
Tumor suppressor genes (TSGs), as emphasized by their name, might prevent cancer development. If their suppressive action is turned off by inactivating mutations of both alleles, malignant transformation is promoted. This is, in brief, the original 2-hit theory of Knudson. In contrast to the original proposition, investigations have demonstrated that some TSGs drive efficiently cancer even when expressed at a reduced dosage (the so-called haploinsufficiency).Citation1 In a similar way, monoallelic inactivation of CDKN1B, the gene encoding p27Kip1 (hereafter defined as p27), has been demonstrated to increase significantly cancer development. Data supporting the importance of CDKN1B haplosinsufficiency came initially from a 1998 study on CDKN1B knockout mice and were then confirmed by others.Citation2
Mechanistically, the p27 capability to bind and inhibit cyclin-dependent kinase (CDK)/cyclin complexes at the G1/S (and S/G2) boundary of cell division cycle has been for several years the only/main function reported for the protein. In accord with such a property, p27 has been functionally considered as an inhibitor of growth and its decrease thought as a factor favoring cancer development. This initial view has been progressively questioned, as frequently occurs in science, by experimental evidence suggesting that only nuclear p27 works as tumor suppressor, while, when the CDK inhibitor (CKI) is in the cytosol, it acts as an oncogene stimulating invasion and metastasization. The importance of compartimentalization also stresses the value of mechanisms (and protein consensus sequences) controlling nucleus/cytosol p27 shuttling. At the same time, an increasing number of p27 interactors were identified (including Jab1, RhoA, Rac, stathmin, citron kinase, Grb2, 14–3–3, Jak2, HIPK2 and HSC70) that enhanced vertiginously the number of the CDK-independent roles of p27.
Suggestions of a connection between human cancerogenesis and p27 derive from the discovery that low CKI levels occur in many human tumors (colon, breast, lung, prostate, gastric, bladder tumors) and are associated to a more aggressive phenotype and a poorer prognosis. p27 deficiency was explained by an increased CKI removal, possibly due to up-regulation of Skp2, a protein participating to p27 ubiquitination/proteasomal degradation. Accordingly, Skp2 overexpression was demonstrated in several aggressive human cancers. Successively, the relevance of an altered p27 re-localization was emphasized. Several investigations underlined that the CKI is, in aggressive cancers, mostly localized in the cytosol and scarcely in the nucleus. In all the instances, and contrarily to the accepted TSGs behavior, CDKN1B was reported as rarely inactivated in human cancers.
As usually occurs, further investigations progressively subvert the initial view. As matter of facts, CDKN1B was demonstrated frequently inactivated in MEN4 (multiple endocrine neoplasias type 4) and in small intestine neuroendocrine tumors.Citation3,4 In addition, CDKN1B was identified as one of the 18 most significantly mutated genes in luminal breast cancer.Citation5 Finally, a study recently published in “Blood” demonstrated that CDKN1B is the second most frequently mutated gene, after BRAF (BRAFV600E mutation), in hairy cell leukemia.Citation6 Thus, CDKN1B mutations occur in several cancers, and in some cases, with high frequency. Two important aspects of CDKN1B alterations should be underlined, namely: the genetic CDKN1B changes are in heterozigosity, and frequently they result in the inactivation of one allele (by deletion or frameshift) causing haploinsufficiency. In a number of instances missense changes have also been identified, but their effects on the protein dosage/function have been scarcely characterized (see ). Intriguingly, the possibility of a dominant negative activity of mutated p27 forms has not been taken into consideration.
Figure 1. The figure shows in the upper part the main p27Kip1 structural domains and their putative roles. At the bottom, the missense mutations so far identified are reported. The corresponding human neoplasia where they have been identified are also listed. mNPAP60, mouse Nuclear Pore Associated Protein. Nuc (nucleus) and Cyt (Cytosol). AIP− (AIP gene mutation-negative). The arrows show the putative interacting residues.
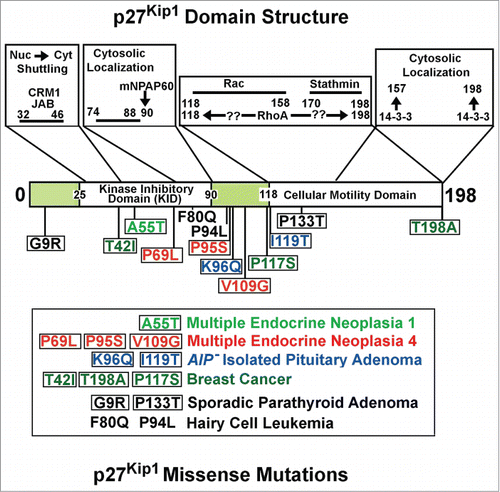
In summary, the occurrence of different mechanisms leading to p27 deficiency allow the conclusion that a fine tuning of p27 level and localization is necessary and that their alterations might favor the development of cancer. Moreover, although the importance of CDKN1B mutations in human cancers is now evident, the mechanism(s) by which these alterations drive cancerogenesis are still obscure.
A pivotal breakthrough on p27 protein has recently emerged. The CKI belongs to the so-called Intrinsically Unstructured Proteins (IUPs, or intrinsically disordered proteins, IDPs) family. Such proteins have a scarce degree of defined tertiary structure and thus their final spatial organization is, at least in part, induced by binding to other proteins. This increases strongly the number of potential targets and functions. On the other hand, Post-Translational Modifications (PTMs) of an unstructured protein, like (but not only) phosphorylations, seems to address the protein toward specific conformations and thus interactions, localization, metabolism and, finally, functions.
In the last few years, we have attempted to critically re-investigate p27 phosphorylations. We considered that several of the approaches employed so far have serious pitfalls. For example, being p27 an IUP, the expression of the CKI tagged at N- or C-terminus might alter significantly its structure/modification/function etc. Similarly, p27 overexpression might misrepresent its interaction and, thus, localization and function. To avoid these biases, we investigated PTMs and interactions of the endogenous CKI by means of 2D-electrophoresis and western blotting using well-characterized anti-phospho antibodies. We named this methodology 2D/immunoblotting. We noticed that the protein is independently monophosphorylated on several residues, but also occurs in bi- and tri-phosphorylated isoforms. Additionally, the pattern of phosphorylations is strongly modified during the cell cycle and differentiation.Citation7 The phosphorylation of specific residues affects the CDK interactions and localization.Citation7 In conclusion, we believe that PTMs characterization of p27 represents a world only partially unraveled and still to be explored.
Coming back to the interplay between cancer and CDKN1B haploinsufficiency, a major future challenge will be to clarify whether and how missense mutations affect the level/PTMs/localization/function of p27. These future studies undoubtedly will give unpredictable information on the structure/function of specific region of the CKI and probably will open novel opportunities for therapeutic strategy. We will expect great surprises by future papers that could help in unravelling the function of a pivotal but yet poorly clarified protein as p27.
References
- Berger AH, Pandolfi PP. J Pathol 2012; 223; 137-46; PMID:21125671; http://dx.doi.org/10.1002/path.2800
- Fero ML, et al. Nature 1998; 396; 177-80; PMID:9823898
- Marinoni L, Pellegata NS. Neuroendocrinology 2011; 93; 19-28; PMID:20980721; http://dx.doi.org/10.1159/000320366
- Francis JM, et al. Nat Genet 2013; 45; 1483-6; PMID:24185511; http://dx.doi.org/10.1038/ng.2821
- Stephens PJ, et al. Nature 2012; 486; 400-4; PMID:22722201; http://dx.doi.org/10.1038/nature11017
- Dietrich S, et al. Blood 2015; 126(8):1005-8; PMID:26065650; http://dx.doi.org/10.1182/blood-2015-04-643361
- Bencivenga D, et al. Cell Cycle 2014; 13; 3768-82; PMID:25483085; http://dx.doi.org/10.4161/15384101.2014.965999