
Abstract
The p53 tumor suppressor is a multifaceted polypeptide that impedes tumorigenesis by regulating a diverse array of cellular processes. Triggered by a wide variety of stress stimuli, p53 transcriptionally regulates genes involved in the canonical tumor suppression pathways of apoptosis, cell-cycle arrest, and senescence. We recently discovered a novel mechanism whereby p53 inhibits cystine uptake through repression of the SLC7A11 gene to mediate ferroptosis. Importantly, this p53-SLC7A11 axis is preserved in the p533KR mutant, and contributes to its ability to suppress tumorigenesis in the absence of the classical tumor suppression mechanisms. Here, we report that wild type p53 can induce both apoptosis and ferroptosis upon reactive oxygen species (ROS)-induced stress. Furthermore, we demonstrate that p53's functional N-terminal domain is required for its capacity to regulate oxidative stress responses and ferroptosis. Notably, activated p53 dynamically modulates intracellular ROS, causing an initial reduction and a subsequent increase of ROS levels. Taken together, these data implicate ferroptosis as an additional component of the cell death program induced by wild type p53 in human cancer cells, and reveal a complex and dynamic role of p53 in oxidative stress responses.
Keywords:
Introduction
Often regarded as the “guardian of the genome,” p53 is a central node for the regulation of a variety of cellular processes including apoptosis, cell-cycle, and metabolism.Citation1 Given p53's unequivocal role as a tumor suppressor through its transcriptional modulation of critical genes, it is unsurprising that this polypeptide is mutated in approximately 50 percent of all human cancers.Citation2,3 p53 primarily exerts its tumor suppressive function through transcriptional regulation of target genes involved in apoptosis and cell-cycle arrest. Recent advancements in the field demonstrate, however, that a subset of p53 transcriptional activities involved in the regulation of apoptosis and growth arrest are dispensable for p53-mediated tumor suppression.Citation4,5 Moreover, the separation-of-function p53 mutant, p533KR, retains its ability to regulate metabolic target genes as well as its capacity to effectively suppress tumorigenesis in vivo in the absence of apoptosis and cell-cycle arrest.Citation4 These observations suggest that p53 functions in a more unconventional manner than previously thought, raising the possibility that p53's metabolic activities serve as additional barriers to tumorigenesis.Citation6,7
Seeking to understand this non-canonical p53-mediated tumor suppression, we recently identified SLC7A11 as a novel p53 repression target through microarray screening.Citation8 SLC7A11 is the active component of the cystine/glutamate antiporter complex (system xc−), which is formed by disulfide-linked heterodimerization of SLC3A2 and SLC7A11.Citation9 System xc− contributes to antioxidant defenses by supplying the rate-limiting substrate, cysteine, for glutathione (GSH) synthesis, and also by maintaining redox balance across the plasma membrane.Citation10 More importantly, system xc−, and thus SLC7A11, is a major facilitator in the negative regulation of ferroptosis.Citation11 Notably, p533KR retains its ability to repress SLC7A11, and sensitizes cells to ferroptosis in response to ROS-induced stress,Citation8 providing mechanistic insight as to how p533KR restrains tumorigenesis in the absence of the classical tumor suppression mechanisms.
Although the novel mechanism of p53-mediated ferroptosis has been uncovered, the dynamic relationships between p53, ROS, and ferroptosis remain to be characterized. Here, we show that p53 dynamically regulates ROS, and that sustained p53 activation results in elevated ROS levels and increased sensitivity to ferroptotic cell death. Moreover, we demonstrate that ferroptosis constitutes part of the cell death program induced by wild type p53 and this relies on a functional p53 N-terminal domain, as p53 harboring mutations in this region fails to repress SLC7A11 expression and displays resistance to ferroptosis.
Results
Ferroptosis is a part of the cell death program induced by wild type p53
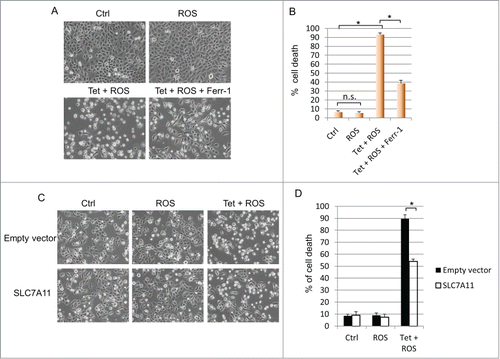
Activation of wild type p53 primarily results in cell-cycle arrest and/or apoptosis.Citation12 It is unclear, however, whether additional cell death mechanisms also contribute to p53's ability to eliminate unsalvageable cells. Our recent finding that p533KR promotes ferroptosisCitation8 prompted us to examine whether wild type p53 also possesses the capacity to induce ferroptotic cell death. H1299 cells containing wild type p53 under the control of a tetracycline-inducible promoter were treated with doxycycline for 16 hours and subjected to ROS-induced stress. While no cell death was detected in cells treated with ROS alone, induction of p53 combined with ROS treatment resulted in massive cell death in which over 90 percent of cells were eliminated. Interestingly, addition of the ferroptosis inhibitor, ferrostatin-1 (Ferr-1), markedly reduced cell death to approximately 40 percent, indicating that both apoptosis and ferroptosis can be induced by wild type p53 upon exposure to oxidative stress (). Since p533KR-mediated ferroptosis occurs through the repression of SLC7A11,Citation8 we investigated whether wild type p53-mediated ferroptosis also relies on the SLC7A11 pathway. Indeed, overexpression of SLC7A11 considerably abrogated ferroptosis caused by combined p53 activation and ROS-induced stress (). Taken together, these data demonstrate that ferroptosis constitutes significant portion of cell death induced by wild type p53, and suggest that both apoptosis and ferroptosis comprise the cell death program of p53 during ROS-induced stress.
Figure 1. Wild type p53 facilitates ferroptosis through SLC7A11 under ROS-induced stress. (A) p53WT tet-on stable line cells were pre-treated with doxycycline (0.1 µg/mL) for 24 hours before ROS (TBH, 60 µM) was added for 3 hours with or without Ferr-1. (B) Quantification of cell death as shown in (A). (C) p53WT tet-on stable line cells were transfected with control plasmid or plasmid overexpressing SLC7A11. Cells were seeded 24 hours later and after attachment, cells were induced by doxycycline (0.1 µg/mL) for 24 hours before addition of ROS (TBH, 30 µM) for another 3 hours. (D) Quantification of cell death as shown in (C). *, P < 0.01, n.s, not significant (Student's t test).
A functional N-terminal domain is required for p53-mediated repression of SLC7A11
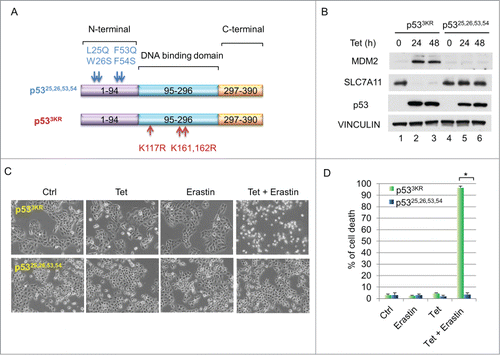
It is well-established that p53's tumor suppression function relies on its transcription activity. While mechanisms of p53-mediated transcriptional activation are well known, the manner in which it represses particular downstream targets remains elusive.Citation13 Specifically, to explore whether N-terminal domain is required for down-regulation of SLC7A11, we generated an H1299 cell line stably expressing a tumor-prone p53,Citation25,26,53,54 mutantCitation5 under a tetracycline-inducible promoter (). Western blot analysis indicated that the protein level of SLC7A11 was markedly decreased upon induction of p533KR, however, induction of p53,Citation25,26,53,54 had no effect on SLC7A11 expression (). To further test whether this abrogated SLC7A11 repression compromises ferroptosis-inducing capability, we induced p53 expression and treated these cells with erastin. While the tetracycline-induced p533KR activation caused massive cell death upon erastin treatment, induction of p53,Citation25,26,53,54 failed to facilitate ferroptosis under the same conditions (). Taken together, these data suggest that a functional N-terminal domain of p53 is required for mediating downregulation of SLC7A11 and facilitating ferroptosis.
Figure 2. (A) Functional N-terminal domain of p53 is required to downregulate SLC7A11 and to promote ferroptosis. (A) Schematic diagram showing the locations and sequences of 2 p53 mutants, p53,Citation25,26,53,54 and p533KR. (B) H1299 tet-on p533KR and p53,Citation25,26,53,54 stable line cells were induced by doxycycline (0.1 µg/mL) and total cell lysate was analyzed by western blots for the expression of MDM2, SLC7A11, p53 and VINCULIN. (C) H1299 tet-on p533KR and p53,Citation25,26,53,54 cells were pre-treated with doxycycline (0.1 µg/mL) for 24 hours and then treated with erastin (10µM); images were taken 40 hours thereafter. (D) Quantification of cell death as shown in (C). *, P <0.01 (Student's t test).
p53 dynamically regulates intracellular ROS
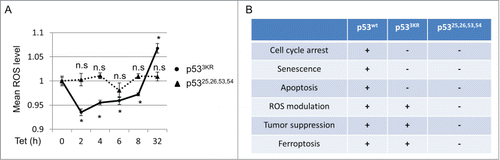
The p53 tumor suppressor possesses both anti- and pro-oxidant functions through regulating a diverse set of downstream effectors.Citation14 For instance, p53 targets like TIGAR, GLS2 and SESN1 have been shown to reduce intracellular ROS by acting on distinct metabolic pathways,Citation15-17 while pro-oxidant functions could be mediated through the activation of PIG3 or PIG6.Citation18,19 Moreover, the execution of ferroptotic cell death requires the accumulation of intracellular ROS and lipid peroxidation.Citation11 Since p533KR retains its capacity to modulate several transcriptional targets involved in metabolic ROS regulation,Citation4,8 we examined the temporal relationship between p533KR activation and intracellular ROS levels. As shown in , activation of p533KR caused an initial reduction in ROS lasting up to 8 hours. Intriguingly, this attenuation was reversed, and an increase in ROS level was observed when p53 stabilization persisted. In contrast, induction of the transcriptionally-defective mutant p53,Citation25,2653,54, which lost the ability to promote ferroptosis, had no significant effect on ROS levels (). The ability of p533KR to temporally alter ROS levels probably reflects its retained capacity to regulate ROS-modulating metabolic targets, including TIGAR, GLS2, and SLC7A11.Citation4,8 These data reveal an intricate balance between p53's anti- and pro-ROS functions, suggesting a complex and dynamic regulation of p53-mediated ROS responses.
Figure 3. p53 dynamically regulates intracellular ROS. (A) p533KR and p53,Citation25,26,53,54 tet-on stable line cells were treated with doxycycline (0.1 µg/mL) for indicated time and ROS levels were determined. (B) Comparison of functional activities among p53WT, p533KR and p53,Citation25,26,53,54. *, P <0.01, n.s, not significant (Student's t test).
Discussion
Cell-cycle arrest and apoptosis serve as important barriers against tumorigenesis. Several lines of evidence indicate, however, that these p53 functions are dispensable for preventing tumor development in vivo.Citation4,5 Our recent findings suggest that an additional route of tumor suppression exists whereby p53 inhibits cystine uptake leading to an attenuation of ROS detoxification and subsequent ferroptosis.Citation8 Here we demonstrate that this critical response relies on p53's N-terminal domain function and its capacity to dynamically modulate ROS levels.
p53's divergent function in ROS regulation stems from its ability to activate or repress target genes with both anti- and pro-oxidant behaviors.Citation14 The various outcomes of this conflicting modulation are likely context specific, but a complete understanding of this paradox is lacking. The dynamic ROS regulation described in the current study () suggests a model whereby p53 mediates an antioxidant response under short term stress, allowing the cell to recover; however if this stress persists, prolonged p53 activation initiates a pro-oxidant response to induce cell death. The rise of ROS at later stage of p53 activation occurs in part from the repression of SLC7A11, although other p53 target genes could also be critical in this cell fate decision with sustained p53 stabilization.
Wild type p53 activation alone appears to primarily induce apoptosis, but our data demonstrate that p53 activation in the presence of ROS-induced stress results in a mixture of cell death that is composed of both apoptosis and ferroptosis (). This suggests an alternative cell death pathway induced by p53 under ROS stress. The precise mechanisms responsible for mediating p53's cell fate decisions are still unclear. With the discovery of p53-mediated ferroptosis, it will be important to decipher how and under what contexts p53 directs the cell toward apoptosis versus ferroptosis. Moreover, it remains to be further investigated in vivo how ferroptosis contributes to tumor suppression and how its function and process relate to those of apoptosis. Potentially, both cell death mechanisms could work in parallel, sequentially, or function as a fail-safe for one another. In any case, how both ferroptosis and apoptosis function together in a physiological setting is likely tissue- and context-dependent.
Cancer cells typically exhibit higher levels of ROS compared to their normal counterparts, and strategies to further increase ROS to induce cell death in transformed cells have been proposed.Citation20 To cope with enhanced ROS levels, however, cancer cells frequently upregulate SLC7A11 thereby strengthening their tolerance to oxidative stress.Citation21,22 As such, inhibitors targeting system xc− have proven to be effective in preventing progression in several types of human cancers.Citation23-28 Thus, utilizing these inhibitors and/or activating p53 to repress SLC7A11 combined with treatments directed at induction of ROS might prove to be an effective therapeutic strategy.
Materials and Methods
Cell culture and stable lines
H1299 cells were previously purchased from ATCC and cultured in DMEM medium supplemented with 10% FBS, 100 units/mL penicillin and 100 µg/mL streptomycin in 37°C incubator with 5% CO2. To generate stable line cells, plasmids were transfected into H1299 cells and selected with puromycin (1 μg/mL) in DMEM medium containing 10% tet-free FBS for 2 weeks.
Plasmids, siRNA and tranfection
Wild type, 3KR (K117R, K161R and K162R) and 25,26,53,54 (L25Q, W26S, F53Q and F54S) mouse p53 cDNA were sequenced and cloned into Tet-on pTRIPZ inducible expression vector (Thermo Open Biosystems). SLC7A11 was amplified from Human Hela Marathon-Ready cDNA (Clonetech) and cloned into TOPO expression vector (Invitrogen). Lipofectamine 2000 (Invitrogen) was used for plasmid transfection according to the manufacturer's protocols.
Western blotting and antibodies
Proteins lysate were prepared using RIPA buffer containing 10 mM Tris-Cl (pH 8.0), 150 mM NaCl, 1% Triton X-100, 1% Na-Deoxycholate, 1mM EDTA, 0.05% SDS and fresh 1X proteinase inhibitor. Equal amount of proteins were loaded and separated in polyacrylamide gels. Proteins were transferred to Hybond ECL membrane (GE healthcare) and incubated overnight with primary antibodies against SLC7A11 (ab37185, abcam), p53 (CM5, Leica biosystems), MDM2 (Ab5, Millipore) and VINCULIN (hVIN-1, Sigma-Aldrich). HRP-conjugated secondary antibodies were used and western signals were detected on autoradiographic films.
Cell death count, drugs and inhibitors
All drugs were ordered from Sigma-Aldrich except otherwise indicated. Ferrostatin-1 was from Xcess Biosciences. Cells were trypsinized and stained with trypan blue followed by counting with a hemocytometer using standard protocol. Cells stained blue were considered as dead cells. Cell death inhibitors were used at the following concentrations: Ferr-1, 2 µM.
ROS treatment and measurement
Reactive oxygen species (ROS) was generated by tert-Butyl hydroperoxide (TBH). Cells were about 50% confluent when medium containing TBH was added. Specific cell death inhibitors were added at the same time. To measure intracellular ROS levels, cells were incubated with CM-H2DCFDA (Invitrogen) for one hour before harvested for FACS analysis.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by the National Cancer Institute of the National Institutes of Health under Award 5R01CA172023, 5RO1CA169246 and 2RO1CA085533 to W.G. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. This work is also supported by the National Science Foundation Graduate Research Fellowship awarded to J.H.H. under Grant No. 11-44155. Any opinion, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the National Science Foundation.
References
- Vousden KH, Prives C. Blinded by the Light: The Growing Complexity of p53. Cell 2009; 137:413-31; PMID:19410540; http://dx.doi.org/10.1016/j.cell.2009.04.037
- Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science (New York, NY) 1991; 253:49-53; http://dx.doi.org/10.1126/science.1905840
- Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature 2000; 408:307-10; PMID:11099028; http://dx.doi.org/10.1038/35042675
- Li T, Kon N, Jiang L, Tan M, Ludwig T, Zhao Y, Baer R, Gu W. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell 2012; 149:1269-83; PMID:22682249; http://dx.doi.org/10.1016/j.cell.2012.04.026
- Brady CA, Jiang D, Mello SS, Johnson TM, Jarvis LA, Kozak MM, Kenzelmann Broz D, Basak S, Park EJ, McLaughlin ME, et al. Distinct p53 transcriptional programs dictate acute DNA-damage responses and tumor suppression. Cell 2011; 145:571-83; PMID: 21565614; http://dx.doi.org/10.1016/j.cell.2011.03.035
- Wang SJ, Gu W. To be, or not to be: functional dilemma of p53 metabolic regulation. Curr Opin Oncol 2014; 26:78-85; PMID:24240177; http://dx.doi.org/10.1097/CCO.0000000000000024
- Maddocks OD, Vousden KH. Metabolic regulation by p53. J Mol Med 2011; 89:237-45; PMID:21340684; http://dx.doi.org/10.1007/s00109-011-0735-5
- Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, Baer R, Gu W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015; 520:57-62; PMID:25799988; http://dx.doi.org/10.1038/nature14344
- Sato H, Tamba M, Ishii T, Bannai S. Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J Biol Chem 1999; 274:11455-8; PMID:10206947; http://dx.doi.org/10.1074/jbc.274.17.11455
- Conrad M, Sato H. The oxidative stress-inducible cystine/glutamate antiporter, system x (c) (−) : cystine supplier and beyond. Amino Acids 2012; 42:231-46. Epub 2011 Mar 16; PMID:21409388; http://dx.doi.org/10.1007/s00726-011-0867-5
- Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 2012; 149:1060-72; PMID:22632970; http://dx.doi.org/10.1016/j.cell.2012.03.042
- Polyak K, Waldman T, He TC, Kinzler KW, Vogelstein B. Genetic determinants of p53-induced apoptosis and growth arrest. Genes Dev 1996; 10:1945-52; PMID:8756351; http://dx.doi.org/10.1101/gad.10.15.1945
- Riley T, Sontag E, Chen P, Levine A. Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol 2008; 9:402-12; PMID:18431400; http://dx.doi.org/10.1038/nrm2395
- Liu B, Chen Y, St Clair DK. ROS and p53: a versatile partnership. Free Radic Biol Med 2008; 44:1529-35; PMID:18275858; http://dx.doi.org/10.1016/j.freeradbiomed.2008.01.011
- Budanov AV, Sablina AA, Feinstein E, Koonin EV, Chumakov PM. Regeneration of peroxiredoxins by p53-regulated sestrins, homologs of bacterial AhpD. Science 2004; 304:596-600; PMID:15105503; http://dx.doi.org/10.1126/science.1095569
- Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, Gottlieb E, Vousden KH. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 2006; 126:107-20; PMID:16839880; http://dx.doi.org/10.1016/j.cell.2006.05.036
- Suzuki S, Tanaka T, Poyurovsky MV, Nagano H, Mayama T, Ohkubo S, Lokshin M, Hosokawa H, Nakayama T, Suzuki Y, et al. Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc Natl Acad Sci U S A 2010; 107:7461-6; PMID:20351271; http://dx.doi.org/10.1073/pnas.1002459107
- Polyak K, Xia Y, Zweier JL, Kinzler KW, Vogelstein B. A model for p53-induced apoptosis. Nature 1997; 389:300-5; PMID:9305847; http://dx.doi.org/10.1038/38525
- Rivera A, Maxwell SA. The p53-induced gene-6 (proline oxidase) mediates apoptosis through a calcineurin-dependent pathway. J Biol Chem 2005; 280:29346-54; PMID:15914462; http://dx.doi.org/10.1074/jbc.M504852200
- Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Dis 2009; 8:579-91; PMID:19478820; http://dx.doi.org/10.1038/nrd2803
- Liu XX, Li XJ, Zhang B, Liang YJ, Zhou CX, Cao DX, He M, Chen GQ, He JR, Zhao Q. MicroRNA-26b is underexpressed in human breast cancer and induces cell apoptosis by targeting SLC7A11. FEBS Lett 2011; 585:1363-7; PMID:21510944; http://dx.doi.org/10.1016/j.febslet.2011.04.018
- Guo W, Zhao Y, Zhang Z, Tan N, Zhao F, Ge C, Liang L, Jia D, Chen T, Yao M, et al. Disruption of xCT inhibits cell growth via the ROS/autophagy pathway in hepatocellular carcinoma. Cancer Lett 2011; 312:55-61; PMID:21906871; http://dx.doi.org/10.1016/j.canlet.2011.07.024
- Chen RS, Song YM, Zhou ZY, Tong T, Li Y, Fu M, Guo XL, Dong LJ, He X, Qiao HX, et al. Disruption of xCT inhibits cancer cell metastasis via the caveolin-1/beta-catenin pathway. Oncogene 2009; 28:599-609; PMID:19015640; http://dx.doi.org/10.1038/onc.2008.414
- Doxsee DW, Gout PW, Kurita T, Lo M, Buckley AR, Wang Y, Xue H, Karp CM, Cutz JC, Cunha GR, et al. Sulfasalazine-induced cystine starvation: potential use for prostate cancer therapy. Prostate 2007; 67:162-71; PMID:17075799; http://dx.doi.org/10.1002/pros.20508
- Chung WJ, Lyons SA, Nelson GM, Hamza H, Gladson CL, Gillespie GY, Sontheimer H. Inhibition of cystine uptake disrupts the growth of primary brain tumors. J Neurosci 2005; 25:7101-10; PMID:16079392; http://dx.doi.org/10.1523/JNEUROSCI.5258-04.2005
- Ishimoto T, Nagano O, Yae T, Tamada M, Motohara T, Oshima H, Oshima M, Ikeda T, Asaba R, Yagi H, et al. CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc(−) and thereby promotes tumor growth. Cancer Cell 2011; 19:387-400; PMID:21397861; http://dx.doi.org/10.1016/j.ccr.2011.01.038
- Gout PW, Buckley AR, Simms CR, Bruchovsky N. Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the x(c)- cystine transporter: a new action for an old drug. Leukemia 2001; 15:1633-40; PMID:11587223; http://dx.doi.org/10.1038/sj.leu.2402238
- Zhang W, Trachootham D, Liu J, Chen G, Pelicano H, Garcia-Prieto C, Lu W, Burger JA, Croce CM, Plunkett W, et al. Stromal control of cystine metabolism promotes cancer cell survival in chronic lymphocytic leukaemia. Nat Cell Biol 2012; 14:276-86; PMID:22344033; http://dx.doi.org/10.1038/ncb2432