
Abstract
Mitotic centromere-associated kinesin (MCAK) is the best characterized member of the kinesin-13 family and plays important roles in microtubule dynamics during mitosis. Its activity and subcellular localization is tightly regulated by an orchestra of mitotic kinases, such as Aurora B. It is well known that serine 196 of MCAK is the major phosphorylation site of Aurora B in Xenopus leavis extracts and that this phosphorylation regulates its catalytic activity and subcellular localization. In the current study, we have addressed the conserved phosphorylation site serine 192 in human MCAK to characterize its function in more depth in human cancer cells. Our data confirm that S192 is the major phosphorylation site of Aurora B in human MCAK and that this phosphorylation has crucial roles in regulating its catalytic activity and localization at the kinetochore/centromere region in mitosis. Interfering with this phosphorylation leads to a delayed progression through prometa- and metaphase associated with mitotic defects in chromosome alignment and segregation. We show further that MCAK is involved in directional migration and invasion of tumor cells, and interestingly, interference with the S192 phosphorylation affects this capability of MCAK. These data provide the first molecular explanation for clinical observation, where an overexpression of MCAK was associated with lymphatic invasion and lymph node metastasis in gastric and colorectal cancer patients.
Abbreviations
| ACA | = | anti-centromere antibody |
| MCAK | = | mitotic centromere-associated kinesin |
| MT | = | microtubule |
| MAP | = | microtubule-associated protein |
| Plk | = | Polo-like kinase |
| Cdk | = | cyclin-dependent kinase |
| HUVEC | = | human umbilical vein endothelial cell |
| PAK | = | p21-activated kinase. |
Introduction
The most crucial and dynamic processes in cells involve microtubules (MTs), structures of highly organized and dynamic α- and β-polymers.Citation1 MTs form the mitotic spindle in mitosis to ensure chromosome congression and segregation, and serve as components of the cytoskeleton responsible for organelle transport, migration and cell motility.Citation2-4 Therefore, microtubule dynamics are tightly regulated by different microtubule-associated proteins (MAPs) and depolymerizing proteins such as the kinesin-13 family. The mitotic centromere-associated protein (MCAK), the best characterized member of the family, couples the energy from its ATP turnover to depolymerize MTs by disassembling tubulin subunits from the polymer end.Citation5,6 By regulating the microtubule dynamics MCAK plays diverse roles in spindle assembly, sister-chromosome separation and error correction.Citation7 Along with MCAK's functions during the cell cycle, it localizes to MTs on the leading edge of endothelial cells and promotes directional migration.Citation8,9 To fulfill these functions, MCAK has to be tempospatially regulated via phosphorylation events by various kinases like Aurora A, Aurora B, cyclin-dependent kinase 1 (Cdk1) and Polo-like kinase 1 (Plk1).Citation10-14 Deregulation of MCAK induces defects in spindle assembly, chromosome congression and segregation in mammalian cells,Citation12-16 leading further to chromosome instability, a hallmark of tumors. In support of this notion, overexpression of MCAK associates with a more invasive and metastatic phenotype and poor prognosis for breast, gastric and colorectal cancer patients.Citation17-20
Aurora B is the major regulator of MCAK by phosphorylating several residues.Citation11,21-23 Previous studies in Xenopus leavis extracts and HeLa cells have reported that Aurora B decreases the catalytic activity of XKCM1, the Xenopus homolog of MCAK, and its localization to the kinetochore region by phosphorylating multiple residues, including serine 196 (S196).Citation11,23 Further studies have demonstrated that MCAK phosphorylation by Aurora B is involved in the correction of merotelic attachments and interfering with these phosphorylation sites lead to mitotic defects in Xenopus leavis extracts.Citation21,22,24,25 Interestingly, Aurora A was also shown to phosphorylate MCAK on S196 at early mitosis to regulate aster organization in Xenopus leavis extracts.Citation10 In addition, the Rac1-Aurora A-MCAK signaling pathway mediated by phosphorylation of S196 promotes endothelial cell polarization and directional migration in HUVEC and MCF-7 cells.Citation9 The inhibitory effect of this phosphorylation has been shown to be mediated by a phospho-conformational switch that reduces the microtubule association of MCAK in vitro.Citation26 To address the function of this phosphorylation residue, numerous studies have been carried out with XKCM1. By contrast, only one study was performed on human MCAK showing that p21-activated kinase 1 (PAK1) phosphorylates serine 192 (S192), the conserved residue in human MCAK, which decreases its MT depolymerization activity in vitro.Citation27 To get a more depth understanding of MCAK's regulation by Aurora B in human cells, we have focused on the major phosphorylation site S192 in MCAK and characterized its function in mitotic progression and cell motility in different human cancer cells. We show here that the phosphorylation of S192 by Aurora B is important for mitotic progression as well as for MCAK's novel role in cell motility.
Results
Phosphorylation at S192 regulates the catalytic activity of MCAK in vivo and in vitro
To investigate the impact of S192 phosphorylation on the catalytic activity of MCAK, GST-tagged human full-length MCAK, the non-phosphorylatable mutant MCAK S192A and the phosphomimetic mutant MCAK S192D were expressed and purified. Aurora B phosphorylates S192 of MCAK (Fig. S1A and B), as reported.Citation11,21-23,25 Using a well-established in vitro depolymerization assay,Citation14 we examined the catalytic activity of MCAK WT and its variants. As expected, MCAK WT efficiently depolymerized MTs (, 1st panel). While MCAK S192D was found to be inactive by showing more stabilized MTs (, 3rd panel), MCAK S192A was hyperactive exhibiting less remained MTs (, 2nd panel). Further analyses showed that MTs were much longer and the number was higher after 15 min incubation with MCAK S192D, whereas MTs were shorter and the number was reduced with MCAK S192A, relative to MCAK WT ( and C). To examine the catalytic activity in cells, an in vivo microtubule depolymerization assay was carried out. HeLa cells were depleted of endogenous MCAK and replaced 24 h later by Flag-tagged MCAK WT, MCAK S192A and MCAK S192D (). HeLa cells depleted of MCAK showed 46% more polymerized tubulin compared to cells treated with control siRNA (). While HeLa cells transfected with S192A contained 30% less polymerized tubulin, S192D transfected cells displayed 22% more polymerized tubulin compared to cells transfected with MCAK WT (). Comparable results were also obtained in osteosarcoma U2OS cells (Fig. S1C and D). To address if this phosphorylation impacts the morphology of the mitotic spindle, we measured the pole-to-pole distance. As depicted in , compared to MCAK WT transfected cells, the spindle length in cells with MCAK S192A was obviously shorter. By contrast, MCAK S192D prolonged the spindle (), suggestive of being inactive in cells. To exclude the possibility that this phosphorylation could alter the subcellular localization and subsequently affect its catalytic activity, we examined HeLa cells transfected with EGFP-tagged MCAK WT and its mutants in more detail. Like MCAK WT, both mutants were localized to the spindle poles as well as kinetochore/centromere region (Fig. S2), indicating that this regulation hardly affects its rough localization in cells. These data suggest that the catalytic activity of MCAK S192D is greatly reduced, both in vivo and in vitro.
Figure 1. For figure legend, see page .
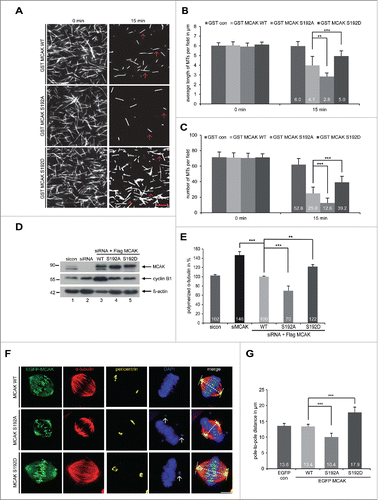
Interfering with the S192 phosphorylation retains cells in metaphase
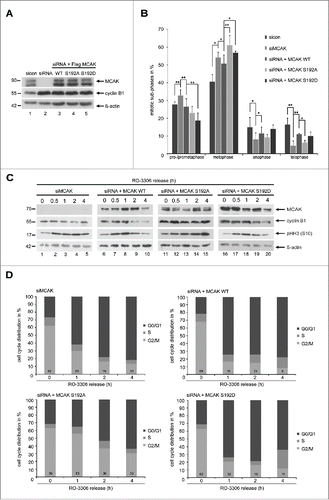
To investigate functional significance, HeLa cells were depleted of endogenous MCAK (, 1st row, lane 2) and rescued with Flag-tagged MCAK WT, MCAK S192A and MCAK S192D at comparable levels (, 1st row, lanes 3–5). Cells were synchronized to the G2 phase using a specific and reversible Cdk1 inhibitor RO-3306Citation28 and released for 1.5 h to allow cells to progress through mitosis. To quantify the distribution of mitotic sub-phases, the cells were stained for MT marker α-tubulin, kinetochore marker Hec1 and centrosome marker pericentrin. HeLa cells treated with siRNA against MCAK showed an accumulation of pro- and prometaphase cells of about 32% (), as previously reported.Citation29,30 Adding back of MCAK WT could efficiently reverse this prophase/prometaphase arrest (). Cells with either of MCAK mutants showed hardly problems in passing through prophase/prometaphase (). However, compared to MCAK WT transfected cells, HeLa cells expressing MCAK S192A and MCAK S192D displayed 22% and 14% more metaphase cells (), indicating that cells rescued with both mutants, especially MCAK S192A, encounter problems in passing through metaphase. A similar distribution of mitotic sub-phases was also observed in colon carcinoma HCT116 cells (Fig. S3A). To corroborate these results, MCAK depleted cells or cells rescued with Flag-tagged MCAK WT, MCAK S192A or MCAK S192D were synchronized to the G2 phase, released into fresh medium and harvested at indicated time points for Western blot analyses. Like MCAK WT expressed cells, those cells rescued with MCAK S192D showed almost normal progression through mitosis, with a peaked phosho-histone H3 (pHH3 (S10)) signal after 1 h (, 3rd row, lane 8 and 18), accompanied by degradation of cyclin B1 (, 2nd row, lanes 9–10 and 19–20). Similar to cells depleted of endogenous MCAK, cells transfected with MCAK S192A displayed a clear mitotic arrest with strong pHH3 (S10) signals even at 4 h after the release (, 3rd row, lane 15), accompanied by a still high amount of cyclin B1 (, 2nd row, lane 15), suggesting the cells with hyperactive non-phosphorylatable form face a severe problem in mitosis. Further flow cytometry analyses underscored this notion by showing a laggard G2/M progression in cells with MCAK S192A (, left bottom panel), even worse than cells depleted of endogenous MCAK demonstrating although an increased amount of the G2/M cells at 1 h yet normalized at 2 h (, left upper panel). Taken together, both mutant expressing cells, especially MCAK S192A expressing cells, are not able to fully restore the function of endogenous MCAK, suggesting this S192 phosphorylation and timely dephosphorylation are crucial for mitotic progression.
Figure 2. Blocking the S192 phosphorylation causes a metaphase arrest. To evaluate the mitotic phase distribution, HeLa cells were transfected with siRNA targeting only endogenous MCAK and followed by the rescue with Flag-tagged MCAK WT and its mutants. Cells were synchronized to the G2 phase with the Cdk1 inhibitor RO-3306 and released into fresh medium for 1.5 h. (A) Control Western blot analysis showing the efficient knockdown of endogenous MCAK and comparable expression levels of Flag MCAK and its mutants. β-actin served as loading control. sicon: control siRNA, siMCAK: siRNA targeting MCAK. (B) Treated HeLa cells were stained for α-tubulin, pericentrin, DAPI and analyzed for distribution of the mitotic sub-phases using immunofluorescence microscopy. The results are displayed as mean ±SD (n = 3). *p < 0.05, **p < 0.01. (C) HeLa cells, depleted of endogenous MCAK and rescued with Flag-tagged MCAK WT or its variants, were synchronized to the G2 phase with RO-3306 and released into fresh medium. At indicated time points cells were harvested for Western blot analyses with indicated antibodies. β-actin served as loading control. (D) Cell cycle distribution at indicated time points by flow cytometry.
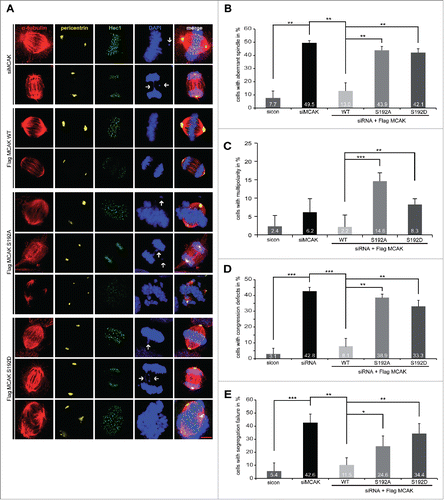
Mitotic defects by perturbing the phosphorylation on serine 192
We were then interested in phenotype of cells expressing MCAK mutants. HeLa cells depleted of endogenous MCAK were transfected with MCAK WT or its variants and stained for α-tubulin, Hec1, pericentrin and DNA. 49.5% of HeLa cells knockdown of MCAK exhibited the typical “hairy” phenotype, forming bipolar spindles with dense and excessively long and disorganized astral microtubules (, 1st and 2nd panel), as described previously.Citation30,31 Moreover, 42.8% of MCAK depleted cells showed defects in chromosome congression, with single or multiple chromosomes not aligned onto the metaphase plate (, 1st panel, DAPI staining, white arrow, and ) and 42.6% had segregation problems in anaphase (, 2nd panel, DAPI staining, white arrow and ). MCAK WT transfected cells could rescue these mitotic defects back to control HeLa cell levels (, 3rd and 4th panel, and ). Cells rescued with MCAK S192A or MCAK S192D could slightly reduce the aberrant spindle formation to 43.9% and 42.1%, respectively, from 49.5% in cells depleted of MCAK (, 5th to 10th panel, and ). Notably, MCAK S192A transfected cells demonstrated thin and less nucleated spindles, whereas cells with MCAK S192D displayed densely polymerized and aberrant spindles (, 5th to 10th panel, α-tubulin staining). Neither MCAK S192A nor MCAK S192D was able to restore an accurate chromosome congression, still with error rates of 38.9% and 33.3%, respectively (, 5th and 8th panel, DAPI staining, white arrows and ). Both mutants also failed to rescue the segregation failures induced by MCAK depletion with 24.6% for MCAK S192A and 34.4% for MCAK S192D compared to 11.5% of MCAK WT cells (, 6th and 9th panel, DAPI Staining, white arrows, and ). In addition, cells expressing the non-phosphorylatable mutant MCAK S192A or the phosphomimetic mutant MCAK S192D displayed 14.6% and 8.3% of multipolar cells, respectively, which could not be observed in control cells, cells depleted of MCAK or cells expressing MCAK WT (, 7th and 10th panel, DAPI Staining and ), in line with previous reports.Citation16,32 Comparable results were also obtained in HCT116 cells (Fig. S3B-E). Thus, neither MCAK S192A nor MCAK S192D could fully restore the phenotype induced by depleting endogenous MCAK. These data indicate that the S192 phosphorylation is an important regulatory event of MCAK and its deregulation links to defective mitosis associated with failure in spindle formation and chromosome movement.
Figure 3. Interfering with MCAK S192 phosphorylation causes multiple mitotic defects. For mitotic phenotype analysis, HeLa cells were transfected with siRNA targeting only endogenous MCAK and followed by the rescue with Flag-tagged MCAK WT or its mutants. Cells were synchronized to the G2 phase using RO-3306 and released into fresh medium for 1.5 h for further analysis. (A) Immunofluorescence staining. To evaluate the mitotic phenotype, treated cells were stained for α-tubulin, pericentrin, Hec1 and DAPI. Representatives are shown. White arrows indicate misaligned or segregated chromosomes. Scale bar: 10 μm. (B) Quantification of aberrant spindles. The results are represented as mean ±SD. **p < 0.01. (C) The frequency of cells showing multipolarity. The data are displayed as mean ±SD. **p < 0.01, ***p < 0.001. (D) Evaluation of chromosome misalignment. The data are displayed as mean ±SD. **p < 0.01, ***p < 0.001. (E) The frequency of cells showing segregation defects. The results are represented as mean ±SD. *p < 0.05, **p < 0.01, ***p < 0.001.
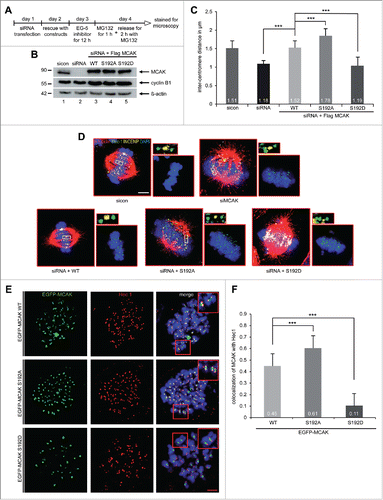
MCAK S192A increases the inter-centromere distance and the kinetochore localization of MCAK
MCAK is involved in creating tension between sister kinetochores by correcting mal-attachments within microtubules and kinetochores, the inter-kinetochore distance is therefore a reliable indicator of MCAK's actitivty.Citation11,33 HeLa cells depleted of endogenous MCAK were rescued with comparable levels of Flag-tagged MCAK WT, MCAK S192A and MCAK S192D (), synchronized to prometaphase using Eg5 inhibitor III and released for 2 h in the presence of MG132 to prevent cell progression beyond metaphase (). Cells were stained for the kinetochore marker Hec1 and the centromere marker INCENP for measuring the inter-kinetochore distance by confocal microscopy. MCAK depleted cells showed a significantly decreased inter-centromere distance compared to cells treated with control siRNA and MCAK WT transfected cells (). Intriguingly, cells transfected with Flag-tagged MCAK S192A displayed a remarkably increased sister kinetochore distance, while the phosphomimetic construct reduced obviously the centromere stretch in HeLa cells, implying that both mutants have deregulated depolymerization activity (). Similar results were also obtained with HCT116 cells (Fig. S4A and B). To examine precisely if the phosphorylation on serine 192 affects the kinetochore/centromere localization of MCAK, a well-established chromosome spreading assay was performed with HeLa cells expressing EGFP-tagged MCAK constructs.Citation14,34 Chromosomes from metaphase cells were spread onto the slides and stained for the kinetochore marker Hec1 and DNA. MCAK WT cells displayed a nearly equal distribution of MCAK localized to kinetochores and centromeres (). By contrast, both mutants showed an altered distribution around the kinetochore-centromere region: the colocalization of the hyperactive mutant MCAK S192A with the kinetochore marker Hec1 was elevated by 26%, whereas the phosphomimetic mutant localization to the kinetochore was decreased by 76% compared to the MCAK WT signal (). The results strengthen the notion that the phosphorylation of serine 192 suppresses the depolymerization activity of MCAK and regulates accurately its location in the kinetochore/centromere region and deregulation of this control could lead to severe failures during mitosis.
Figure 4. For figure legend, see page .
Involvement of S192 in directional migration and invasion of tumor cells
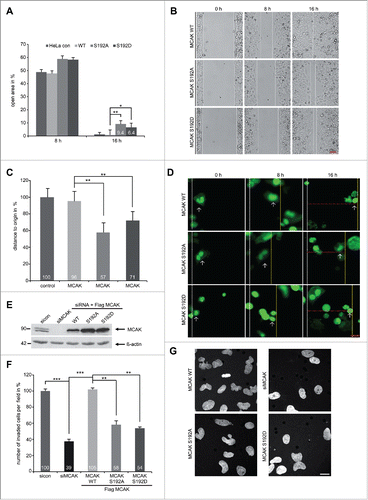
It has been reported that MCAK promotes endothelial cell polarity and directional migration by regulating MT dynamics.Citation9 As the serine 192 is an important regulatory residue of MCAK, we were wondering if this phosphorylation event confers additional functions beyond regulating mitosis. To address this, a wound-healing/migration assay was performed with HeLa cells expressing EGFP-tagged MCAK WT, MCAK S192A or MCAK S192D. After 16 h, EGFP MCAK WT cells were able to close the cell free gap to 0.6% comparable to HeLa control cells with 1.2% (). In contrast, both mutants inhibited cell migration: cells with MCAK S192A or with MCAK S192D displayed an increase in the cell free area of 9.4% and 6.4%, respectively, at 16 h (). To exclude deviations caused by varied transfection efficiencies, single EGFP-tagged cells were tracked during the wound-healing assay. Again relative to EGFP MCAK WT cells, both mutants slowed migration (). Moreover, to assess if this reduced migration is related to the invasive capability, HeLa cells depleted of endogenous MCAK were added back with Flag-tagged MCAK WT, MCAK S192A and MCAK S192D for invasion assay (Fig. 5E). While HeLa cells treated with siRNA against MCAK showed a potent reduction of invading cells to 39%, both MCAK S192A and MCAK 192D reduced also significantly invading cells with 58% and 54%, respectively (). These results could be corroborated in HCT116 cells (Fig. S4D and E). Thus, these observations support a novel role for MCAK in directional migration by remodeling MT dynamics in the cytoskeleton and the phosphorylation on S192 plays an important role in this process.
Figure 5. For figurelegend, see page .
Discussion
Since deregulated MCAK leads to chromosome instability and is associated with invasiveness, metastasis and poor prognosis of cancer patients,Citation35 the precise regulation of MCAK by kinases is fundamentally crucial. In fact, numerous important studies have revealed the importance of the phosphorylation sites in MCAK to delineate the regulatory mechanisms responsible for its functions.Citation10-14,27 We have shown that the phosphorylation at S621 by Plk1 facilitates MCAK's recognition by the ubiquitin/proteasome dependent APC/CCdc20 degradation pathwayCitation13 and phosphorylation at SS632/633 by Plk1 augments its catalytic activity during mitosis.Citation14 In contrast, the phosphorylation at T537 by Cdk1 attenuates MCAK's catalytic activity and initiates its release from the centrosomes in pro-/prometaphase.Citation12 Among all revealed phosphorylation sites in MCAK, however, S192 (S196 in Xenopus leavis), the major phosphorylation site by Aurora B/Aurora A, is the most important regulatory residue for MCAK's activity and localization and has thus attracted the most attention. As Aurora A as well as Aurora B is deregulated in various tumor cells,Citation36-39 we have set up to study the function of the S192 phosphorylation in cancer cells in more depth.
In the current work, we show that this phosphorylation on S192 decreases MCAK's catalytic activity in vitro and in mitotic tumor cells, in line with the results derived from Xenopus leavis extracts as well as in interphase cells.Citation22,27 We demonstrate further that the non-phosphoryltable MCAK S192A is hyperactive evidenced by reduced spindle length and increased inter-centromere distance, whereas the phosphomimetic mutant MCAK S192D is inactive by showing prolonged spindle and shortened inter-centromere distance. These results emphasize the role of Aurora B in regulating the catalytic activity of human MCAK by phosphorylating S192 of MCAK. This catalytic regulation by Aurora B is likely mediated by a phospho-conformation change inside MCAK shown in vitro.Citation26 Upon phosphorylation MCAK shifts to a more “open” state characterized by a reduced affinity for tubulin dimers, interestingly with a hardly changed MT on-rate, which was previously assumed to play a role by interfering with the positive charged region in the neck-domain.Citation26,40
Furthermore, our data indicate a delayed mitotic progression by interfering with the phosphorylation of Aurora B on S192. This mitotic delay is caused by the deregulation of MCAK's catalytic activity and consequently defects in spindle assembly and chromosome movement. While MCAK S192A, the hyperactive form, induced thin abnormal spindles, the inactive variant MCAK S192D displayed densely polymerized microtubules similar to that in cells depleted of endogenous MCAK. Moreover, both mutants MCAK S192A and MCAK S192D were not able to rescue the assembly of aberrant spindles and to restore properly the chromosome alignment and the sister-chromatid segregation. Additionally, a part of mutant transfected was not able to establish a bipolar spindle, leading to multipolar spindles, possibly resulted from deregulated microtubule nucleation or defective cytokinesis due to severely altered MCAK's activity.Citation16,32 We show further that this phosphorylation of MCAK regulates its localization on the kinetochore/centromere region in cells: the hyperactive MCAK S192A favored the kinetochores, whereas the inactive form MCAK S192D preferred the centromere region. These data suggest that mitotic failures could be ascribed to altered catalytic activity as well as to deregulated localization of MCAK at the centromere/kinetochore region, where a precise balance between active and inactive MCAK is required to correct defective kinetochore-MT attachments.Citation41 These observations highlight the importance of a proper regulation of S192 in MCAK and its phosphorylation and dephosphorylation must be regulated in space and time, to maintain a precisely controlled activity of MCAK throughout mitosis. This is substantial to ensure MCAK's function in regulating the microtubule nucleation, spindle formation, chromosome alignment and separation.Citation42 Our data also indicate that deregulated MCAK could be involved in chromosome instability facilitating tumor development and therapy resistance, as Aurora kinases are very often overexpressed in tumor cells.Citation36-39
We reveal further that MCAK is important for migration and invasion of tumor cells, which is in line with previous observation,Citation8 possibly via its involvement in modulating MT dynamics in the cytoskeleton. Interestingly, interfering with S192 phosphorylation hampers migration and invasiveness of tumor cells, indicating that this residue plays a role in cell motility. This function might be regulated by the Rac1-Aurora A-MCAK signaling pathway, as suggested.Citation9 Importantly, invasive cervix carcinoma HeLa cells and colon carcinoma HCT116 cells showed a remarkably reduced invasion capability by knockdown of endogenous MCAK, which could be restored by wild type MCAK but not by either of both mutants. These data provide the first molecular indication for clinical studies, where an overexpression of MCAK was associated with lymphatic invasion and lymph node metastasis in gastric and colorectal cancer patients.Citation18,19 Further investigations are required to explore the molecular mechanisms by which overexpression of MCAK increases mobility and invasiveness in cancer cells.
Materials and Methods
Cell culture, synchronization and cell cycle analysis
HeLa, HCT116 and U2OS cells were cultured as instructed (DSMZ, Braunschweig). Cells were synchronized to the G2 phase with the Cdk1 inhibitor RO-3306 (9 µM) for 16 h and to prometaphase with nocodazole treatment (50 ng/ml).Citation28 To prevent progression of cells beyond metaphase, cells were treated with the specific proteasome inhibitor MG132 (10 μM, Sigma-Aldrich, Hamburg). Cell lysis was performed with RIPA buffer.Citation43,44
Kinase assay in vitro, Western blot and cell cycle analysis
Recombinant full-length and S192 mutants of MCAK were induced and expressed in Escherichia coli BL21 (DE3, Codon Plus) at 37°C for 2 h by addition of 1 mM IPTG and purified using Glutathione-Sepharose 4B beads (GE Healthcare, Hamburg). The GST-tagged MCAK constructs were incubated with Aurora B kinase (Biomol, Hamburg) in the presence of 1 μCi [γ32P] ATP and 100 μM non-radioactive ATP at 37°C for 30 min. The reaction was terminated by adding sample buffer and boiling for 5 min, loaded onto 10% SDS PAGE, separated, stained with Coomassie brilliant blue and scanned as input control. The phosphorylation of Aurora B was quantified by measuring the incorporation of γ32P using Image J software.
Western blots were carried out as describedCitation45,46 and the following primary antibodies were used: mouse monoclonal antibodies against anti-Kif2c (MCAK), Plk1 and cyclin B1 (Santa Cruz biotechnology, Heidelberg), mouse monoclonal antibody against β-actin (Sigma-Aldrich), rabbit polyclonal antibody against phospho-histone H3 (Merck Millipore, Lake Placid). Cell cycle was analyzed as described.Citation47
siRNA knockdown and MCAK rescue experiments
Small interfering RNA (siRNA) (25 nM) targeting the 3′-untranslated region of MCAK was synthesized by Sigma-Aldrich against position 1666 to 1686. siRNA was transiently transfected using the transfection reagent Oligofectamine (Invitrogen, CA) as described.Citation46,48 For rescue experiments, HeLa cells depleted of endogenous MCAK were transfected with Flag-tagged MCAK WT, MCAK S192A or MCAK S192D by using FuGene-HD transfection reagent (Promega, Madisson). All rescue experiments were independently performed at least 3 times.
Immunofluorescence staining and measurement of inter-centromere and inter-centrosome distance
For immunofluorescence staining cells were transfected with MCAK constructs and fixed for 8–10 min with methanol at −20°C or with 4% paraformaldehyde containing 0.1% Triton X-100 for 15 min at room temperature. The following primary antibodies were used: rat polyclonal antibody against α-tubulin (Biozol, Eching), rabbit polyclonal antibodies against pericentrin and INCENP and mouse monoclonal antibody against Hec1 (Abcam, Darmstadt), human ACA (anti-centromere antibody) (Jackson Immunoresearch, Newmarket), rabbit polyclonal antibody against Aurora B (Cell signaling, Frankfurt) and mouse monoclonal antibody against Plk1 (Santa Cruz biotechnology). FITC-, Cy3- and Cy5 conjugated secondary antibodies were obtained from Jackson Immunoresearch. DNA was stained using DAPI (4′,6-diamidino-2-phenylindole-dihydrochloride) (Roche, Mannheim).
For measuring the distance of the inter-centromere and inter-centrosome, cells were depleted of endogenous MCAK, added back of Flag-tagged MCAK and its variants, and synchronized to prometaphase using Eg5 inhibitor III (1 µM, Sigma Aldrich) for 12 h, the proteasome inhibitor MG132 was provided to the medium containing Eg5 inhibitor for the last hour, and cells were then released into fresh medium with MG132 for 2 h. Both distances were measured by using a confocal laser scanning microscope (CLSM, Leica CTR 6500) with LAS AF software (Leica, Heidelberg). Images were processed using Adobe Photoshop.
Construction of MCAK mutants
The cDNA of full-length human MCAK was obtained from RZPD (IRATp970F0111D, Berlin) and was cloned into BamH1/EcoR1 sites of pGEX 5x-3 (GE healthcare), into EcoR1/BamH1 sites of p3xFLAG-CMV7.1 (Invitrogen) and into BamH1/ EcoR1 sites of pEGFP-C2 (Invitrogen). Single point mutations were generated as previously describedCitation48 using the following primers: S192A sense: 5′-gaactcagttcggaggaaagcatgtcttgtgaaggaag-3′, S192A antisense: 5′-cttccttcacaagacatgctttcctccgaactgagttc-3′, S192D sense: 5′-ctgtgaactcagttcggagg-aaagattgtcttgtgaaggaagtgg-3′, S192D antisense: 5′-ccacttccttcacaagacaatctttcctccgaactgagttcacag-3′. All mutant constructs were confirmed by DNA sequencing.
Microtubule depolymerization assay in vitro and in vivo
Recombinant human GST-tagged MCAK and its mutants were expressed and purified as described.Citation13 The in vitro MT depolymerization assay was performed and evaluated with the AxioVision SE64 Re. 4.9 software as reported.Citation14 For in vivo depolymerization assay, cells were depleted of endogenous MCAK, rescued with Flag-tagged MCAK or its mutants, and synchronized with nocodazole and mitotic shake-off cells were collected. For analysis of cellular microtubule polymer content, mitotic cells were released for 1.5 h in fresh medium, then extracted, fixed and stained for α-tubulin for fluorescence flow cytometry (Becton Dickinson, Heidelberg) as described previously.Citation29 Fluorescence intensity was evaluated using the Cell Quest software (Becton Dickinson). Cells transfected with MCAK WT were assigned as microtubule polymer content 100%. The experiments were independently performed 3 times and each time was in triplicate.
Chromosome spreading assay with metaphase cells
HeLa cells were rescued with EGFP-tagged MCAK WT, MCAK S192A and MCAK S192D after depleting endogenous MCAK with siRNA. On day 2 cells were synchronized to prometaphase using nocodazole and released into fresh medium supplemented with the proteasome inhibitor MG132 to retract cells in metaphase. Harvested cells were subjected to hypotonic buffer (10 mM Hepes-Na pH 7.0, 30 mM glycerol, 1.0 mM CaCl2 and 0.8 mM MgCl2), centrifuged onto an object slide by using a cyto-centrifuge with 3000 rpm for 5 min, followed by fixation and staining for Hec1 and DNA as described.Citation34 The chromosome spreads were further examined by a confocal laser scanning microscope (Leica CTR 6500). The colocalization of EGFP-MCAK with Hec1 was quantified using the Pearsons's coefficient and analyzed with the software ImageJ with the plugin Coloc2.Citation49
Migration and invasion assay
Cell migration assays were performed with culture-inserts from ibidi (Martinsried). Culture-inserts (cell free gap of 500 µm) were placed in a 24-well culture dish and both wells of each insert were filled with cell suspension. HeLa cells (7.5 × 104) transfected with EGFP-MCAK or its mutants were seeded in each well of the culture-inserts. They were gently removed after at least 8 h. The cells were acquired and imaged at indicated time points with a fluorescence microscope. Five pictures of each insert were taken and the experiments were performed in duplicate. The open area was measured using the AxioVision SE64 Re. 4.9 software (Zeiss). For single cell tracking of EGFP-transfected cells, the cells were treated as described above. Cell movement was followed using a time lapse module of the AxioVision SE64 Re. 4.9 software. This module took every 4 hours pictures of 2five defined positions, which enabled the measurement of the same cell during different time points. The migrated distance of single cells was measured by using the AxioVision SE64 Re. 4.9 software (n = 50 cells for each condition). The experiments were independently performed 2 times and each time was in duplicate.
For invasion assays, cells depleted of endogenous MCAK were rescued with Flag-tagged MCAK WT, MCAK S192A or MCAK S192D and seeded in 24-well transwell matrigel chambers (Cell Biolabs Inc., San Diego) according to the manufacturer's instructions and as previously reported.Citation50 Briefly, cells (7.5 × 104) were seeded into the upper chamber of the transwell in 500 µl serum-free medium and the lower chamber was filled with 750 µl serum-free medium. After 12 h the medium of both chambers was discarded and the invasion assay was started by adding medium containing 10% fetal bovine serum for the next 24 h. Cells were then fixed with ethanol and stained with DAPI. Invaded cells were counted with a microscope. The experiments were independently performed 3 times.
Statistical analysis
Student's t-test (2 tailed and paired or homoscedastic) was used to evaluate the significance of difference between different groups. Difference was considered as statistically significant when p < 0.05.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
1068481_supplemental_files.zip
Download Zip (18.6 MB)Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
Additional information
Funding
Related Research Data
Reference
- Shelanski ML, Taylor EW. Properties of the protein subunit of central-pair and outer-doublet microtubules of sea urchin flagella. J Cell Biol 1968; 38:304-15; PMID:5664206; http://dx.doi.org/10.1083/jcb.38.2.304
- Gadde S, Heald R. Mechanisms and molecules of the mitotic spindle. Curr Biol 2004; 14:R797-805; PMID:15380094; http://dx.doi.org/10.1016/j.cub.2004.09.021
- Vale RD, Milligan RA. The way things move: looking under the hood of molecular motor proteins. Science 2000; 288:88-95; PMID:10753125; http://dx.doi.org/10.1126/science.288.5463.88
- Tanaka E, Ho T, Kirschner MW. The role of microtubule dynamics in growth cone motility and axonal growth. J Cell Biol 1995; 128:139-55; PMID:7822411; http://dx.doi.org/10.1083/jcb.128.1.139
- Ogawa T, Nitta R, Okada Y, Hirokawa N. A common mechanism for microtubule destabilizers-M type kinesins stabilize curling of the protofilament using the class-specific neck and loops. Cell 2004; 116:591-602; PMID:14980225; http://dx.doi.org/10.1016/S0092-8674(04)00129-1
- Oguchi Y, Uchimura S, Ohki T, Mikhailenko SV, Ishiwata S. The bidirectional depolymerizer MCAK generates force by disassembling both microtubule ends. Nat Cell Biol 2011; 13:846-52; PMID:21602793; http://dx.doi.org/10.1038/ncb2256
- Walczak CE, Gayek S, Ohi R. Microtubule-depolymerizing kinesins. Annu Rev Cell Dev Biol 2013; 29:417-41; PMID:23875646; http://dx.doi.org/10.1146/annurev-cellbio-101512-122345
- Moore AT, Rankin KE, von Dassow G, Peris L, Wagenbach M, Ovechkina Y, Andrieux A, Job D, Wordeman L. MCAK associates with the tips of polymerizing microtubules. J Cell Biol 2005; 169:391-7; PMID:15883193; http://dx.doi.org/10.1083/jcb.200411089
- Braun A, Dang K, Buslig F, Baird MA, Davidson MW, Waterman CM, Myers KA. Rac1 and Aurora A regulate MCAK to polarize microtubule growth in migrating endothelial cells. J Cell Biol 2014; 206:97-112; PMID:25002679; http://dx.doi.org/10.1083/jcb.201401063
- Zhang X, Ems-McClung SC, Walczak CE. Aurora A phosphorylates MCAK to control ran-dependent spindle bipolarity. Mol Biol Cell 2008; 19:2752-65; PMID:18434591; http://dx.doi.org/10.1091/mbc.E08-02-0198
- Andrews PD, Ovechkina Y, Morrice N, Wagenbach M, Duncan K, Wordeman L, Swedlow JR. Aurora B regulates MCAK at the mitotic centromere. Dev Cell 2004; 6:253-68; PMID:14960279; http://dx.doi.org/10.1016/S1534-5807(04)00025-5
- Sanhaji M, Friel CT, Kreis NN, Kramer A, Martin C, Howard J, Strebhardt K, Yuan J. Functional and spatial regulation of mitotic centromere-associated kinesin by cyclin-dependent kinase 1. Mol Cell Biol 2010; 30:2594-607; PMID:20368358; http://dx.doi.org/10.1128/MCB.00098-10
- Sanhaji M, Ritter A, Belsham HR, Friel CT, Roth S, Louwen F, Yuan J. Polo-like kinase 1 regulates the stability of the mitotic centromere-associated kinesin in mitosis. Oncotarget 2014; 5:3130-44; PMID:24931513
- Ritter A, Sanhaji M, Steinhauser K, Roth S, Louwen F, Yuan J. The activity regulation of the mitotic centromere-associated kinesin by Polo-like kinase 1. Oncotarget 2015; 6:6641-55; PMID:25504441
- Maney T, Hunter AW, Wagenbach M, Wordeman L. Mitotic centromere-associated kinesin is important for anaphase chromosome segregation. J Cell Biol 1998; 142:787-801; PMID:9700166; http://dx.doi.org/10.1083/jcb.142.3.787
- Zhang L, Shao H, Huang Y, Yan F, Chu Y, Hou H, Zhu M, Fu C, Aikhionbare F, Fang G, et al. PLK1 phosphorylates mitotic centromere-associated kinesin and promotes its depolymerase activity. J Biol Chem 2011; 286:3033-46; PMID:21078677; http://dx.doi.org/10.1074/jbc.M110.165340
- Shimo A, Tanikawa C, Nishidate T, Lin ML, Matsuda K, Park JH, Ueki T, Ohta T, Hirata K, Fukuda M, et al. Involvement of kinesin family member 2C/mitotic centromere-associated kinesin overexpression in mammary carcinogenesis. Cancer Sci 2008; 99:62-70; PMID:17944972
- Nakamura Y, Tanaka F, Haraguchi N, Mimori K, Matsumoto T, Inoue H, Yanaga K, Mori M. Clinicopathological and biological significance of mitotic centromere-associated kinesin overexpression in human gastric cancer. Br J Cancer 2007; 97:543-9; PMID:17653072; http://dx.doi.org/10.1038/sj.bjc.6603905
- Ishikawa K, Kamohara Y, Tanaka F, Haraguchi N, Mimori K, Inoue H, Mori M. Mitotic centromere-associated kinesin is a novel marker for prognosis and lymph node metastasis in colorectal cancer. Br J Cancer 2008; 98:1824-9; PMID:18506187; http://dx.doi.org/10.1038/sj.bjc.6604379
- Gnjatic S, Cao Y, Reichelt U, Yekebas EF, Nolker C, Marx AH, Erbersdobler A, Nishikawa H, Hildebrandt Y, Bartels K, et al. NY-CO-58/KIF2C is overexpressed in a variety of solid tumors and induces frequent T cell responses in patients with colorectal cancer. Int J Cancer 2010; 127:381-93; PMID:19937794
- Knowlton AL, Lan W, Stukenberg PT. Aurora B is enriched at merotelic attachment sites, where it regulates MCAK. Curr Biol 2006; 16:1705-10; PMID:16950107; http://dx.doi.org/10.1016/j.cub.2006.07.057
- Lan W, Zhang X, Kline-Smith SL, Rosasco SE, Barrett-Wilt GA, Shabanowitz J, Hunt DF, Walczak CE, Stukenberg PT. Aurora B phosphorylates centromeric MCAK and regulates its localization and microtubule depolymerization activity. Curr Biol 2004; 14:273-86; PMID:14972678; http://dx.doi.org/10.1016/j.cub.2004.01.055
- Ohi R, Sapra T, Howard J, Mitchison TJ. Differentiation of cytoplasmic and meiotic spindle assembly MCAK functions by Aurora B-dependent phosphorylation. Mol Biol Cell 2004; 15:2895-906; PMID:15064354; http://dx.doi.org/10.1091/mbc.E04-02-0082
- Zhang X, Lan W, Ems-McClung SC, Stukenberg PT, Walczak CE. Aurora B phosphorylates multiple sites on mitotic centromere-associated kinesin to spatially and temporally regulate its function. Mol Biol Cell 2007; 18:3264-76; PMID:17567953; http://dx.doi.org/10.1091/mbc.E07-01-0086
- Shao H, Ma C, Zhang X, Li R, Miller AL, Bement WM, Liu XJ. Aurora B regulates spindle bipolarity in meiosis in vertebrate oocytes. Cell Cycle 2012; 11:2672-80; PMID:22751439; http://dx.doi.org/10.4161/cc.21016
- Ems-McClung SC, Hainline SG, Devare J, Zong H, Cai S, Carnes SK, Shaw SL, Walczak CE. Aurora B inhibits MCAK activity through a phosphoconformational switch that reduces microtubule association. Curr Biol 2013; 23:2491-9; PMID:24291095; http://dx.doi.org/10.1016/j.cub.2013.10.054
- Pakala SB, Nair VS, Reddy SD, Kumar R. Signaling-dependent phosphorylation of mitotic centromere-associated kinesin regulates microtubule depolymerization and its centrosomal localization. J Biol Chem 2012; 287:40560-9; PMID:23055517; http://dx.doi.org/10.1074/jbc.M112.399576
- Vassilev LT, Tovar C, Chen S, Knezevic D, Zhao X, Sun H, Heimbrook DC, Chen L. Selective small-molecule inhibitor reveals critical mitotic functions of human CDK1. Proc Natl Acad Sci U S A 2006; 103:10660-5; PMID:16818887; http://dx.doi.org/10.1073/pnas.0600447103
- Holmfeldt P, Zhang X, Stenmark S, Walczak CE, Gullberg M. CaMKIIgamma-mediated inactivation of the Kin I kinesin MCAK is essential for bipolar spindle formation. EMBO J 2005; 24:1256-66; PMID:15775983; http://dx.doi.org/10.1038/sj.emboj.7600601
- Hedrick DG, Stout JR, Walczak CE. Effects of anti-microtubule agents on microtubule organization in cells lacking the kinesin-13 MCAK. Cell Cycle 2008; 7:2146-56; PMID:18635958; http://dx.doi.org/10.4161/cc.7.14.6239
- Stout JR, Rizk RS, Kline SL, Walczak CE. Deciphering protein function during mitosis in PtK cells using RNAi. BMC Cell Biol 2006; 7:26; PMID:16796742; http://dx.doi.org/10.1186/1471-2121-7-26
- Holmfeldt P, Stenmark S, Gullberg M. Differential functional interplay of TOGp/XMAP215 and the KinI kinesin MCAK during interphase and mitosis. EMBO J 2004; 23:627-37; PMID:14749730; http://dx.doi.org/10.1038/sj.emboj.7600076
- Tanenbaum ME, Medema RH. Localized Aurora B activity spatially controls non-kinetochore microtubules during spindle assembly. Chromosoma 2011; 120:599-607; PMID:21786106; http://dx.doi.org/10.1007/s00412-011-0334-9
- Stenman S, Rosenqvist M, Ringertz NR. Preparation and spread of unfixed metaphase chromosomes for immunofluorescence staining of nuclear antigens. Exp Cell Res 1975; 90:87-94; PMID:164358; http://dx.doi.org/10.1016/0014-4827(75)90360-2
- Sanhaji M, Friel CT, Wordeman L, Louwen F, Yuan J. Mitotic centromere-associated kinesin (MCAK): a potential cancer drug target. Oncotarget 2011; 2:935-47; PMID:22249213
- Takeshita M, Koga T, Takayama K, Ijichi K, Yano T, Maehara Y, Nakanishi Y, Sueishi K. Aurora-B overexpression is correlated with aneuploidy and poor prognosis in non-small cell lung cancer. Lung Cancer 2013; 80:85-90; PMID:23313006; http://dx.doi.org/10.1016/j.lungcan.2012.12.018
- Zhang Y, Jiang C, Li H, Lv F, Li X, Qian X, Fu L, Xu B, Guo X. Elevated Aurora B expression contributes to chemoresistance and poor prognosis in breast cancer. Int J Clin Exp Pathol 2015; 8:751-7; PMID:25755770
- Xu J, Wu X, Zhou WH, Liu AW, Wu JB, Deng JY, Yue CF, Yang SB, Wang J, Yuan ZY, et al. Aurora-A identifies early recurrence and poor prognosis and promises a potential therapeutic target in triple negative breast cancer. PLoS One 2013; 8:e56919; PMID:23437271; http://dx.doi.org/10.1371/journal.pone.0056919
- Lo Iacono M, Monica V, Saviozzi S, Ceppi P, Bracco E, Papotti M, Scagliotti GV. Aurora Kinase A expression is associated with lung cancer histological-subtypes and with tumor de-differentiation. J Transl Med 2011; 9:100; PMID:21718475; http://dx.doi.org/10.1186/1479-5876-9-100
- Tanenbaum ME, Medema RH, Akhmanova A. Regulation of localization and activity of the microtubule depolymerase MCAK. Bioarchitecture 2011; 1:80-7; PMID:21866268; http://dx.doi.org/10.4161/bioa.1.2.15807
- Kline-Smith SL, Khodjakov A, Hergert P, Walczak CE. Depletion of centromeric MCAK leads to chromosome congression and segregation defects due to improper kinetochore attachments. Mol Biol Cell 2004; 15:1146-59; PMID:14699064; http://dx.doi.org/10.1091/mbc.E03-08-0581
- Huang H, Feng J, Famulski J, Rattner JB, Liu ST, Kao GD, Muschel R, Chan GK, Yen TJ. Tripin/hSgo2 recruits MCAK to the inner centromere to correct defective kinetochore attachments. J Cell Biol 2007; 177:413-24; PMID:17485487; http://dx.doi.org/10.1083/jcb.200701122
- Sanhaji M, Kreis NN, Zimmer B, Berg T, Louwen F, Yuan J. p53 is not directly relevant to the response of Polo-like kinase 1 inhibitors. Cell Cycle 2012; 11:543-53; PMID:22262171; http://dx.doi.org/10.4161/cc.11.3.19076
- Kreis NN, Sommer K, Sanhaji M, Kramer A, Matthess Y, Kaufmann M, Strebhardt K, Yuan J. Long-term downregulation of Polo-like kinase 1 increases the cyclin-dependent kinase inhibitor p21(WAF1/CIP1). Cell Cycle 2009; 8:460-72; PMID:19177004; http://dx.doi.org/10.4161/cc.8.3.7651
- Kreis NN, Sanhaji M, Kramer A, Sommer K, Rodel F, Strebhardt K, Yuan J. Restoration of the tumor suppressor p53 by downregulating cyclin B1 in human papillomavirus 16/18-infected cancer cells. Oncogene 2010; 29:5591-603; PMID:20661218; http://dx.doi.org/10.1038/onc.2010.290
- Muschol-Steinmetz C, Friemel A, Kreis NN, Reinhard J, Yuan J, Louwen F. Function of survivin in trophoblastic cells of the placenta. PLoS One 2013; 8:e73337; PMID:24069188; http://dx.doi.org/10.1371/journal.pone.0073337
- Sanhaji M, Louwen F, Zimmer B, Kreis NN, Roth S, Yuan J. Polo-like kinase 1 inhibitors, mitotic stress and the tumor suppressor p53. Cell Cycle 2013; 12:1340-51; PMID:23574746; http://dx.doi.org/10.4161/cc.24573
- Yuan J, Yan R, Kramer A, Eckerdt F, Roller M, Kaufmann M, Strebhardt K. Cyclin B1 depletion inhibits proliferation and induces apoptosis in human tumor cells. Oncogene 2004; 23:5843-52; PMID:15208674; http://dx.doi.org/10.1038/sj.onc.1207757
- Sharma S, Quintana A, Findlay GM, Mettlen M, Baust B, Jain M, Nilsson R, Rao A, Hogan PG. An siRNA screen for NFAT activation identifies septins as coordinators of store-operated Ca2+ entry. Nature 2013; 499:238-42; PMID:23792561; http://dx.doi.org/10.1038/nature12229
- Hehlgans S, Petraki C, Reichert S, Cordes N, Rodel C, Rodel F. Double targeting of Survivin and XIAP radiosensitizes 3D grown human colorectal tumor cells and decreases migration. Radiother Oncol 2013; 108:32-9; PMID:23830189; http://dx.doi.org/10.1016/j.radonc.2013.06.006