
Abstract
Chromothripsis is a recently recognized mode of genetic instability that generates chromosomes with strikingly large numbers of segmental re-arrangements. While the characterization of these derivative chromosomes has provided new insights into the processes by which cancer genomes can evolve, the underlying signaling events and molecular mechanisms remain unknown. In medulloblastomas, chromothripsis has been observed to occur in the context of mutational inactivation of p53 and activation of the canonical Hedgehog (Hh) pathway. Recent studies have illuminated mechanistic links between these 2 signaling pathways, including a novel PTCH1 homolog that is regulated by p53. Here, we integrate this new pathway into a hypothetical model for the catastrophic DNA breakage that appears to trigger profound chromosomal rearrangements.
The comprehensive genetic analysis of many types of tumors has revealed a continuum of alterations to the cancer cell genome, ranging from single nucleotide substitutions to a variety of complex chromosomal rearrangements.Citation1 Perhaps the most curious finding has been the observation of chromosomes that harbor an inordinate number of translocations.Citation2,3 So extreme is the extent of structural rearrangement in such chromosomes that they appear to be derived from the randomly reassembled shards of forebears that were shattered at clustered breakpoints. Evidence of chromothripsis (from the Greek ‘chromo’, which means color and refers to chromosomes, and ‘thripsis’, which means shattering into pieces) is typically limited to one or a few chromosomes, and is therefore found in cells that also retain structurally normal chromosomes.
Detailed evaluation of these ‘shattered’ chromosomes has revealed several interesting features that set them apart from the derivative chromosomes more routinely observed in cancers. Sequence analysis of shattered chromosomes reveals a chaotic rearrangement of segments that joined at clustered, non-random breakpoints. The sequences along these chromosomes consistently exist in just 2 distinct and alternating copy number states.Citation4 Derivative chromosomes created by chromothripsis are therefore a patchwork of alternating regions of lost heterozygosity and preserved heterozygosity. This pattern contrasts with other, more conventional complex rearrangements observed to occur in the context of region amplification. Rearrangements associated with region amplification are understood to arise via DNA replication-based mechanisms, and therefore develop in a stepwise manner over many cell division cycles.
While it is impossible to infer a conclusive evolutionary history from a single cytogenetic snapshot, the evidence to date suggests that chromothripsis occurs as an “all-at-once” form of genomic instability – the consequence of a singular, catastrophic breakage event closely followed by a chaotic reassembly of chromosome fragments.Citation3,4 This premise challenges prevailing views of genomic instability in cancer. The better-understood forms of genetic instability cause incremental changes to DNA sequences and chromosome structures. Most critical features of cancer genomes thus evolve gradually, as the attendant alterations accumulate over many rounds of cell division. Chromothripsis appears to be something very different.
Hh signaling causes DNA stand breakage
Presumably, chromothripsis is caused by catastrophic chromosomal breakage, but how might such an event be precipitated? Ionizing radiation, telomere dysfunction, abortive apoptosis, replication stress and mitotic compaction of incompletely replicated chromosomes have all been suggested as potential triggers for the clustered chromosomal breaks that underlie chromothripsis.Citation5 While these possibilities are attractive for a variety of reasons, there is presently no consensus mechanistic explanation for chromosome shattering and reassembly. In particular, the contemporaneous genesis of a large number – tens to hundreds – of DNA double strand breaks that appear to underlie this phenomenon remains rooted in mystery. To contribute to this ongoing discussion, we summarize several recent observations and integrate them into a potentially testable hypothetical model that is admittedly highly speculative.
Since the first observation of chromothripsis by Stephens et al.Citation2 in the tumor genome of a patient with chronic lymphocytic leukemia, this phenomenon has been found in a wide variety of solid and liquid tumors, including common cancers in the lung, prostate, colorectum and brain.Citation4 One potentially important clue to the etiology of chromothripsis emerged from a comprehensive analysis of medulloblastomas. Medulloblastoma has 4 distinct molecular subtypes, defined by highly characteristic spectra of genetic alterations and gene expression profiles.Citation6 Rausch et al.Citation7 demonstrated that chromothripsis exclusively occurred in the subtype, the molecular subtype defined by the activation of Hedgehog (Hh) signaling. The Hh signaling pathway is a key regulator of development orthologous to the Sonic hedgehog pathway originally described in Drosophila. Medulloblastomas with active Hh signaling are accordingly referred to as SHH-medulloblastomas. Many other types of tumor exhibit evidence of Hh signaling, but the basis of pathway activation is best understood in SHH-medulloblastomas and other tumors that commonly harbor pathway-specific mutations.
Hh signaling is normally involved in developmental morphogenesis. In many adult tissues, activation of Hh signaling can initiate or otherwise promote neoplastic growth.Citation8 While the canonical Hh signaling pathway is understood to ultimately stimulate transcription by the Gli family of oncoproteins, a number of additional targets, pathways and downstream effects have been reported.Citation9
Among the incompletely understood consequences of Hh pathway activation is the induction of genetic instability. Wetmore, Karnitz and colleagues described Citation10 an inhibitory relationship between Gli1, the primary downstream effector of canonical Hh signaling, and Chk1, an essential checkpoint kinase that suppresses DNA strand breakage. Increased expression of Gli1 was found to abolish the interaction between Chk1 and its binding partner Claspin, and to thereby attenuate Chk1 activation. The Chk1-Claspin interaction is inducible by DNA damage and essential for robust activation of the DNA damage response.Citation11 During S-phase, Chk1 is activated by the upstream kinase ATR in response to stalled DNA replication forks. Chk1-null mutants are inviable, suggesting that this response to stalled replication forks may be an essential component of normal cell proliferation.Citation12,13 Heterozygous cells (Chk1+/−) from humans and mice are haploinsufficient, and express genetic instability phenotypes related to mitotic dysfunction.Citation14,15 Transient knockdown of Chk1 by RNAi can trigger DNA double strand breakage Citation16,17 and, in the context of partial DNA replication inhibition, can induce breaks at non-random loci known as common fragile sites.Citation17 Similarly, the inhibition of Chk1 activation by Gli1 expression sensitizes cells to ionizing radiation and increases the frequency of chromosome aberrations.Citation10 One interpretation of these findings is that Hh activation triggers a level of DNA breakage that is quantitatively additive to that caused by ionizing radiation, and thus potentiates radiation effects.
In light of the inhibitory effects of Gli1 on Chk1, it is interesting that induction of double strand DNA breaks accelerates SHH-medulloblastoma initiation. As a model of basal cell nevus syndrome (BCNS, also known as Gorlin syndrome), Ptch1+/− mice develop Shh-medulloblastoma with an incidence of 5–10%.Citation18 The rate limiting genetic step for spontaneous tumor initiation in Ptch1+/− mice - as in humans with BCNS - is the loss of the remaining functional Ptch1 allele. Tumor incidence in heterozygous mice is greatly accelerated - to 50–80% - by irradiation.Citation19-21 The timing of DNA damage is critical; ionizing radiation is only an effective accelerant of tumorigenesis if applied during the perinatal period when progenitor cells are still proliferative, an interval that ends several days after birth. Perinatal irradiation does not appear to enhance the rate of Ptch1 allelic loss per se, but rather triggers the growth of preneoplastic lesions that subsequently lose the remaining wild type Ptch1 allele.Citation22 Such preneoplastic lesions do not arise in tumors with 2 Ptch1 alleles, suggesting haploinsufficiency in the heterozygous state. These studies provide further evidence that activation of Hh signaling and DNA strand breaks are interdependent factors that are together required for the robust initiation of SHH-medulloblastomas. It would seem plausible that the DNA strand breaks that are so central to tumor development could also initiate chromothripsis.
Interactions between p53 and Hh signaling
Among SHH-medulloblastomas, chromothripsis is restricted to those tumors that harbor mutant TP53.Citation7 More generally, chromothripsis tends to occur in cancers that also have a high frequency of TP53 mutations.Citation4,23 The tight association of chromothripsis with p53 loss-of-function is probably not incidental. p53 is highly responsive to DNA damage, and the loss of this response has been shown to contribute to several forms of chromosomal and DNA sequence instability, including regional amplification and translocations.Citation23 When considering this well-known role for p53, it would seem intuitive that p53 can suppress chromothripsis. But how exactly is such suppression implemented?
As a suppressor of genetic instability, p53 can play 2 roles. Most obviously, p53 can reduce the overall level of genetic instability in a proliferative cell population by selecting against the expansion of unstable clones. In the case of SHH-medulloblastoma, p53 would likely be activated by the double strand DNA breaks associated with Hh activation, and would turn on the downstream pathways to cell cycle arrest or apoptosis that prevent such cells from proliferating. However, p53 is also known to act more directly to enforce genome stabilization, for example by increasing the expression of DNA repair proteins.Citation24 In the case of SHH-medulloblastomas, and perhaps other types of tumors, p53 could function to suppress Hh signaling, thereby proactively preventing the appearance of DNA strand breaks.
Several observations suggest that p53 does in fact suppress Hh signaling. p53 has been shown to directly affect the localization, stabilization and phosphorylation of Gli1.Citation25 Several intermediaries have recently been described. Gli1 is a substrate of the p53-induced phosphatase Wip1.Citation26 Via a separate pathway, p53 can promote the degradation of Gli1 protein by transcriptional induction of the acetyltransferase p300/CBP-associated factor, an E3 ubiquitin ligase.Citation27 Whether either of these pathways is active in the cellular progenitors of SHH-medulloblastoma is unknown. Also unknown is whether these effects on Gli1 can affect its regulation of Chk1-Claspin complex assembly.
The interaction between p53 and Hh signaling is further supported by mouse genetics. Double mutant (Ptch1+/− Tp53−/−) mice exhibit a markedly increased incidence of tumors, including SHH-medulloblastomas, compared to single mutant (Ptch1+/−) mice.Citation28 In fact, the rate of SHH-medulloblastoma incidence in the double mutant mice is similar to what is observed after perinatal irradiation of Ptch1+/− mice. Whether the single or double mutant pups have detectable endogenous DNA damage in the precursor cell populations that give rise to medulloblastoma is an interesting question.
A new connection closes the circle
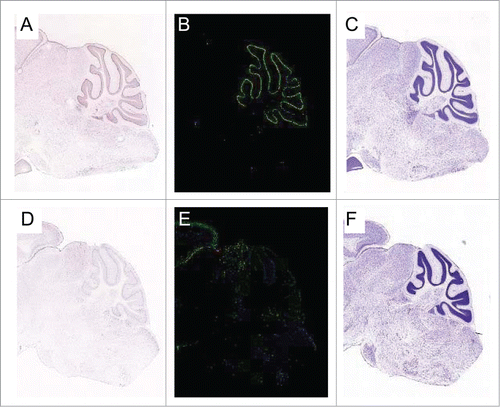
Our laboratory has recently identified a novel mammalian Patched gene that is robustly induced by p53 and which antagonizes canonical Hh signaling.Citation29 This gene, PTCH53, is a structural and functional homolog of PTCH1, the tumor suppressor in the canonical Hedgehog pathway. Unlike many other p53 target genes that are induced by DNA damage, PTCH53 is expressed in a p53-dependent manner, even in the absence of exogenous DNA damage. As a result, PTCH53 ranks among a very select group of genes that are dependent upon p53 for their basal expression in vivo.Citation29 In the brain of the adult mouse, Ptch53 RNA is expressed in the internal granular layer of the cerebellum, which originates from the same precursor cells as medulloblastoma ().
Figure 1. Expression of PTCH53 in the mouse cerebellum. (A) Assessed by in situ hybridization (ISH), PTCH53 RNA is localized in the cerebellum of the C57Bl/6 mouse. Staining is heavily concentrated in the purkinje cell layer and also visible in the internal granular layer, which originates from the external granular layer during the postnatal proliferative phase. Medulloblastomas arise from this precursor cell population. (B) PTCH53 heat map. (C) Nissl staining of the corresponding section. (D) The evolutionarily related protein PTCHD2 does not appreciably localize to these layers. (E) PTCHD1 heat map. (F) Nissl staining of the corresponding section. Images are from the Allen Brain Atlas Project.Citation30
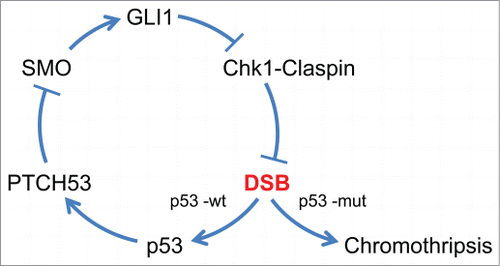
The p53-PTCH53 pathway provides a potential mechanism for the suppression of DNA strand breaks during the early stages of SHH-medulloblastoma growth. In our hypothetical model (), p53 responds to the DNA damage caused by Hh activation by dampening Hh signals via the upregulation of PTCH53. In nascent tumors that inactivate p53, this dampening effect would be lost and the damage caused by unmitigated Gli1 would be more extensive. It is possible that such a sudden, unopposed source of DNA damage could trigger chromothripsis. Our model would predict that the DNA breaks resulting from Chk1-Claspin inhibition would be preferentially located within common fragile sites, specific regions of the genome with low origin density and high rates of DNA replication stalling. It has been noted that expression of these fragile sites as a result of replication stress could explain the clustering of breakpoints that is characteristic of chromothripsis.Citation5
Figure 2. A model for the suppression of chromothripsis by p53. The oncogenic signals that arise upon activation of the canonical Hh pathway are generated by the mammalian ortholog of Smoothened (SMO). Active SMO triggers the upregulation of the transcription factor GLI1, which is itself a transcriptional target of the Hh pathway. The robust activation of GLI1 then disrupts the Chk1-Claspin interaction and thereby triggers chromosome breakage, possibly at common fragile sites. In cells that retain functional p53, the DNA strand breaks (DSB) generated by GLI1 trigger expression of PTCH53, which antagonizes SMO. Thus, p53 acts to dampen a cycle that could otherwise rapidly lead to extensive numbers of DNA strand breaks. Chromothripsis could arise in a stochastic manner if this feedback loop were to be interrupted by mutational loss of p53.
Several features of this model are obviously highly speculative as many aspects of PTCH53 function remain unclear. There are also many fundamental gaps in our knowledge regarding chromothripsis, such as its precise timing and how the shards are reassembled. Much experimentation and new genetic models are needed to rigorously test whether and when endogenous DNA strand breaks occur in cerebellar precursor cells as a result of activation of Hh signaling and inactivation of the p53-Ptch53 pathway. The next question is whether such breaks could conceivably give rise to the shattered chromosomes of chromothripsis.
It is unclear if our proposed model, even if validated in SHH-medulloblastomas, would apply to other cancers. While p53 and Hh signaling are recurrently altered in many tumor types, the convergence of these pathways during the very early evolution of SHH-medulloblastomas might create unique conditions for catastrophic DNA damage and repair that do not exist in other tumor types. The exact sets of conditions and molecular scenarios that facilitate chromosome shattering and relegation during neoplastic growth could therefore be distinct in different types of tissues. In this case, there would be many pathways to chromothripsis.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr, Kinzler KW. Cancer genome landscapes. Science 2013; 339:1546-58; PMID:23539594; http://dx.doi.org/10.1126/science.1235122
- Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, Pleasance ED, Lau KW, Beare D, Stebbings LA, et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 2011; 144:27-40; PMID:21215367; http://dx.doi.org/10.1016/j.cell.2010.11.055
- Meyerson M, Pellman D. Cancer genomes evolve by pulverizing single chromosomes. Cell 2011; 144:9-10; PMID:21215363; http://dx.doi.org/10.1016/j.cell.2010.12.025
- Zhang CZ, Leibowitz ML, Pellman D. Chromothripsis and beyond: rapid genome evolution from complex chromosomal rearrangements. Genes Dev 2013; 27:2513-30; PMID:24298051; http://dx.doi.org/10.1101/gad.229559.113
- Jones MJ, Jallepalli PV. Chromothripsis: chromosomes in crisis. Dev Cell 2012; 23:908-17; PMID:23153487; http://dx.doi.org/10.1016/j.devcel.2012.10.010
- Jones DT, Jager N, Kool M, Zichner T, Hutter B, Sultan M, Cho YJ, Pugh TJ, Hovestadt V, Stutz AM, et al. Dissecting the genomic complexity underlying medulloblastoma. Nature 2012; 488:100-5; PMID:22832583; http://dx.doi.org/10.1038/nature11284
- Rausch T, Jones DT, Zapatka M, Stutz AM, Zichner T, Weischenfeldt J, Jager N, Remke M, Shih D, Northcott PA, et al. Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. Cell 2012; 148:59-71; PMID:22265402; http://dx.doi.org/10.1016/j.cell.2011.12.013
- Barakat MT, Humke EW, Scott MP. Learning from jekyll to control hyde: hedgehog signaling in development and cancer. Trends Mol Med 2010; 16:337-48; PMID:20696410; http://dx.doi.org/10.1016/j.molmed.2010.05.003
- Jenkins D. Hedgehog signalling: emerging evidence for non-canonical pathways. Cell Signal 2009; 21:1023-34; PMID:19399989; http://dx.doi.org/10.1016/j.cellsig.2009.01.033
- Leonard JM, Ye H, Wetmore C, Karnitz LM. Sonic hedgehog signaling impairs ionizing radiation-induced checkpoint activation and induces genomic instability. J Cell Biol 2008; 183:385-91; PMID:18955550; http://dx.doi.org/10.1083/jcb.200804042
- Liu S, Song N, Zou L. The conserved C terminus of claspin interacts with Rad9 and promotes rapid activation of Chk1. Cell Cycle 2012; 11:2711-6; PMID:22732499; http://dx.doi.org/10.4161/cc.21041
- Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K, Luo G, Carattini-Rivera S, DeMayo F, Bradley A, et al. Chk1 is an essential kinase that is regulated by atr and required for the G(2)/M DNA damage checkpoint. Genes Dev 2000; 14:1448-59.; PMID:10859164; http://dx.doi.org/10.1101/gad.840500
- Wilsker D, Petermann E, Helleday T, Bunz F. Essential function of Chk1 can be uncoupled from DNA damage checkpoint and replication control. Proc Natl Acad Sci U S A 2008; 105:20752-7; PMID:19091954; http://dx.doi.org/10.1073/pnas.0806917106
- Wilsker D, Chung JH, Bunz F. Chk1 suppresses bypass of mitosis and tetraploidization in p53-deficient cancer cells. Cell Cycle 2012; 11:1564-72; PMID:22433954; http://dx.doi.org/10.4161/cc.19944
- Lam MH, Liu Q, Elledge SJ, Rosen JM. Chk1 is haploinsufficient for multiple functions critical to tumor suppression. Cancer Cell 2004; 6:45-59; PMID:15261141; http://dx.doi.org/10.1016/j.ccr.2004.06.015
- Syljuasen RG, Sorensen CS, Hansen LT, Fugger K, Lundin C, Johansson F, Helleday T, Sehested M, Lukas J, Bartek J. Inhibition of human Chk1 causes increased initiation of DNA replication, phosphorylation of ATR targets, and DNA breakage. Mol Cell Biol 2005; 25:3553-62; PMID:15831461; http://dx.doi.org/10.1128/MCB.25.9.3553-3562.2005
- Durkin SG, Arlt MF, Howlett NG, Glover TW. Depletion of CHK1, but not CHK2, induces chromosomal instability and breaks at common fragile sites. Oncogene 2006; 25:4381-8; PMID:16732333; http://dx.doi.org/10.1038/sj.onc.1209466
- Goodrich LV, Milenkovic L, Higgins KM, Scott MP. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science 1997; 277:1109-13; PMID:9262482; http://dx.doi.org/10.1126/science.277.5329.1109
- Pazzaglia S, Mancuso M, Atkinson MJ, Tanori M, Rebessi S, Majo VD, Covelli V, Hahn H, Saran A. High incidence of medulloblastoma following X-ray-irradiation of newborn Ptc1 heterozygous mice. Oncogene 2002; 21:7580-4; PMID:12386820; http://dx.doi.org/10.1038/sj.onc.1205973
- Pazzaglia S, Tanori M, Mancuso M, Rebessi S, Leonardi S, Di Majo V, Covelli V, Atkinson MJ, Hahn H, Saran A. Linking DNA damage to medulloblastoma tumorigenesis in patched heterozygous knockout mice. Oncogene 2006; 25:1165-73; PMID:16407852; http://dx.doi.org/10.1038/sj.onc.1209032
- Hahn H, Wojnowski L, Zimmer AM, Hall J, Miller G, Zimmer A. Rhabdomyosarcomas and radiation hypersensitivity in a mouse model of gorlin syndrome. Nat Med 1998; 4:619-22; PMID:9585239; http://dx.doi.org/10.1038/nm0598-619
- Pazzaglia S, Tanori M, Mancuso M, Gessi M, Pasquali E, Leonardi S, Oliva MA, Rebessi S, Di Majo V, Covelli V, et al. Two-hit model for progression of medulloblastoma preneoplasia in patched heterozygous mice. Oncogene 2006; 25:5575-80; PMID:16636673; http://dx.doi.org/10.1038/sj.onc.1209544.
- Zhu C, Mills KD, Ferguson DO, Lee C, Manis J, Fleming J, Gao Y, Morton CC, Alt FW. Unrepaired DNA breaks in p53-deficient cells lead to oncogenic gene amplification subsequent to translocations. Cell 2002; 109:811-21; PMID:12110179; http://dx.doi.org/10.1016/S0092-8674(02)00770-5
- Amundson SA, Patterson A, Do KT, Fornace AJ, Jr. A nucleotide excision repair master-switch: p53 regulated coordinate induction of global genomic repair genes. Cancer Biol Ther 2002; 1:145-9; PMID:12170774; http://dx.doi.org/10.4161/cbt.59
- Stecca B, Ruiz i Altaba A. A GLI1-p53 inhibitory loop controls neural stem cell and tumour cell numbers. EMBO J 2009; 28:663-76; PMID:19214186; http://dx.doi.org/10.1038/emboj.2009.16
- Pandolfi S, Montagnani V, Penachioni JY, Vinci MC, Olivito B, Borgognoni L, Stecca B. WIP1 phosphatase modulates the hedgehog signaling by enhancing GLI1 function. Oncogene 2013; 32:4737-47; PMID:23146903; http://dx.doi.org/10.1038/onc.2012.502
- Mazza D, Infante P, Colicchia V, Greco A, Alfonsi R, Siler M, Antonucci L, Po A, De Smaele E, Ferretti E, et al. PCAF ubiquitin ligase activity inhibits hedgehog/Gli1 signaling in p53-dependent response to genotoxic stress. Cell Death Differ 2013; 20:1688-97; PMID:24013724; http://dx.doi.org/10.1038/cdd.2013.120
- Wetmore C, Eberhart DE, Curran T. Loss of p53 but not ARF accelerates medulloblastoma in mice heterozygous for patched. Cancer Res 2001; 61:513-6; PMID:11212243
- Chung JH, Larsen AR, Chen E, Bunz F. A PTCH1 homolog transcriptionally activated by p53 suppresses hedgehog signaling. J Biol Chem 2014; 289:33020-31; PMID:25296753; http://dx.doi.org/10.1074/jbc.M114.597203
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature 2007; 445:168-76; PMID:17151600; http://dx.doi.org/10.1038/nature05453