
Abstract
Profilin-1 (Pfn1) is an important regulator of actin polymerization that is downregulated in human breast cancer. Previous studies have shown Pfn1 has a tumor-suppressive effect on mesenchymal-like triple-negative breast cancer cells, and Pfn1-induced growth suppression is partly mediated by upregulation of cell-cycle inhibitor p27kip1 (p27). In this study, we demonstrate that Pfn1 overexpression leads to accumulation of p27 through promoting AMPK activation and AMPK-dependent phosphorylation of p27 on T198 residue, a post-translational modification that leads to increased protein stabilization of p27. This pathway is mediated by Pfn1-induced epithelial morphological reversion of mesenchymal breast cancer through cadherin-mediated restoration of adherens junctions. These findings not only elucidate a potential mechanism of how Pfn1 may inhibit proliferation of mesenchymal breast cancer cells, but also highlight a novel pathway of cadherin-mediated p27 induction and therefore cell-cycle control in cells.
Introduction
Oncogenic transformation of cells is accompanied by alteration in actin cytoskeleton. In general, tumor cells display less organized actin cytoskeleton than their normal counterparts.Citation1 In some cases, filamentous actin density is inversely correlated with malignant characteristic of tumor cells, suggesting that alteration in actin cytoskeleton has a functional significance in tumor progression.Citation2 There are numerous instances of dysregulation of actin-binding proteins and/or signaling mediators of actin cytoskeletal control in various types of cancer. Importantly, in certain cases, there are causal connections between altered expression of actin cytoskeletal regulators and cancer progression.Citation3,4 Along this line, profilin-1 (Pfn1), a phylogenetically conserved actin-monomer binding protein that also interacts with membrane phosphoinositides and a wide range of other proteins bearing poly-L-proline (PLP) motifs, has been reported to be downregulated in human breast cancer.Citation5,6 Reduced level of Pfn1 promotes malignant features of breast cancer cells including extracellular matrix degradation, ECM invasion and dissemination.Citation6,7
At least in 2 triple-negative (lacks expression of estrogen-receptor (ER), progesterone receptor (PR) and HER2) human breast cancer cell lines of mesenchymal phenotype including MDA-MB-231 (MDA-231) and CAL51, overexpression of Pfn1 has a pronounced tumor-suppressive effect in vivo.Citation5,8 While the underlying molecular mechanisms of Pfn1's tumor-suppressive action in these cell lines are still unclear, proteomic studies in MDA-231 cells have shown that Pfn1 overexpression is associated with alteration in expression of many biomarkers of cell proliferation and survival.Citation9 Thus, it is likely that tumor-suppressive action of Pfn1 results from perturbation of multiple regulatory pathways governing tumor growth.
Many tumor-suppressor proteins interfere with G1-to-S phase progression of cell cycle. Cell cycle progression is tightly regulated by the activation of cyclin/cyclin-dependent kinase (CDK) complexes. Interactions between cyclins and CDKs are inhibited by the action of cyclin kinase inhibitors (CKI). P27kip1 (p27) is a prominent member of the CKI family which specifically binds to and inhibits cyclinE/CDK2 complex activity, causing cell-cycle arrest in G1 phase. Downregulation in expression and/or cytoplasmic mislocalization of p27 have been reported in a substantial number of human epithelial cancers (breast, prostate, lung, colon, head and neck).Citation10 We previously reported that stable overexpression of Pfn1 in MDA-231 breast cancer cells leads to p27 accumulation with concomitant induction of cell-cycle arrest in G0/G1 phase. Silencing p27 expression partly relieves the proliferation defect of Pfn1 overexpressing cells further suggesting that elevating Pfn1 expression causes cell cycle arrest, at least, in part through p27 induction.Citation11 Therefore, misregulation of p27 expression could be one of the potential pathways by which Pfn1 elicits its tumor-suppressive action in certain types of breast cancer cells.
While p27 expression can be controlled at all levels of gene expression including transcription, translation and post-translation, in cancer it is most often deregulated at post-translational level that involves accelerated proteolysis.Citation10 Protein stability as well as sub-cellular (i.e. nuclear vs cytoplasmic) localization of p27 are critically regulated by its phosphorylation on serine and threonine residues.Citation12 Hyperactivation of PI3K-AKT pathway has been most prominently linked to p27 deregulation in cancer. AKT can directly phosphorylate p27 on multiple residues (S10 and T157) leading to its nuclear exclusion.Citation13,14 AKT can also regulate the activity of skp2, a key component of the E3 ligase for p27 ubiquitination.Citation15 P27 can be also phosphorylated on T198 by AMPK (AMP-activated protein kinase – a kinase that is activated under conditions of metabolic stress e.g. when the AMP:ATP ratio rises in cells). Upon nutrient deprivation, AMPK-mediated phosphorylation confers increased stability to p27.Citation16 Therefore, AMPK-dependent phosphorylation of p27 is a major mechanism that links nutrient deprivation to cell-cycle control.
In this study, we have established a novel mechanistic link between Pfn1 and p27 in mesenchymal human breast cancer cells that involves AMPK activation secondary to epithelial morphological reversion.
Results
We previously reported that stable overexpression of Pfn1 leads to increased protein stability of p27 in MDA-231 cells,Citation11 suggesting that cellular changes induced by Pfn1 elevation are linked to post-translational regulation of p27. To determine whether differential protein stability of p27 solely accounts for Pfn1-dependent change in p27 expression, we analyzed the effect of MG-132 (a proteasome inhibitor) on the relative levels p27 expression in isogenic sublines of MDA-231 cells stably overexpressing either GFP-Pfn1 or GFP (control). Under DMSO (vehicle control) treated condition, Pfn1 overexpressing cells have markedly higher p27 expression than GFP expressors as expected but the p27 differential is nullified when the proteasomal degradation pathway is inhibited (). These data demonstrate that elevating Pfn1 expression upregulates p27 in MDA-231 cells through retarding its protein turnover.
Figure 1. Pfn1 overexpression causes p27 accumulation in MDA-231 cells through inhibiting its protein turnover. (A) P27 immunoblot of total cell extracts prepared from sub-confluent cultures of GFP- and GFP-Pfn1-expressing MDA-231 cells following treatment with 10 μM of either MG-132 (a proteasome inhibitor) or DMSO (vehicle control) for 10 hours. Tubulin blot serves as a loading control. (B) P27 and skp2 immunoblots of cytosolic and nuclear extracts prepared from GFP- and GFP-Pfn1-expressors (tubulin and histone blots served as loading control for cytosolic and nuclear fractions, respectively). These data are representative of 2–3 independent experiments.
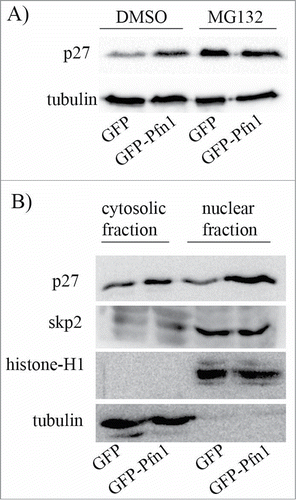
P27 is subjected to ubiquitin-proteasome-mediated degradation in both cytoplasmic and nuclear compartments of cells. Subcellular fractionation analyses revealed that Pfn1 overexpression results in p27 elevation only modestly in the cytoplasmic compartment but very dramatically in the nucleus (), further indicating that Pfn1 causes p27 accumulation in MDA-231 cells primarily through misregulation of p27 proteolysis in the nucleus. Poly-ubiquitination and subsequent degradation of p27 in the nucleus is mediated by SCF-skp2 (Skp-Cullin-F-box) E3 ubiquitin ligase complex.Citation17 Subcellular fractionation analyses showed no difference in either cytoplasmic or nuclear content of skp2 between control and Pfn1 overexpressing cells (). Thus, Pfn1 overexpression does not lead to nuclear accumulation of p27 through dysregulation of either the overall expression or subcellular localization of skp2.
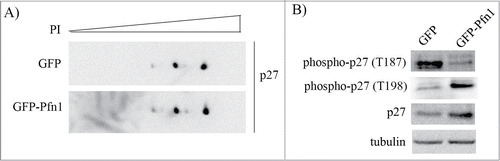
Phosphorylation status of p27 is a key determinant of protein stability of p27. For example, phosphorylation of p27 on T187 residue is a key prerequisite for skp2:p27 interaction and skp2-mediated p27 degradation.Citation18 By contrast, phosphorylation of p27 on T198 residue confers increased protein stability to p27.Citation19 To determine whether Pfn1 overexpression influences the overall phosphorylation pattern of p27, we resolved total extracts from control and Pfn1 overexpressing cells by 2D gel electrophoresis and performed immunoblotting with anti-p27 antibody. Note that as Pfn1 overexpressors have a higher total p27 level, we loaded higher amounts of total extract from control cells in order to normalize the total p27 level between the 2 groups and faithfully assess Pfn1-induced post-translational changes of p27, if any. We found no discernible difference in the overall post-translational modification pattern of p27 between control and Pfn1 overexpressing cells (). These data suggest that p27's ability to become phosphorylated per se is not affected by Pfn1. However, consistent with increased protein stability of p27, Pfn1 overexpressors exhibited reduced and elevated phosphorylation on T187 and T198 residues, respectively (). These data demonstrate that Pfn1 overexpression has site-specific effects on p27 phosphorylation.
Figure 2. Pfn1 overexpression has site-specific effects on p27 phosphorylation in MDA-231 cells. (A) Total lysates prepared from sub-confluent cultures of GFP- and GFP-Pfn1-expressing MDA-231 cells were separated by 2D-gel electrophoresis and immunoblotted with p27 antibody. Higher amount of total protein was loaded for the GFP group to normalize the total p27 level between 2 groups. (B) Total cell extracts of GFP- and GFP-Pfn1 expressors were separated on an SDS-PAGE and immunoblotted with the indicated antibodies (tubulin blot serves as a loading control). These data are representative of 2–3 independent experiments.
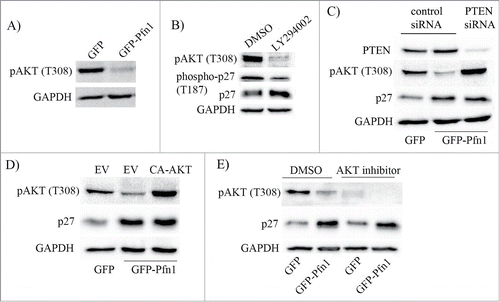
Hyperactivation of PI3K/AKT signaling is a major driving force behind increased proteolysis and/or cytoplasmic mislocalization of p27 in cancer cells.Citation20 For example, AKT can directly phosphorylate p27 on the serine 10 (S10) and threonine 157 (T157) residues. Nuclear export of p27 is facilitated by S10 phosphorylation while its nuclear import is inhibited by T157 phosphorylation, and therefore, either of these 2 phosphorylations can promote cytoplasmic mislocalization of p27. Although p27 cannot be directly phosphorylated by AKT on the T187 residue, AKT may indirectly promote T187 phosphorylation through activation of cyclinE-CDK2 (a kinase that is responsible for p27 phosphorylation on T187) complex.Citation21 We previously showed that loss of Pfn1 expression enhances accumulation of PI(3,4)P2 (a phosphoinositide that is generated downstream of activated PI3K) in MDA-231 cells in response to acute activation of various receptor tyrosine kinases (RTKs).Citation22 Conversely, elevating Pfn1 expression greatly suppresses RTK-induced generation of PtdIns(3,4,5)P3 (another PI3K-generated phosphoinositide) and the downstream AKT activation.Citation23 Even in a regular culture setting i.e., without acute activation of any particular RTK pathway, Pfn1 overexpressors exhibit a marked suppression of AKT activation (as measured by the level of AKT phosphorylation on T308) when compared to control cells (). These data are suggestive of Pfn1s inhibitory effect on PI3K/AKT signaling in MDA-231 cells. Since pharmacological inhibition of PI3K by LY294002 mimics the effect of Pfn1 overexpression in terms of attenuation of T187 phosphorylation with concomitant elevation of p27 expression (), we further asked whether Pfn1-induced p27 accumulation is due to deficient PI3K/AKT signaling. To address this question, we performed 2 types of rescue experiments. First, we studied the effect of silencing of PTEN (phosphatase and tensin homolog 10 – a dual lipid-protein phosphatase that antagonizes PI3K/AKT signaling by dephosphorylating PI(3,4,5)P3 to PtdIns(4,5)P2) on Pfn1-induced change in p27 expression. PTEN silencing was able to rescue Pfn1 overexpressing cells from defect in AKT activation but surprisingly had no effect on p27 expression (). Likewise, when AKT was directly hyperactivated in Pfn1 overexpressing cells by expressing a constitutively active mutant of AKT (CA-AKT: it harbors phosphomimetic T308D and S473D mutations), p27 accumulation could also not be reversed (). Furthermore, when AKT function was blocked by an inhibitor, the p27 differential between control and Pfn1 overexpressors was still maintained (). Collectively, these results demonstrate that Pfn1-induced elevation of p27 in MDA-231 cells is not due to deficiency in AKT activation.
Figure 3. Deficiency in PI3K/AKT signaling does not account for Pfn1-induced accumulation of p27 in MDA-231 cells. (A) Total lysates prepared from sub-confluent cultures of GFP- and GFP-Pfn1-expressing MDA-231 cells were run on an SDS-PAGE and immunoblotted with phospho-AKT (T308) antibody. (B) Total extracts prepared from GFP-expressing cells following treatment with 25 μM of either LY294002 or DMSO (vehicle control) for 24 hours were resolved by SDS-PAGE and immunoblotted with the indicated antibodies. (C) Total extracts of GFP- and GFP-Pfn1 expressors transfected with 100 nM of either non-targeting control or PTEN-siRNAs were resolved by SDS-PAGE and immunoblotted for PTEN and p27. (D) P27 and pAKT (T308) immunoblots of total lysates prepared from GFP- and GFP-Pfn1 expressors transfected with either empty vector (EV) or constitutively active AKT (CA-AKT) encoding plasmids. (E) Phospho-AKT (T308) and p27 immunoblots of extracts prepared from GFP- and GFP-Pfn1 overexpressors following treatment with 25 μM of either AKT inhibitor or DMSO (vehicle control) for 24 hours. GAPDH blot served as the loading control in all experiments. These data are representative of 2–3 independent experiments.
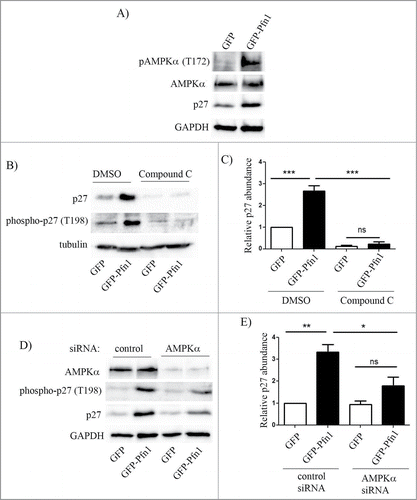
As we found that Pfn1 overexpression causes elevated T198 phosphorylation of p27, a post-translational modification that also increases the protein stability of p27, we asked whether Pfn1 overexpression causes p27 elevation through affecting AMPK (a candidate kinase for T198 phosphorylation of p27) activity. Phospho-specific immunoblot analyses of total cell extracts showed that Pfn1 overexpression increases T172–phosphorylated form of AMPK (an indicator for the activated form of AMPK) while the total expression level of AMPK was unaffected (). Next, we performed rescue experiments involving suppression of AMPK activity using both pharmacological and molecular strategies. Treating cells with AMPK antagonist Compound C dramatically reduced T198 phosphorylation of p27 and completely reversed Pfn1-induced elevation of p27 ( – in control experiments, DMSO treatment preserved higher levels of T198-phosphorylated form and total p27 in Pfn1 overexpressors relative to control GFP expressing cells as expected). Quantification showed that Compound C treatment led to an average fold10- reduction in p27 level in Pfn1 expressing cells. As a complementary approach, we inhibited AMPK function through silencing of AMPKα, an essential component of AMPK holoenzyme complex. Similar to the trends of Compound C experimental results, silencing of AMPKα also led to downregulation of T198 phosphorylation and fold2- reduction in the overall expression of p27 expression in Pfn1 overexpressing cells, completely reversing Pfn1-induced p27 accumulation in MDA-231 cells (). We noted that compound C treatment lowered even the basal p27 expression in control GFP-expressing cells which AMPKα knockdown failed to do, and this likely explains the differences in fold-change in p27 expression in Pfn1 overexpressors between the settings of molecular and pharmacological approaches of AMPK inhibition. This was not totally surprising as AMPKα knockdown was not 100% in our experiments and it has been reported that compound C can have off-target effects on several other kinases besides AMPK,Citation24 which may also impact p27 expression. Nonetheless, the overall similarity between the effects of AMPK silencing and compound C treatment demonstrate that Pfn1 overexpression leads to p27 accumulation in MDA-231 cells through stimulating AMPK-dependent phosphorylation on the T198 residue.
Figure 4. Pfn1 overexpression upregulates p27 in MDA-231 cells through AMPK activation. (A) Immunoblots of total extracts show the relative levels of T172-phosphorylated- AMPK, total AMPK and p27 between GFP- and GFP-Pfn1 expressors. (B, C) Representative immunoblots showing relative levels of T198-phosphorylated and total p27 between GFP and GFP-Pfn1 expressors following treatment with 10 μM of AMPK antagonist Compound C or DMSO (vehicle) for 24 hours. D-E) Representative immunoblots showing relative levels of AMPKα, T198-phosphorylated and total p27 between GFP and GFP-Pfn1 expressors 72 hours after transfection with 100 nM of either non-targeting control or AMPKα-specific siRNAs. The bar graph on the right summarizes the data from 3 independent experiments. GAPDH and tubulin blots served as the loading controls (***: p<0.001, **: p<0.01, *: p<0.05; ns: not significant).
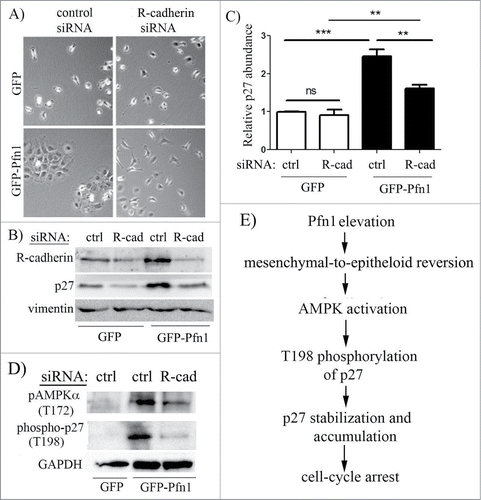
We next investigated whether Pfn1overexpression induced p27 accumulation is sensitive to culture conditions. We found that elevation of T198 phosphorylation and overall accumulation of p27 in Pfn1 overexpressors is much more pronounced in confluent than in sub-confluent culture conditions ( – note that degree of confluence has no effect on p27 expression in control cells). Based on this observation, we speculated that cell-cell interaction may play a role in Pfn1-induced p27 accumulation in MDA-231 cells. The MDA-231 cell line is null for expression of most of the major cadherin family of adherens junction (AJ) proteins including E-, P-, and N-cadherin and displays all characteristics of post-epithelial-to-mesenchymal transition. MDA-231 cells however express R-cadherin (retinal cadherin) and we previously reported that stable Pfn1 overexpression is able to morphologically transform this cell line into an epithelial phenotype through upregulating R-cadherin and formation of R-cadherin-based AJs.Citation25 A similar Pfn1-dependent epithelial phenotypic induction was also reported by another group in CAL-51,Citation26 an otherwise mesenchymal breast cancer cell line that loses its tumorigenic ability upon Pfn1 overexpression.Citation5 Note that at least in the case of MDA-231 cells, we confirmed that Pfn1-induced morphological transformation is not a classical mesenchymal-to-epithelial transition (MET) as it was not accompanied by loss of vimentin (a marker for mesenchymal cells) and induction of E-cadherin (a marker for epithelial cells) expressions.Citation25 Since stable Pfn1 overexpression in ER+/PR+ epithelial-type breast cancer cell lines (BT474, MCF-7) does not lead to p27 elevation (Fig. S1), it is likely that Pfn1 does not have a direct mechanism of regulating p27 expression. This prompted us to further query whether epithelial phenotypic transformation plays a role in Pfn1-dependent upregulation of p27 in MDA-231 cells. To test this, we reverted Pfn1 overexpressing cells to a mesenchymal phenotype through silencing of R-cadherin expression (). Downregulation of R-cadherin expression did not completely abrogate but reversed Pfn1-induced p27 accumulation by 1.5-fold in MDA-231 cells (; note that R-cadherin silencing did not have any effect on p27 expression in control GFP expressors). R-cadherin depletion also resulted in a prominent suppression of AMPK activation and T198 phosphorylation of p27 in Pfn1 overexpressing cells (). Overall, these data demonstrate that epithelial morphological reversion plays a key role in AMPK-dependent p27 accumulation upon overexpression of Pfn1 in mesenchymal breast cancer cells ().
Figure 5. Pfn1-induced p27 upregulation in MDA-231 cells increases with cell confluence. GFP- and GFP-Pfn1- expressors were either maintained in sub-confluent condition or grown to confluency. Cell extracts prepared under these conditions were resolved by SDS-PAGE and immunoblotted for the relative levels of p27 (panel A) and T198-phosphorylated p27 (panel B) between the 2 sublines (GAPDH and tubulin blots served as the loading control).
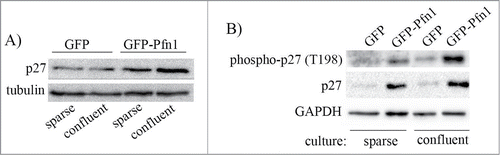
Figure 6. Epithelial reversion plays a role in Pfn1-induced p27 upregulation in MDA-231 cells. Phase contrast micrographs of GFP and GFP-Pfn1 expressors following transfection with either control or R-cadherin-specific siRNAs demonstrate that Pfn1 overexpression can induce epithelioid morphological reversion (as revealed by clustering of cells) in mesenchymal MDA-231 cells which can be reversed by silencing of R-cadherin. (B, C) Representative immunoblots (B) of total extracts from GFP- and GFP-Pfn1 expressors transfected with the indicated siRNAs (ctrl: control, R-cad: R-cadherin) showing changes in p27 expression specifically in Pfn1 overexpressors upon R-cadherin depletion (vimentin blot: loading control). The bar graph on the right (C) summarizes the data from 5 independent experiments (***: p<0.001, **: p<0.01, ns: not significant). (D) Representative immunoblots of total lysates of GFP and GFP-Pfn1 overexpressors transfected with either control or R-cadherin siRNAs show downregulation of AMPKα- and p27-phosphorylations on T172 and T198 residues, respectively, upon R-cadherin depletion (GAPDH blot: loading control). (E) A proposed model depicting that Pfn1 overexpression elevates p27 accumulation in mesenchymal breast cancer cells through impacting AMPK pathway as a consequence of cadherin-dependent epithelioid reversion.
Discussion
Cell-cell adhesion plays a critical role in embryonic development, differentiation and maintenance of tissue architecture. Disruption of cell-cell adhesion which occurs during EMT through downregulation of junctional components promotes cell migration and proliferation. Although EMT was originally described as a morphogenetic process that occurs during normal embryonic development, some aspects of EMT are also recapitulated during metastatic progression of epithelial-derived tumors. Loss of E-cadherin (the central player in the makeup of AJ in epithelial cells) function, a hallmark of EMT, promotes invasiveness of carcinoma cells.Citation27-31 Conversely, experimental restoration of E-cadherin suppresses the tumorigenic ability and invasive phenotype of E-cadherin negative cancer cells,Citation32 pointing to a critical role of cell-cell adhesion in the regulation of tumor initiation and malignant progression. E-cadherin engagement causes growth suppression of epithelial tumors at least partly through increasing the cellular level of p27.Citation33 In this study, we demonstrated a parallel mechanism involving R-cadherin, an important but under-studied member of cadherin-family proteins that mediates mesenchymal-to-epithelial morphological transformationCitation34,35,36 and plays a key role in Pfn1-induced elevation of p27 expression in mesenchymal breast cancer cells.Citation11 R-cadherin expression is often associated with induction of EMT in epithelial cells most likely owing to its ability to displace E-cadherin from AJsCitation37 and accordingly several types of carcinoma cells have lower R-cadherin expression than their normal counterparts.Citation38 However, in cancer cells that have already lost E-cadherin expression (such as MDA-231 cells), R-cadherin induces MET-like morphological transformation similar to the action of E-cadherin as shown previously and regulates p27 as demonstrated herein, suggesting some degrees of functional similarity between the 2 cadherin family of AJ proteins.
How cadherin engagement upregulates p27 is not clearly understood. It has been previously shown that PTEN (a negative regulator of skp2 and AKT) can be recruited to AJs in epithelial cells through its binding to MAGI (an AJ adaptor protein), and this causes stabilization and increase in the overall expression level of PTEN.Citation39 As actions of skp2 and AKT promote downregulation of p27, PTEN-mediated skp2/AKT modulation could certainly serve as the mechanistic basis for cadherin-dependent p27 regulation in cells. While Pfn1 overexpression in MDA-231 cells also causes elevation of PTEN and suppression of AKT activation (),Citation23 surprisingly, we found no evidence of AKT's involvement in Pfn1-dependent accumulation of p27. Rather, our study demonstrated that Pfn1 overexpression causes p27 accumulation primarily through cadherin-dependent upregulation of AMPK activation and p27 phosphorylation on T198. Therefore, our findings now reveal a new pathway of cadherin-mediated p27 regulation in cells. LKB1, a tumor suppressor that is linked to Peutz-Jeghers syndrome, phosphorylates and activates AMPK.Citation40 While in some cases activation of LKB1-AMPK pathway causes p27 upregulation,Citation16 there are also examples where hypeactivation of this pathway can cause cell cycle arrest at G1 phase without affecting the level of p27Citation41 therefore suggesting context specificity of LKB1-AMPK pathway in p27 regulation. Recently, it has been shown that LKB1 is able to co-localize with E-cadherin at AJs, and LKB1-mediated activation of AMPK requires maturation of AJ,Citation42 a finding that is consistent with our observation of reduction of AMPK activation upon disruption of AJ in Pfn1 overexpressing cells. It would be interesting to determine in the future whether Pfn1 regulates AMPK activity through R-cadherin-dependent modulation of LKB1. As LKB1 is also required for maintaining epithelial integrity,Citation43 it is possible that cadherin and LKB1 act through a positive feedback loop to negatively regulate cell cycle via AMPK-mediated phosphorylation of p27.
Is AMPK-mediated T198 phosphorylation of p27 the sole mechanism for Pfn1-induced p27 upregulation in MDA-231 cells? At least 2 other alternative mechanisms can be considered. First, there is now evidence that in triple-negative breast cancer cells, p27 expression is transcriptionally downregulated through cooperation of TEAD4 [transcription enhancer factor domain 4: a transcription factor partner of YAP (yes-associated protein) and KLF5 (kruppel like factor)].Citation44 Since we previously showed that Pfn1 overexpression does not change the mRNA level of p27,Citation11 we think it is unlikely that alteration in upstream signaling affecting TEAD4 and/or KLF5 contributes to Pfn1-dependent regulation of p27. Second, AMPK activation also downregulates mTOR (mammalian target of rapamycin) signaling. MTOR-raptor complex promotes SGK1 (serum- and glucocorticoid-inducible kinase-1) activation and SGK1-dependent phosphorylation of p27 at T157 residue leading to cytoplasmic mislocalization of p27.Citation45 It is now known that contact-inhibition can lead to suppression of mTOR activation and p27 induction in epithelial cells.Citation46 Therefore, it is also possible that Pfn1 overexpression causes nuclear accumulation of p27 partly through AMPK-dependent suppression of mTOR activity and in turn inhibiting SGK1-mediated T157 phosphorylation of p27.
We previously showed that actin-binding of Pfn1 is critical for its ability to induce R-cadherin mediated epithelial morphological transformation in MDA-231 cells.Citation25 A causal connection between epithelial reversion and p27 upregulation upon Pfn1 overexpression in our cell line is consistent with the requirement for actin-binding of Pfn1 for its tumor-suppressive effect as demonstrated previously in another mesenchymal breast cancer cell line.Citation8,26 Given that Pfn1 overexpression does not have any effect on p27 expression in epithelial breast cancer cell lines, we speculate that p27 as a potential intermediary molecule for Pfn1's tumor-suppressive action, if true, would be applicable to only certain subtypes (e.g., basal triple-negative) of breast cancer cells. It will be interesting to extend our findings to other triple-negative breast cancer cell lines of mesenchymal vs epithelial types in the future for a more definitive answer. Additionally, a recent study has shown that cytoplasmic mislocalization of p27 in HER2-positive breast cancer cells causes resistance to lapatinib, a dual EGFR-HER2 inhibitor.Citation47 Therefore, it will be also interesting to determine in the future whether Pfn1 expression has any effect on p27 localization and therapeutic resistance in HER2-positive breast cancer cells.
Finally, our finding that Pfn1 overexpression stimulates AMPK activation has much broader implications in the context of cancer beyond and above p27 regulation. AMPK activation is known to promote autophagy which has opposing effects on cancer progression.Citation48 On one hand, autophagy causes cell death and suppression of primary tumor. On the other hand, autophagy promotes cancer cell survival in the face of metabolic stress/hypoxia ultimately resulting in metastatic progression and therapeutic resistance. Therefore, it will be interesting to explore in the future whether autophagy plays any role in Pfn1s regulation of tumor growth and chemo-sensitivity of breast cancer cells.
Materials and Methods
Antibody and reagents
Monoclonal GAPDH and tubulin antibodies are products of Sigma-Aldrich. Monoclonal p27 antibody was obtained from BD biosciences. Polyclonal phospho-AKT (T308), AMPK, phospho-AMPK (T172) and monoclonal skp2 antibodies were purchased from Cell Signaling Technologies. Monoclonal Histone H1 antibody was from Santa Cruz Biotechnology. Polyclonal phospho-p27 (T187) antibody was purchased from Invitrogen. Phospho-p27 (T198) was a product from R&D systems. Monoclonal Pfn1 antibody was purchased from Novus Biological. R-cadherin antibody was a generous gift of Dr. Rachel Hazan (Albert Einstein College of Medicine, NY). LY294002 compound was purchased from Cell Signaling Technologies. MG132 was a product of Merck Millipore. Compound C was a product from Enzo Life Sciences. Akt Inhibitor (Akti-1/2) was a product from Abcam. All cell culture reagents were products of Invitrogen.
Cell culture and transfection
Generation and culture of MDA-MB-231 cell lines stably expressing GFP and GFP-Pfn1 have been described previously.Citation8 GFP and GFP-Pfn1 were also subcloned into pQCXIP retroviral vector (Clontech). Retrovirus packaging and subsequent infection of BT474 and MCF-7 cells were carried out according to the manufacturer's instructions. Infected cells were selected for puromycin resistance (250 ng/ml) and finally, stable cells were sorted based on their GFP-fluorescence before experimental use. BT474 and MCF-7 cells were cultured in RPMI1640 and DMEM medium supplemented with 10% FBS, respectively. Constitutively active HA-AKT1 plasmid was obtained through Addgene. Plasmid transfection was preformed performed using Lipofectamine LTX/Plus reagent (Invitrogen) according to manufacturer's instruction. In gene silencing experiments, cells were transfected with pooled non-targeting control or gene-targeted (AMPKα, R-Cadherin, PTEN) siRNAs obtained from Santa Cruz Biotechnology using transfection reagent from Dharmacon according to the manufacturer's instructions.
Protein extraction, inmmunoblot
Total cell lysate was prepared by extracting cells with modified RIPA buffer (50 mM Tris-HCl [pH = 7.5], 150 mM NaCl, 1% NP-40, 0.25% sodium deoxycholate, 0.1% SDS, 2 mM EDTA) supplemented with 50 mM NaF, 1 mM sodium pervanadate, and protease inhibitors. Subcellular fractionation was performed according to our published protocol.Citation11 Immunoblotting concentrations for different antibodies were: 1:500 (Histone H1, phospho-p27 (T187, T198)), 1:1000 (R-cadherin, AMPK, phospho-AMPK (T172), phospho-AKT (T308), skp2), 1:2000 (p27kip1) and 1:4000 (Pfn1, GAPDH and tubulin).
2D gel electrophoresis
Cells were washed twice with ice-cold DPBS followed by 2 washes with ice-cold Tris/Sucrose Buffer [10 mM Tris, 250 mM Sucrose (Invitrogen)] and maintained on ice throughout the extraction process. Dishes were placed at oblique for 5 minutes to collect residual fluid. Cells were scraped in the presence of 2D lysis Buffer [2 M Urea (Fisher Scientific), 7 M Thiourea (Invitrogen), 4% (w/v) CHAPS (Sigma), 50 mM DTT (Roche)] and collected into a Bead-beater tube (Biospec) containing 50 mg Glass Beads (Sigma). Lysates were pulsed 4 times for 20 seconds each in a Mini Beadbeater (Biospec) with 2 minutes on ice between pulses. 0.1 U Benzoase Nuclease (Sigma) and 2 mM MgCl2 (Fisher Scientific) were then added and solution incubated on ice for 30 minutes. The lysates were collected into microcentrifuge tubes and subjected to 2 rounds of centrifugation. The first round was 3500 RPM for 5 minutes to separate glass beads, where the supernatant was then subjected to 13000 RPM for 20 minutes to remove debris. The protein concentration of the supernatant was measured using the RC DC Protein Assay Kit (Bio-Rad) and aliquots stored at −80°C or immediately subjected to isoelectric focusing.
Isoelectric focusing was performed using the Zoom IPGRunner System (Invitrogen) and carried out per manufacturer's instructions with modification. Equal masses of proteins were used to make Rehydration Buffer [final concentrations: 2 M Urea, 7 M Thiourea, 4% (w/v) CHAPS, 50 mM DTT, 0.5% (v/v) Carrier Ampholytes (Invitrogen), 0.005% (w/v) Bromophenol Blue (Fisher Scientific), 50–150 μg Proteins]. Rehydration buffer was loaded into a Zoom IPG Runner Cassette (Invitrogen) and incubated with ZOOM IPG Strips (Invitrogen) for 1 hour at room temperature. In all cases, the pH range of the Carrier Ampholytes and IPG Strip were identical. Greater than 95% of the rehydration buffer was taken up by the IPG strip. IPG strips underwent isoelectric focusing using the following program: 175 V for 30 minutes; linear ramp 175–2000 V over 45 minutes; 2000 V for 105 minutes. IPG strips were then stored at −80°C or immediately equilibrated. IPG strips were incubated with Equilibration Buffer [6 M Urea, 2% (w/v) SDS, 50 mM Tris pH = 8.8, 20% (v/v) Glycerol, 2% (w/v) DTT] for 25 minutes at room temperature with gentle agitation. IPG strips were briefly washed with Running Buffer [25 mM Tris pH 8.3, 192 mM Glycine, 0.1% (w/v) SDS] and sealed on Tris-HCl polyacrylimide gels with running buffer containing 0.5% Agarose (Invitrogen) and 0.005% (w/v) Bromophenol Blue. These gels were subjected to Western blot.
Statistical analysis
Statistical differences were assessed by one-way ANOVA followed by Tukey post-hoc test, and a p-value of less than 0.05 was considered significant.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
1069929_Supplemental_Material.zip
Download Zip (874.4 KB)Funding
This study was supported by grants from the National Institute of Health (2R01-CA 108607 to PR, R01 DK075048 to KRH and P30 DK079307, the Pittsburgh Center for Kidney Research to KRH and HL).
Related Research Data
References
- Yamaguchi H, Condeelis J. Regulation of the actin cytoskeleton in cancer cell migration and invasion. Biochim Biophys Acta 2007; 1773:642-52; PMID:16926057; http://dx.doi.org/10.1016/j.bbamcr.2006.07.001
- Mouneimne G, Hansen SD, Selfors LM, Petrak L, Hickey MM, Gallegos LL, Simpson KJ, Lim J, Gertler FB, Hartwig JH, et al. Differential remodeling of actin cytoskeleton architecture by profilin isoforms leads to distinct effects on cell migration and invasion. Cancer Cell 2012; 22:615-30; PMID:23153535; http://dx.doi.org/10.1016/j.ccr.2012.09.027
- Stevenson RP, Veltman D, Machesky LM. Actin-bundling proteins in cancer progression at a glance. J Cell Sci 2012; 125:1073-9; PMID:22492983; http://dx.doi.org/10.1242/jcs.093799
- Gross SR. Actin binding proteins: their ups and downs in metastatic life. Cell Adh Migr 2013; 7:199-213; PMID:23302954; http://dx.doi.org/10.4161/cam.23176
- Janke J, Schluter K, Jandrig B, Theile M, Kölble K, Arnold W, Grinstein E, Schwartz A, Estevéz-Schwarz L, Schlag PM, et al. Suppression of tumorigenicity in breast cancer cells by the microfilament protein profilin 1. J Exp Med 2000; 191:1675-86; PMID:10811861; http://dx.doi.org/10.1084/jem.191.10.1675
- Ding Z, Joy M, Bhargava R, Gunsaulus M, Lakshman N, Miron-Mendoza M, Petroll M, Condeelis J, Wells A, Roy P. Profilin-1 downregulation has contrasting effects on early vs late steps of breast cancer metastasis. Oncogene 2014; 33:2065-74; PMID:23686314; http://dx.doi.org/10.1038/onc.2013.166
- Valenzuela-Iglesias A, Sharma VP, Beaty BT, Ding Z, Gutierrez-Millan LE, Roy P, Condeelis JS, Bravo-Cordero JJ. Profilin1 regulates invadopodium maturation in human breast cancer cells. Eur J Cell Biol 2015; 94:78-89; PMID:25613364; http://dx.doi.org/10.1016/j.ejcb.2014.12.002
- Zou L, Jaramillo M, Whaley D, Wells A, Panchapakesa V, Das T, Roy P. Profilin-1 is a negative regulator of mammary carcinoma aggressiveness. Br J Cancer 2007; 97:1361-71; PMID:17940506; http://dx.doi.org/10.1038/sj.bjc.6604038
- Coumans JV, Gau D, Poljak A, Wasinger V, Roy P, Moens PD. Profilin-1 overexpression in MDA-MB-231 breast cancer cells is associated with alterations in proteomics biomarkers of cell proliferation, survival, and motility as revealed by global proteomics analyses. OMICS 2014; 18:778-91; PMID:25454514; http://dx.doi.org/10.1089/omi.2014.0075
- Chu IM, Hengst L, Slingerland JM. The Cdk inhibitor p27 in human cancer: prognostic potential and relevance to anticancer therapy. Nat Rev Cancer 2008; 8:253-67; PMID:18354415; http://dx.doi.org/10.1038/nrc2347
- Zou L, Ding Z, Roy P. Profilin-1 overexpression inhibits proliferation of MDA-MB-231 breast cancer cells partly through p27kip1 upregulation. J Cell Physiol 2010; 223:623-9; PMID:20143334
- Vervoorts J, Luscher B. Post-translational regulation of the tumor suppressor p27(KIP1). Cell Mol Life Sci 2008; 65:3255-64; PMID:18636226; http://dx.doi.org/10.1007/s00018-008-8296-7
- Nacusi LP, Sheaff RJ. Akt1 sequentially phosphorylates p27kip1 within a conserved but non-canonical region. Cell Div 2006; 1:11; PMID:16780593; http://dx.doi.org/10.1186/1747-1028-1-11
- Larrea MD, Liang J, Da Silva T, Hong F, Shao SH, Han K, Dumont D, Slingerland JM. Phosphorylation of p27Kip1 regulates assembly and activation of cyclin D1-Cdk4. Mol Cell Biol 2008; 28:6462-72; PMID:18710949; http://dx.doi.org/10.1128/MCB.02300-07
- Gao D, Inuzuka H, Tseng A, Chin RY, Toker A, Wei W. Phosphorylation by Akt1 promotes cytoplasmic localization of Skp2 and impairs APCCdh1-mediated Skp2 destruction. Nat Cell Biol 2009; 11:397-408; PMID:19270695; http://dx.doi.org/10.1038/ncb1847
- Liang J, Shao SH, Xu ZX, Hennessy B, Ding Z, Larrea M, Kondo S, Dumont DJ, Gutterman JU, Walker CL, et al. The energy sensing LKB1-AMPK pathway regulates p27(kip1) phosphorylation mediating the decision to enter autophagy or apoptosis. Nat Cell Biol 2007; 9:218-24; PMID:17237771; http://dx.doi.org/10.1038/ncb1537
- Tsvetkov LM, Yeh KH, Lee SJ, Sun H, Zhang H. p27(Kip1) ubiquitination and degradation is regulated by the SCF(Skp2) complex through phosphorylated Thr187 in p27. Curr Biol 1999; 9:661-4; PMID:10375532; http://dx.doi.org/10.1016/S0960-9822(99)80290-5
- Hershko DD. Oncogenic properties and prognostic implications of the ubiquitin ligase Skp2 in cancer. Cancer 2008; 112:1415-24; PMID:18260093; http://dx.doi.org/10.1002/cncr.23317
- Schiappacassi M, Lovisa S, Lovat F, Fabris L, Colombatti A, Belletti B, Baldassarre G. Role of T198 modification in the regulation of p27(Kip1) protein stability and function. PloS One 2011; 6:e17673; PMID:21423803; http://dx.doi.org/10.1371/journal.pone.0017673
- Shanmugasundaram K, Block K, Nayak BK, Livi CB, Venkatachalam MA, Sudarshan S. PI3K regulation of the SKP-2/p27 axis through mTORC2. Oncogene 2013; 32:2027-36; PMID:22733130; http://dx.doi.org/10.1038/onc.2012.226
- Maddika S, Ande SR, Wiechec E, Hansen LL, Wesselborg S, Los M. Akt-mediated phosphorylation of CDK2 regulates its dual role in cell cycle progression and apoptosis. J Cell Sci 2008; 121:979-88; PMID:18354084; http://dx.doi.org/10.1242/jcs.009530
- Bae YH, Ding Z, Das T, Wells A, Gertler F, Roy P. Profilin1 regulates PI(3,4)P2 and lamellipodin accumulation at the leading edge thus influencing motility of MDA-MB-231 cells. Proc Natl Acad Sci U S A 2010; 107:21547-52; PMID:21115820; http://dx.doi.org/10.1073/pnas.1002309107
- Das T, Bae YH, Wells A, Roy P. Profilin-1 overexpression upregulates PTEN and suppresses AKT activation in breast cancer cells. J Cell Physiol 2009; 218:436-43; PMID:18937284; http://dx.doi.org/10.1002/jcp.21618
- Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, Klevernic I, Arthur JS, Alessi DR, Cohen P. The selectivity of protein kinase inhibitors: a further update. Biochem J 2007; 408:297-315; PMID:17850214; http://dx.doi.org/10.1042/BJ20070797
- Zou L, Hazan R, Roy P. Profilin-1 overexpression restores adherens junctions in MDA-MB-231 breast cancer cells in R-cadherin-dependent manner. Cell Motil Cytoskeleton 2009; 66:1048-56; PMID:19593789; http://dx.doi.org/10.1002/cm.20407
- Wittenmayer N, Jandrig B, Rothkegel M, Schlüter K, Arnold W, Haensch W, Scherneck S, Jockusch BM. Tumor suppressor activity of profilin requires a functional actin binding site. Mol Biol Cell 2004; 15:1600-8; PMID:14767055; http://dx.doi.org/10.1091/mbc.E03-12-0873
- Berx G, Cleton-Jansen AM, Nollet F, de Leeuw WJ, van de Vijver M, Cornelisse C, van Roy F. E-cadherin is a tumour/invasion suppressor gene mutated in human lobular breast cancers. EMBO J 1995; 14:6107-15; PMID:8557030
- Hennig G, Behrens J, Truss M, Frisch S, Reichmann E, Birchmeier W. Progression of carcinoma cells is associated with alterations in chromatin structure and factor binding at the E-cadherin promoter in vivo. Oncogene 1995; 11:475-84; PMID:7630631
- Hirohashi S. Inactivation of the E-cadherin-mediated cell adhesion system in human cancers. Am J Pathol 1998; 153:333-9; PMID:9708792; http://dx.doi.org/10.1016/S0002-9440(10)65575-7
- Yokoyama K, Kamata N, Hayashi E, Hoteiya T, Ueda N, Fujimoto R, Nagayama M. Reverse correlation of E-cadherin and snail expression in oral squamous cell carcinoma cells in vitro. Oral Oncol 2001; 37:65-71; PMID:11120485; http://dx.doi.org/10.1016/S1368-8375(00)00059-2
- Yoshiura K, Kanai Y, Ochiai A, Shimoyama Y, Sugimura T, Hirohashi S. Silencing of the E-cadherin invasion-suppressor gene by CpG methylation in human carcinomas. Proc Natl Acad Sci U S A 1995; 92:7416-9; PMID:7543680; http://dx.doi.org/10.1073/pnas.92.16.7416
- Birchmeier W, Behrens J. Cadherin expression in carcinomas: role in the formation of cell junctions and the prevention of invasiveness. Biochim Biophys Acta 1994; 1198:11-26; PMID:8199193
- St Croix B, Sheehan C, Rak JW, Florenes VA, Slingerland JM, Kerbel RS. E-Cadherin-dependent growth suppression is mediated by the cyclin-dependent kinase inhibitor p27(KIP1). J Cell Biol 1998; 142:557-71; PMID:9679152; http://dx.doi.org/10.1083/jcb.142.2.557
- Agiostratidou G, Li M, Suyama K, Badano I, Keren R, Chung S, Anzovino A, Hulit J, Qian B, Bouzahzah B, et al. Loss of retinal cadherin facilitates mammary tumor progression and metastasis. Cancer Res 2009; 69:5030-8; PMID:19491271; http://dx.doi.org/10.1158/0008-5472.CAN-08-4007
- Dahl U, Sjodin A, Larue L, Radice GL, Cajander S, Takeichi M, Kemler R, Semb H. Genetic dissection of cadherin function during nephrogenesis. Mol Cell Biol 2002; 22:1474-87; PMID:11839813; http://dx.doi.org/10.1128/MCB.22.5.1474-1487.2002
- Goto S, Yaoita E, Matsunami H, Kondo D, Yamamoto T, Kawasaki K, Arakawa M, Kihara I. Involvement of R-cadherin in the early stage of glomerulogenesis. J Am Soc Nephrol 1998; 9:1234-41; PMID:9644633
- Maeda M, Johnson E, Mandal SH, Lawson KR, Keim SA, Svoboda RA, Caplan S, Wahl JK 3rd, Wheelock MJ, Johnson KR. Expression of inappropriate cadherins by epithelial tumor cells promotes endocytosis and degradation of E-cadherin via competition for p120(ctn). Oncogene 2006; 25:4595-604; PMID:16786001; http://dx.doi.org/10.1038/sj.onc.1209396
- Miotto E, Sabbioni S, Veronese A, Calin GA, Gullini S, Liboni A, Gramantieri L, Bolondi L, Ferrazzi E, Gafà R, et al. Frequent aberrant methylation of the CDH4 gene promoter in human colorectal and gastric cancer. Cancer Res 2004; 64:8156-9; PMID:15548679; http://dx.doi.org/10.1158/0008-5472.CAN-04-3000
- Kotelevets L, van Hengel J, Bruyneel E, Mareel M, van Roy F, Chastre E. Implication of the MAGI-1b/PTEN signalosome in stabilization of adherens junctions and suppression of invasiveness. FASEB J 2005; 19:115-7; PMID:15629897
- Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol 2012; 13:251-62; PMID:22436748; http://dx.doi.org/10.1038/nrm3311
- Liang X, Wang P, Gao Q, Tao X. Exogenous activation of LKB1/AMPK signaling induces G(1) arrest in cells with endogenous LKB1 expression. Mol Med Rep 2014; 9:1019-24; PMID:24469340
- Sebbagh M, Santoni MJ, Hall B, Borg JP, Schwartz MA. Regulation of LKB1/STRAD localization and function by E-cadherin. Curr Biol 2009; 19:37-42; PMID:19110428; http://dx.doi.org/10.1016/j.cub.2008.11.033
- Partanen JI, Tervonen TA, Myllynen M, Lind E, Imai M, Katajisto P, Dijkgraaf GJ, Kovanen PE, Mäkelä TP, Werb Z, et al. Tumor suppressor function of Liver kinase B1 (Lkb1) is linked to regulation of epithelial integrity. Proc Natl Acad Sci U S A 2012; 109:E388-97; PMID:22308451; http://dx.doi.org/10.1073/pnas.1120421109
- Wang C, Nie Z, Zhou Z, Zhang H, Liu R, Wu J, Qin J, Ma Y, Chen L, Li S, et al. The interplay between TEAD4 and KLF5 promotes breast cancer partially through inhibiting the transcription of p27Kip1. Oncotarget 2015; 6:17685-17697; PMID:25970772.
- Hong F, Larrea MD, Doughty C, Kwiatkowski DJ, Squillace R, Slingerland JM. mTOR-raptor binds and activates SGK1 to regulate p27 phosphorylation. Mol Cell 2008; 30:701-11; PMID:18570873; http://dx.doi.org/10.1016/j.molcel.2008.04.027
- Leontieva OV, Demidenko ZN, Blagosklonny MV. Contact inhibition and high cell density deactivate the mammalian target of rapamycin pathway, thus suppressing the senescence program. Proc Natl Acad Sci U S A 2014; 111:8832-7; PMID:24889617; http://dx.doi.org/10.1073/pnas.1405723111
- Zhao H, Faltermeier CM, Mendelsohn L, Porter PL, Clurman BE, Roberts JM. Mislocalization of p27 to the cytoplasm of breast cancer cells confers resistance to anti-HER2 targeted therapy. Oncotarget 2014; 5:12704-14; PMID:25587029
- Hardie DG. AMP-activated protein kinase: an energy sensor that regulates all aspects of cell function. Genes Dev 2011; 25:1895-908; PMID:21937710; http://dx.doi.org/10.1101/gad.17420111