 An increase in the overall number of replication sites during transition from early S to mid S and a decrease in transition to late S was also reflected in the surface areas of the histograms in .
In cells exposed to Tpt for 30 min in early, mid or late S the sites of replication active during the last 15 min of exposure to the drug were generally located within or very close (less than 300 nm) to the original sites, suggesting that under conditions of stress active replication was maintained in the same regions ( (images), also compare quantitative data in vs. ). However, it is important to note that at the end of a 0.5 h exposure to Tpt, which commenced in early S, a number of replication foci were activated at a distance from the original ones (; compare the right side of the histogram with ). This ability to recruit new replicating regions was not maintained at the end of longer, 2 and 4 h exposures to Tpt (). The general tendency to continue replication at the original sites is also illustrated by the behavior of cells exposed to Tpt in mid S. After 0.5 h with Tpt replication continued in the same regions (). At the end of a 2 h and 4 h exposure to Tpt, corresponding to a time point when untreated cells began to exhibit replication at new sites distant from the original ones (, ), the treated cells still continued to replicate predominantly in the originally active regions (, ,M vs. ). The cells exposed to Tpt in late S followed the same trend – they continued to replicate in the originally active sites (), and this process lasted for several hours, while the untreated cells had already finished replication (). This observation reinforces the notion that, under conditions of stress, there was a general tendency to continue replication in the originally activated areas rather than activate new distant regions. The only exception was early S, when the cells exposed to Tpt for 0.5 h (but not 2 or 4 h) demonstrated an ability to activate many new distant replication sites.
When the exposure to Tpt lasted 4 h, the number of still active replication regions was significantly lower than in untreated cells (see below, ). The amount of the incorporated precursor was also lower (see below, ). This suggests that replication persisted in some, but not all originally active regions, and that either the number of replication forks or the rate of precursor incorporation was lower than in the untreated cells.
Figure 6. For figure legend, see page 2642.
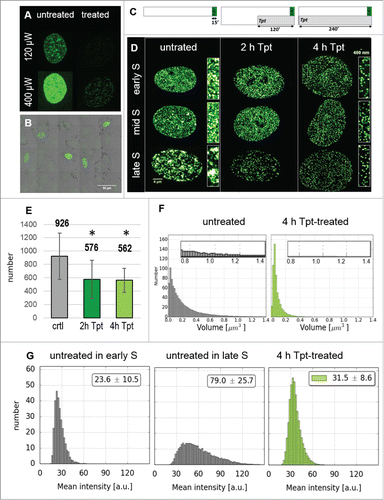
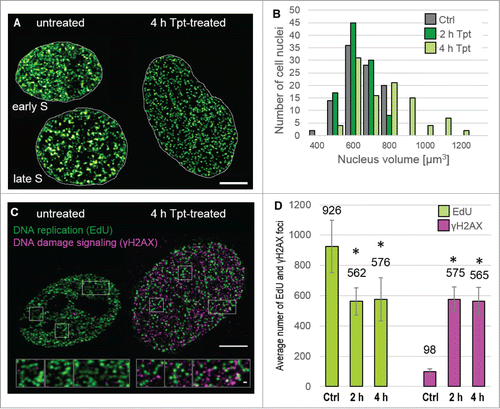
Figure 7. Nuclei volumes and DNA damage signaling following Tpt-induced replication stress (the outline of the experiment as in ). (A) Replication patterns in nuclei of untreated cells in early and late S phase, and in an enlarged nucleus of a Tpt-treated cell. Scale bar 5 μm, (B) Distribution of nuclear volumes within a cell population (untreated, 2 and 4 h exposure to Tpt) demonstrating a growing proportion of cells with enlarged nuclei, (C) Patterns of replication and γH2AX foci in untreated and Tpt-treated (4 h) cells. Scale bars: nuclei 5 μm, insets 500 nm. (D) The numbers of replication and γH2AX foci, demonstrating a decrease of the number of replicating regions, and a concomitant dramatic increase of the number of DDR foci. Approximately 100 γH2AX foci/nucleus represent endogenous damage signaling, while an increase to approximately 550 foci represents Tpt-induced damage, most likely double-strand breaks.
Spectrum of cellular reactions to replication stress
The histograms shown in . depict the distribution of distances between the replication sites active under and prior to stress. Such histograms represent the data averaged over the whole cell population. These data do not reveal, however, the total numbers of replication sites in individual cells, nor do they show the spectrum of reactions of various cells within the whole population. We noticed that the number of replication foci activated after 2 h of Tpt treatment appeared to differ widely within the cell population. For instance, we saw cells that reacted to a 2 h stress by a dramatic increase in the number of replication sites, but we also detected cells in which the number of replication foci decreased significantly. In contrast, no such differences in the number of replication sites were noted after a 4 h Tpt treatment. In this case more than 90% of cells responded in a similar manner, i.e. a global halt to replication and activation of some new origins was observed. Therefore we measured the numbers of cells in which the number of replication foci changed by a given factor under conditions of stress, and displayed this information in . This graph is intended as a means of visual interpretation of the spectrum of changes of replication activity in a cell population under conditions of stress.
After a 0.5 h Tpt treatment, which started in early and mid S (), all cells increased the number of recognizable replication foci in comparison with untreated cells. When the exposure to Tpt started in late S (), a slight increase in the number of replication foci occurred in most cells, but a decrease occurred in some cells. At the same time in the untreated population a majority of cells showed a decrease of the number of replication foci. In other words, within the population of Tpt-treated cells, the subpopulation which activated new replication foci was greater than in untreated cells.
When a 2 h Tpt treatment was applied in early S (), a great majority of cells showed an increase in the number of replication sites. In the case of a 2 h Tpt treatment starting in mid S, a decrease was seen in most cells, though a small portion of cells showed no change in the number of replication foci (). At the same time the untreated cells showed a strong decrease in the number of replication sites (). If the exposure to Tpt started in late S, within 2 h all treated cells had less active replication foci than before the drug exposure (). The untreated cells had completed replication by that time.
In general, these data are consistent with the observation that a 0.5 h exposure to Tpt induced an overall increase in the number of replication foci in early and mid S, as compared to untreated cells, and demonstrates that the reactions of individual cells within a population vary significantly. Activation of new origins was not a uniform reaction. Although a significant number of cells showed an increase, others, under the same stress conditions, showed a decrease in the number of recognizable replication sites.
Rates of replication under stress
So far we have demonstrated that most cells reacted to Tpt-induced replication stress (0.5 h exposure) by increasing the number of replication sites, although the spectrum of these reactions was very wide (). A longer exposure (2 or 4 h) resulted in less replicating regions (). However, in order to gain a complete picture of the reaction of a cell to drug-induced replication stress, a number of issues need to be clarified, including the rate of DNA synthesis within replication factories (the amount of the precursor incorporated in individual active replication regions) and the number of active replication forks in individual recognizable replication regions. Some of this information can be gained by analyzing the intensity of fluorescence of the incorporated nucleotide precursors and the size of replication regions ().
The experiments described so far were focused on accurate detection of the number of replication sites in untreated and stressed cells. To this end, the instrumental parameters used for each sample were adjusted for maximum sensitivity. Since the intensity of the exciting light and instrumental gain had to be optimized, quantitative analysis of the amounts of the precursors incorporated within replicating areas in different samples was cumbersome (). An accurate detection of the amount of incorporated precursors was also complicated by the fact that the intensities of fluorescence encountered in different cells within the same population and among various replication foci within one nucleus differed widely, and often the range of fluorescence intensities exceed the dynamic range of the detector. An example is given in , which demonstrates that 48 hours after exposure to Tpt many cells still replicate DNA intensively, while others incorporate only small amounts of EdU. Thus, we performed independent experiments, in which we measured the amounts of the incorporated precursors on the basis of the integrated intensity of fluorescence signals associated with individual replication foci. Keeping in mind the technical limitations described above we analyzed the numbers, volumes and fluorescence intensities of replication regions in untreated and Tpt-treated cells. The outline of the experiment is depicted in , and the images of replication regions detected in cells exposed to Tpt for 2 or 4 h are shown in . The total number of replication foci (detected via EdU incorporation) in cells exposed to Tpt for 2 or 4 h reached only approximately 60% (62.1% and 60.7%) of the number detected in untreated cells (). However, within this population, the number of small replication foci (<0.1 μmCitation3) was much higher in Tpt-treated than in untreated cells. These small foci may have embraced fewer replication forks than the larger ones. Larger foci (volume of 0.8 to 1.4 μmCitation3; enlarged parts inside histograms) were only detected in untreated cells; no such large replication foci were found in cells exposed to Tpt for 4 h. demonstrates that in untreated cells the fluorescence intensities of individual replication foci increased as the cells moved from early to late S (this agrees with the PCNA imaging shown in ) suggesting that more precursor molecules were incorporated per replication focus in late S then in early S. In cells exposed to Tpt, however, the fluorescence intensities of individual replication foci were significantly lower and did not differ appreciably between cells, regardless of their position in the cell cycle at the time of exposure to Tpt (). Apparently the total amounts of the precursors incorporated in various replication foci in cells under stress were similar. These amounts were much lower than in untreated cells. This would be expected if the number of active replication forks embraced by one factory was indeed reduced under the influence of Tpt.
As demonstrated above, Tpt caused stalling of replication. Interference with replication forks must have led to DNA damage (DSBs). As DNA damage has been shown to lead to chromatin unfolding and expansion, we reasoned that such chromatin expansion resulting from a large number of DSBs might be responsible for enlargement of nuclei after 4 h of Tpt (). We measured the increase of nuclear volumes and imaged phosphorylation of histone H2AX on serine 139 in order to relate nuclear size to the level of DNA damage signaling induced by Tpt. The number of γH2AX foci increased fivefold after 2h and 4 h with Tpt (the number of replication foci decreased by ca. 40% within this time, ). There was no detectable difference in the distribution of nuclear sizes before and after 2 h of Tpt (), but the 4 h exposure to Tpt resulted in a dramatic increase in the nuclear volumes. At this stage many nuclei were approximately 2 times larger than the untreated ones (exhibiting a volume of over 1000 μm3, while the volume of nuclei in untreated cells was approximately 500–600 μmCitation3) (). The Tpt-induced growth in nuclear volume may have been related to chromatin expansion following damage.Citation25-27 However, it is interesting to note that the nuclear volume was not directly proportional to the number of γH2AX foci.
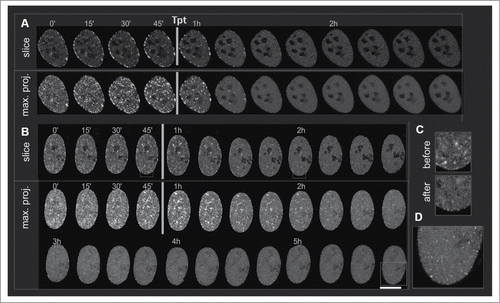
As described above, drug-induced replication stress caused an initial increase, followed by a decrease of the number of replication foci. This decrease might be due to a halt of active fork progression but without dismantling of the replication complex, or it can be due to a complete detachment of the replication complex from the DNA strand. We investigated this issue by time-lapse imaging of PCNA in replication foci in cells exposed to Tpt. Cells expressing EGFP-tagged PCNA were imaged for 1 h in the absence of Tpt. Subsequently replication stress was induced by adding Tpt, and the cells were imaged for 2 to 5 hours (, Suppl. Movie SM2). The two examples shown in demonstrate that the intensity of fluorescence of EGFP-PCNA in individual replication foci decreased significantly within 15 minutes after adding the drug, while the intensity of the uniformly distributed, unbound PCNA increased (). This observation agrees with the notion that PCNA detaches from DNA under the influence of Tpt, implying a lower number of active replication forks. It is worth noting that small PCNA foci were still present even 4–5 hours after exposure to Tpt ().
Figure 8. Live cell imaging of GFP-PCNA in replication of cells exposed to Tpt. 3D stacks were acquired every 15 minutes; cells were imaged for at least 1 hour before Tpt was added to culture medium. Confocal slices and 3D maximum intensity projections are shown. Scale bar 10 μm. (A) A gradual loss of PCNA from replication foci after adding Tpt in late S, (B) As in (B), following Tpt exposure commencing in mid S. PCNA persists in some foci even for 2h, and the mid S replication pattern is maintained, (C) The subnuclear distribution of PCNA before and after Tpt treatment differs – the signals from replication foci decrease while the signal of the mobile fraction of PCNA increases (see also Suppl. Movie SM2), (D) after 4–5 h with Tpt PCNA in new replication sites becomes visible.
Discussion
Low stress levels and activation of new replication factories
The evidence presented in this report indicates that replication stress induced by Tpt and the consequent halting of many replication forks does not result in global cessation of replication. Moreover, cells respond to replication stress by activating new replication regions. Imaging of replication sites before and at the end of 0.5 h Tpt exposure shows that there is no significant change in the general sub-nuclear pattern of replication (., .), despite the fact that the number of replication foci increase significantly in the case of early and mid S-phase (). This observation testifies to the general rule that under conditions of low stress the cell activates new replication origins readily without a global halt to replication. Importantly, in early S, when the stress is moderate (0.5 h with Tpt), the new replication sites are activated both near and far from the original ones. In all other cases (0.5 h Tpt in mid or late S and 2–4 h Tpt in all sub-phases of S) replication continues only in the immediate vicinity of the originally active sites.
The fact that the number of replication foci (both near and far from the original foci) increases under low stress conditions in early S but not in late S may indicate that the number of licensed replication origins that can be activated decreases as the cell progresses through S-phase. This is consistent with earlier observations demonstrating that replication cannot occur in already replicated stretches of DNA.Citation20 It also indicates that, just as under optimal growth conditions, cells experiencing replication stress do not over-replicate their DNA. Finally, these observations suggest that activation of new replication origins from the pool of dormant origins within individual replication clusters, under conditions of mild stress, is generally restricted to the clusters (factories) active at a time of stress initiation, while activation of distant origins and new replication factories is suppressed.
High stress levels and a global halt to replication
After a longer Tpt exposure (2 h or 4 h, all sub-phases of S) the rate of replication as well as the total number of replication factories decreased (), but new replication sites were still activated. The majority of these new sites were located close to the original ones. This observation is consistent with a general rule stipulating that replication under stress is generally continued within the originally active regions. Here, under conditions of heavy stress, the rate of nucleotide incorporation was much slower and replication continued mostly in the originally activated factories. Activation of new regions was rarer than in early S. In other words, under high stress the cells maintained their global replication pattern, and replication slowed down but persisted. At the same time cells, that did not experience replication stress, proceeded with typical changes of successive replication patterns.
The question arises of whether Tpt-induced stress caused the active forks to continue their movement at a lower pace or induced new replication forks within the originally active factories. Obviously, when replication is detected in the same region before and during the exposure to Tpt, as described above, 2 possible mechanisms need to be considered - (i) a continuation of movement of some replication forks that had been active prior to adding Tpt, and (ii) activation of new forks immediately adjacent to the previously active ones. Confocal microscopy cannot distinguish between the 2 possibilities due to insufficient spatial resolution, but it can determine the positions of local maxima in EdU incorporation images with a precision of approximately 100 nm.Citation14 The images shown in have sufficient quality to be analyzed both visually and by our quantitative image cytometry approach in order to precisely determine the positions of newly activated replication sites.Citation14 The quantitative analysis of over a hundred 3D images suggests that new forks are indeed activated within or very close to the existing replication factories ().
Rate of replication under stress
The number of replicons embraced by a replication focus which can be resolved by confocal microscopy is probably on the order of 10.Citation28,29 The replication foci imaged after a 2 and 4 h long exposure to Tpt are less numerous than in untreated cells and have a uniform, smaller size. This suggests that when replication forks are stalled, new ones are activated in the immediate vicinity, but eventually the number of replication forks active under stress in each of these foci is lower. It also hints at a mechanism whereby the cell is struggling to complete replication within the region where it was initiated, before activating replication in distant areas.
Interestingly, replication foci that appear under stress conditions (4 h Tpt) often form strings (enlarged images in and ) resembling the structures formed by Pol II foci.Citation30 Such strings of replication foci are also seen in images of nuclei of untreated cells (however they are relatively more difficult to image due to the high density of actively replicating regions, ). A characteristic pattern of foci arranged along a curved line may arise from positioning at the surface of structures like 1Mbp domains, as would be expected if the replication process were performed at the edges of such domains.Citation31,32
Global DNA damage response and the size of cell nuclei
Exposure to Tpt leads to induction of DSBs. It has been shown previously that DNA damage leads to loosening of chromatin structure and opening of regions of high DNA density. This phenomenon may be at the origin of a dramatic increase of nuclear size after 4 h with Tpt. Although a roughly 2-fold increase in the size of the nucleus is typically observed during transition from G1 to G2 phase of the cell cycle and associated with doubling of the genome, the Tpt-induced increase occurred under conditions of inhibited DNA replication after 4 h with Tpt. Thus, this increase could not be a consequence of the increasing DNA content. We note that the 2 h exposure to Tpt did not result in a detectable increase in nuclear size. The increase in nuclear size may be a consequence of widespread phosphorylation of histone H2AX and electrostatic repulsion between numerous new negative charges acquired by the surface of chromatin fibers. However, a dramatic increase in the size of cell nuclei after 4 h with Tpt cannot be linked directly to the level of DNA damage and the subsequent histone H2AX phosphorylation. We have established that 0.5 h of incubation with Cpt (0.2 μM) results in approximately 225 γH2AX foci/nucleus (data not shown) while 2 h and 4 h exposures result in approximately 550 γH2AX foci/nucleus () and a significant decrease in the number of replication foci. Therefore the increase in nuclear volumes, even if related, was not a linear function of the number of induced DNA lesions. This suggests that the Tpt-induced increase in nuclear volume, which we putatively associate with the significant increase in the concentration of γH2AX, may be occurring above a certain threshold of the DNA damage level.
PCNA is rapidly detached from replisomes
The stress-induced decrease of PCNA fluorescence in previously active replication foci, and an increased concentration of soluble PCNA, indicates that PCNA is detached from DNA upon exposure to Tpt. It is thus reasonable to assume that when replication forks encounter the Tpt-DNA-topoisomerase complex, the replication complex is dismantled, and all the components including the sliding clamp are detached from the DNA. Based on these and earlier observations described above we postulate that, in agreement with the previous conclusion, there is a lower number of active replication forks in cells exposed to Tpt, replication is generally resumed in the immediate vicinity of the stalled fork, but at a lower local concentration of PCNA. The issue is complicated by the fact that PCNA is also involved in repair of DNA damage, including post-replication repair.Citation33 Thus some of the PCNA detached from replication sites is expected to be recruited to Tpt-induced damage within minutes.Citation34,35
Conclusion
It is known that large eu- and heterochromatin regions are replicated in a well defined sequence, however activation of individual replication origins within individual replication clusters is thought to be largely stochastic (Blow et al. 2011). Taken together, our data speak in favor of a notion that under conditions of mild stress activation of individual origins of replication within one replication cluster remains stochastic, while a preset order of activating of replication in large chromatin regions remains unaltered. This would explain why new replicating regions appear always (with some exceptions in early S) within or adjacent to the replication foci active prior to stress. This phenomenon is not only a feature of A549 cells, since our preliminary experiments involving HeLa cells demonstrated similar proximity between replication foci active prior and under topotecan-induced stress (data not shown). Studies of the mechanisms of replication stress in cancer cells may have practical implications in designing new treatment strategies.Citation39,40
Materials and Methods
Cell cultures
A549 human lung adenocarcinoma cells were obtained from ATCC and cultured in Nutrient Mixture Ham F-12 (Sigma-Aldrich, cat. no. N6760) supplemented with 10% fetal bovine serum (Sigma-Aldrich, cat. no. F2442) and antibiotics. Cells were plated on 0.17 mm thick glass coverslips (Menzel-Glasser, cat no. CB00220RA1), placed in 40 mm diameter tissue culture dishes and grown under standard conditions. Exposures to topotecan (Sigma-Aldrich, cat. no. T2705) or camptothecin (Sigma-Aldrich, cat. no. C9911) were commenced 24–48 h after seeding, when cells were in the exponential phase of growth and reached approximately 70% confluency.
Pulse-labeling of nascent DNA – single pulse experiments (EdU)
In order to fluorescently label DNA synthesized in cells before treatment with camptothecin (Cpt, 0.2 μM, 1 h exposure) a DNA precursor (EdU, 10 μM) was added to culture medium 30 minutes before exposure to the drug. To label DNA which was synthesized under the conditions of replication stress, during the final stage of exposure to topotecan (0.2 μM Tpt for 2 h or 4 h) EdU (20 μM) was added to a culture medium for the last 15 minutes during the drug treatment. At the end of a drug exposure cells were fixed with formaldehyde (4%, methanol free, Electron Microscopy Sciences, cat. no. 15710-S), permeabilized with 0.5% Triton-X 100 (Sigma-Aldrich, cat. no. T8787), and a ‘click’ reaction with a fluorescent label was performed (Click-iT® EdU Alexa Fluor® 488 or 555 Imaging Kit; Invitrogen/Molecular Probes, cat. no. C10338). The labeling procedure was carried out according to the manufacturer instructions.
Pulse-labeling of nascent DNA – double pulse experiments (EdU and BrdU)
In order to label separately the DNA synthesized prior and during replication stress an asynchronous population of A549 cells cultured on glass coverslips was exposed to 2 pulses of DNA precursors. EdU (20 µM) was added to culture medium for 15 minutes prior to drug treatment, subsequently the cell culture was rinsed several times, and fresh medium supplemented with topotecan (0.2 µM) was added. At the end of a 0.5 h, 2 h or 4 h drug treatment BrdU was added for the last 15 minutes of the period of drug exposure. Subsequently the cell cultures were fixed with formaldehyde (4%, methanol free), permeabilized with 0.1% Triton-X 100, and denatured with 4 M HCl. After blocking with 5% BSA cells were incubated with primary anti-BrdU antibodies BrdU, clone MoBU-1 (1 h, 1:100 dilution, Invitrogen/Molecular Probes, cat. no. B35128), rinsed and incubated with a secondary antibody (1 h, 1:1000 dilution, Atto594 goat anti-mouse Sigma-Aldrich cat. no. 76085). EdU was labeled due to manufacturer instructions (Click-iT® EdU Imaging Kit; Invitrogen/Molecular Probes with Atto488 azide dye, ATTO-TEC GmbH, Germany, cat no. AD488-101).
Detecting double strand DNA breaks
DNA double strand breaks (DSBs) were detected by fluorescence immunostaining of γH2AX.Citation36 After 1 h blocking with BSA (3%) a phosphospecific γH2AX mAb (1 h, 1:350 dilution, Millipore, cat no. 05–636) was used, followed by a secondary antibody (1 h, 1:1000 dilution, Alexa Fluor® 568 goat anti-mouse, Invitrogen/Molecular Probes, cat. no. A11004).
Visualizing replication sites in living cells
Cells were transiently transfected with PCNA-EGFP plasmid, originally obtained from Dr. Cristina Cardoso and modified as described previously,Citation37 using FuGENE® HD Transfection Reagent (Promega, cat. no. E2311). Cell cultures growing on coverslips and immersed in F12 culture medium buffered for contact with air, without Phenol Red (Sigma, cat. no. D5030), supplemented with 2% fetal bovine serum, were placed on a microscope stage. Topotecan was added to medium and cells were imaged at 37°C. Microscope Temperature Control System “The Cube & The Box” (Life Imaging Services, Switzerland) was used.
Confocal imaging
Leica TCS SP5 confocal system (Leica Microsystems GmbH, Wetzlar, Germany) was used to image live and fixed cells. The following instrumental parameters were used: 63x HCX PL APO CS NA 1.4 oil immersion lens, confocal iris set at 1 Airy disc, excitation 488 (Ar laser) and 561 nm (HeNe laser), emission detection bands 500–550 nm for AlexaFluor488 (EdU or EGFP-PCNA) and 600–660 nm for AlexaFluor568 (immunofluorescence, γH2AX or BrdU), registration in sequential mode, scanning rate 8000 Hz (resonant scanner), 8 bit dynamic range, with 8–16 averaged frames for one confocal plane. One 3D stack consisted of at least 80 confocal slices, single image with 512x512 pixels (pixel size 60nm) spaced every 130 nm along z axis. In time-lapse imaging of live cells in each time point a stack of 40 horizontal planes was collected. The images were registered at 15 minutes intervals for at least 6 hours.
Image processing and quantitative image analysis
3D images were deconvolved using Huygens Deconvolution & Analysis Software (Scientific Volume Imaging B.V., Hilversum, Netherlands). Quantitative analysis of the deconvolved images representing cell nuclei with replication or DSB foci was used to determine the position, number, volume and mean fluorescence intensity of the biologically relevant fluorescence maxima representing replication foci. The analysis was carried out with the use of algorithms developed under ImageJ macro language and Python. Briefly, local maxima within regions (foci) were delineated using 3D max finder software. The first stage of the maxima finding in 3D space was making an estimation of the background level and the area of the maxima. In the second stage positions of the maxima were determined in each XY slice of the stack with the use of Michael Schmidt's ImageJ plugin ‘Find Maxima’. The noise tolerance parameter was based on calculations made at the first stage. Another search was made in orthogonal XZ slices. The conjunction of the resulting points was considered to represent the positions of the maxima of fluorescence intensities in 3D. The final result consisted of positions (coordinates) of the barycenters of the merged and individual spots obtained with the use of S. Bolte's ImageJ plugin „3D Object Counter.”Citation38
Estimations of the volumes of foci associated with each maximum, which was determined at the previous stage, was performed with the use of a Python script and was based on an iterative flooding of the maxima in 3D space. Each spot was expanded by an adjacent voxel so long as it was not already assigned to the other nearby spot, nor was its gray level lower than the one given in the current iterative step. The expansion ceased when the user-defined gray level was reached. The mean intensity of each spot was calculated as the integrated gray level of the voxels assigned to the spot, divided by its volume.
An independent student's T-test was used in statistical analysis, using a value of t < 0.05 as a measure of significance. The number of nuclei analyzed in was over 20, and over 100 for .
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Author Contributions
PR, AW planned and executed the experiments and participated in writing the manuscript, AH and ŁB wrote the algorithms for assessing spatial relations between subnuclear foci, and analysis of nuclear volumes, and intensities and volumes of subnuclear foci, PR and AH analyzed the data using the algorithms, JWD planned and supervised the experiments, and wrote the manuscript.
1064566_Movie_S2.avi
Download Microsoft Video (AVI) (133.5 KB)1064566_Movie_S1.avi
Download Microsoft Video (AVI) (258.6 KB)Funding
This work was supported by a National Center for Science grant 2011/01/B/NZ3/00609 and 2013/11/B/NZ3/00189. PR and AH are a recipients of SET doctoral studentship from Jagiellonian University. Faculty of Biochemistry, Biophysics and Biotechnology is a partner of the Leading National Research Center (KNOW) supported by the Ministry of Science and Higher Education in Warsaw. Confocal instrumentation was purchased through EU structural funds program BMZ (POIG.02.01.00-12-064/08).
Related Research Data
References
- Cayrou C, Coulombe P, Méchali M. Programming DNA replication origins and chromosome organization. Chromosome Res 2010; 18:137-45; PMID:20066560; http://dx.doi.org/10.1007/s10577-009-9105-3
- Ma H, Samarabandu J, Devdhar RS, Acharya R, Cheng PC, Meng C, Berezney R. Spatial and temporal dynamics of DNA replication sites in mammalian cells. J Cell Biol 1998; 143:1415-25; PMID:9852140; http://dx.doi.org/10.1083/jcb.143.6.1415
- Cseresnyes Z, Schwarz U, Green CM. Analysis of replication factories in human cells by super-resolution light microscopy. BMC Cell Biol 2009; 10:88; PMID:20015367; http://dx.doi.org/10.1186/1471-2121-10-88
- Jackson DA, Pombo A. Replicon Clusters Are Stable Units of Chromosome Structure: Evidence That Nuclear Organization Contributes to the Efficient Activation and Propagation of S Phase in Human Cells. J Cell Biol 1998; 140:1285-95; PMID:9508763; http://dx.doi.org/10.1083/jcb.140.6.1285
- Gilbert DM. Replication origin plasticity, Taylor-made: inhibition vs recruitment of origins under conditions of replication stress. Chromosoma 2007; 116:341-7; PMID:17404750; http://dx.doi.org/10.1007/s00412-007-0105-9
- Yekezare M, Gómez-González, B, Diffley JFX. Controlling DNA replication origins in response to DNA damage – inhibit globally, activate locally. J Cell Sci 2013; 126:1297-306; PMID:23645160; http://dx.doi.org/10.1242/jcs.096701
- Taylor JH. Increase in DNA replication sites in cells held at the beginning of S phase. Chromosoma 1977; 62:291-300; PMID:142621; http://dx.doi.org/10.1007/BF00327029
- Merrick CJ, Jackson, D, Diffley JFX. Visualization of altered replication dynamics after DNA damage in human cells. J Biol Chem 2004; 279:20067-75; PMID:14982920; http://dx.doi.org/10.1074/jbc.M400022200
- Courbet S, Gay S, Arnoult N, Wronka G, Anglana M, Brison O, Debatisse M. Replication fork movement sets chromatin loop size and origin choice in mammalian cells. Nature 2008; 455:557-60; PMID:18716622; http://dx.doi.org/10.1038/nature07233
- Ge XQ, Blow JJ. Chk1 inhibits replication factory activation but allows dormant origin firing in existing factories. J Cell Biol 2010; 191:1285-97; PMID:21173116; http://dx.doi.org/10.1083/jcb.201007074
- Petermann E, Woodcock M, Helleday T. Chk1 promotes replication fork progression by controlling replication initiation. Proc Natl Acad Sci 2010; 107:16090-5; http://dx.doi.org/10.1073/pnas.1005031107
- Guilbaud G, Rappailles A, Baker A, Chen C-L, Arneodo A, Goldar A, D'Aubenton-Carafa Y, Thermes C, Audit B, Hyrien O. Evidence for sequential and increasing activation of replication origins along replication timing gradients in the human genome. PLoS Comput Biol 2011; 7:e1002322; PMID:22219720; http://dx.doi.org/10.1371/journal.pcbi.1002322
- Huang X, Okafuji M, Traganos F, Luther E, Holden E, Darzynkiewicz Z. Assessment of histone H2AX phosphorylation induced by DNA topoisomerase I and II inhibitors topotecan and mitoxantrone and by the DNA cross-linking agent cisplatin. Cytometry A 2004; 58:99-110; http://dx.doi.org/10.1002/cyto.a.20018
- Berniak K, Rybak P, Bernas T, Zarebski M, Biela E, Zhao H, Darzynkiewicz Z, Dobrucki JW. Relationship between DNA Damage Response, initiated by camptothecin or oxidative stress, and DNA replication, analyzed by quantitative image analysis. Cytometry A 2013; 83:913-24
- Ockey CH, Saffhill R. The comparative effects of short-term DNA inhibition on replicon synthesis in mammalian cells. Exp Cell Res 1976; 103:361-73; PMID:137123; http://dx.doi.org/10.1016/0014-4827(76)90272-X
- Anglana M, Apiou F, Bensimon A, Debatisse M. Dynamics of DNA replication in mammalian somatic cells: nucleotide pool modulates origin choice and interorigin spacing. Cell 2003; 114:385-94; PMID:12914702; http://dx.doi.org/10.1016/S0092-8674(03)00569-5
- Henry-Mowatt J, Jackson D, Masson J-Y, Johnson PA, Clements PM, Benson FE, Thompson LH, Takeda S, West SC, Caldecott KW. XRCC3 and Rad51 modulate replication fork progression on damaged vertebrate chromosomes. Mol Cell 2003; 11:1109-17; PMID:12718895; http://dx.doi.org/10.1016/S1097-2765(03)00132-1
- Pasero P, Shimada K, Duncker BP. Multiple Roles of Replication Forks in S Phase Checkpoints. Cell Cycle 2003; 2(6):568-72; PMID:14512770; http://dx.doi.org/10.4161/cc.2.6.577
- Nedelcheva-Veleva MN, Krastev DB, Stoynov SS. Coordination of DNA synthesis and replicative unwinding by the S-phase checkpoint pathways. Nuclear Acids Res 2006; 34:4138-46; http://dx.doi.org/10.1093/nar/gkl528
- Petermann E, Orta ML, Issaeva N, Schultz N, Helleday T. Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol Cell 2010; 37:492-502; PMID:20188668; http://dx.doi.org/10.1016/j.molcel.2010.01.021
- Chagin VO, Stear JH, Cardoso MC. Organization of DNA replication. Cold Spring Harb Perspect Biol 2010; 2:1-13; http://dx.doi.org/10.1101/cshperspect.a000737
- Leonhardt H, Rahn H, Weinzierl P, Sporbert A, Cremer T, Zink D, Cardoso MC. Dynamics of DNA Replication Factories in Living Cells. J Cell Biol 2000; 149:271-9; PMID:10769021; http://dx.doi.org/10.1083/jcb.149.2.271
- Pommier Y, Leo E, Zhang H, Marchand C. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem Biol 2010; 17:421-33; PMID:20534341; http://dx.doi.org/10.1016/j.chembiol.2010.04.012
- Seiler JA, Conti C, Syed A, Aladjem MI, Pommier Y. The intra-S-phase checkpoint affects both DNA replication initiation and elongation: single-cell and -DNA fiber analyses. Mol Cell Biol 2007; 27(16):5806-18; http://dx.doi.org/10.1128/MCB.02278-06
- Kruhlak MJ, Celeste A, Dellaire G, Fernandez-Capetillo O, Müller WG, McNally JG, Bazett-Jones DP, Nussenzweig A. Changes in chromatin structure and mobility in living cells at sites of DNA double-strand breaks. J Cell Biol 2006; 172:823-34; PMID:16520385; http://dx.doi.org/10.1083/jcb.200510015
- Halicka DH, Zhao H, Podhorecka M, Traganos F, Darzynkiewicz Z. Cytometric detection of chromatin relaxation, an early reporter of DNA damage response. Cell Cycle 2009; 8:2233-7; PMID:19502789; http://dx.doi.org/10.4161/cc.8.14.8984
- Burgess RC, Burman B, Kruhlak MJ, Misteli T. Activation of DNA Damage Response Signaling by Condensed Chromatin. Cell Rep 2014; 9:1703-17; PMID:25464843; http://dx.doi.org/10.1016/j.celrep.2014.10.060
- Gillespie PJ, Blow JJ. Clusters, factories and domains: The complex structure of S-phase comes into focus. Cell Cycle 2010; 9:3218-26; PMID:20724827; http://dx.doi.org/10.4161/cc.9.16.12644
- Aparicio OM. Location, location, location: it's all in the timing for replication origins. Gen Dev 2013; 27:117-28; PMID:23348837; http://dx.doi.org/10.1101/gad.209999.112
- Zhao ZW, Roy R, Gebhardt JCM, Suter DM, Chapman AR, Xie XS. Spatial organization of RNA polymerase II inside a mammalian cell nucleus revealed by reflected light-sheet superresolution microscopy. Proc Natl Acad Sci U S A 2014; 111:681-6; PMID:24379392; http://dx.doi.org/10.1073/pnas.1318496111
- Hozák P, Jackson DA, Cook PR. Replication factories and nuclear bodies: the ultrastructural characterization of replication sites during the cell cycle. J Cell Sci 1994; 107(8):2191-202
- Koberna K, Ligasová A, Malínský J, Pliss A, Siegel AJ, Cvacková Z, Fidlerová H, Masata M, Fialová M, Raska I, et al. Electron microscopy of DNA replication in 3-D: evidence for similar-sized replication foci throughout S-phase. J Cell Biochem 2005; 94:126-38; PMID:15523671; http://dx.doi.org/10.1002/jcb.20300
- Lehmann AR, Fuchs RP. Gaps and forks in DNA replication: Rediscovering old models. DNA Repair 2006; 5:1495-8; PMID:16956796; http://dx.doi.org/10.1016/j.dnarep.2006.07.002
- Essers J, Theil, AF, Baldeyron, C, Wiggert, A, Cappellen, V, Houtsmuller, AB, Kanaar, R, Cappellen, WA Van. Nuclear Dynamics of PCNA in DNA Replication and Repair. Mol Gen Genet 2005; 25:9350-9
- Balajee AS, Geard CR. Chromatin-bound PCNA complex formation triggered by DNA damage occurs independent of the ATM gene product in human cells. Nuclear Acids Res 2001; 29:1341-51; http://dx.doi.org/10.1093/nar/29.6.1341
- Rogakou EP, Pilch D, Orr A, Ivanova V, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem 1998; 273:5858-68; PMID:9488723; http://dx.doi.org/10.1074/jbc.273.10.5858
- Trembecka-Lucas DO, Dobrucki JW. A heterochromatin protein 1 (HP1) dimer and a proliferating cell nuclear antigen (PCNA) protein interact in vivo and are parts of a multiprotein complex involved in DNA replication and DNA repair Do not distribute. Cell Cycle 2012; 11:2170-5; PMID:22617335; http://dx.doi.org/10.4161/cc.20673
- Bolte S, Cordelières FP. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc 2006; 224:213-32; PMID:17210054; http://dx.doi.org/10.1111/j.1365-2818.2006.01706.x
- Da-Rè C, Halazonetis TD. DNA replication stress as an Achilles' heel of cancer. Oncotarget. 2015;6(1):1-2
- Tripathi K, Hussein UK, Anupalli R, Barnett R, Bachaboina L, Scalici J, Rocconi RP, Owen LB, Piazza GA, Palle K. Allyl isothiocyanate induces replication-associated DNA damage response in NSCLC cells and sensitizes to ionizing radiation. Oncotarget. 2015;6(7):5237-52; PMID:25742788