
Genome instability, chromosomal structural aberrations, and copy number variation are hallmarks of cancer.Citation1 Recent advances in sequencing technology have uncovered a large amount of intra-tumoral heterogeneity in both gene expression and chromosomal copy number across a range of tumors.Citation2 These aberrations are often thought of as integrated and inherited genetic events within tumors. However, new analyses are beginning to demonstrate that some copy number heterogeneity may not be stably inherited.
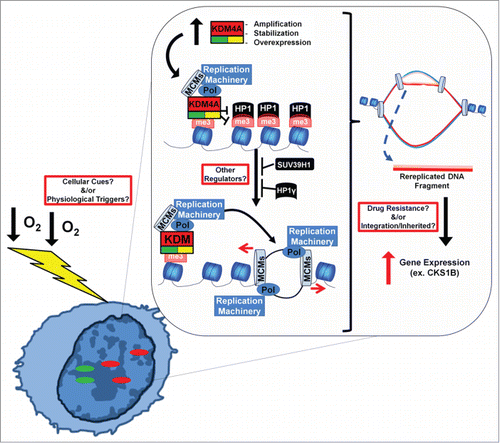
We previously demonstrated that the lysine demethylase KDM4A/JMJD2A (present on 1p34) was amplified in ∼20% of the tumors analyzed within The Cancer Genome Atlas (TCGA).Citation3 Amplification of KDM4A in these tumors significantly correlated with copy gain of specific cytogenetic bands, especially chromosome 1q12–22. We demonstrated that modest overexpression of KDM4A, which mimics the increased expression observed in amplified tumors, was sufficient to promote site-specific copy gains of the same 1q12–22 regions in cultured cells. Overexpression of KDM4A increased recruitment of the DNA replication machinery and promoted rereplication of specific chromosomal regions through alterations in the chromatin structure. In fact, directly interfering with H3K36 or H3K9 methylation also promoted site-specific copy gain, while overexpression of the H3K9 methyltransferase Suv39H1 or heterochromatin protein HP1γ could suppress the gain (). This study demonstrated for the first time that copy gain events are dependent on the chromatin microenvironment and directly regulated by enzymatic activity. Therefore, comparable pathways could exist that regulate other frequently acquired amplified regions in both developmental processes and cancer.Citation3
Figure 1. A model by which hypoxia and KDM4A could promote TSSGs. Increased expression or stabilization of KDM4A (i.e., hypoxia) promotes re-replication and copy gain through altering the chromatin environment. Hypoxia also promoted re-replication and increased expression of a drug resistant oncogene, CKS1B. Additional work is necessary to uncover factors that regulate the generation and stability of TSSG (outlined boxes).
Importantly, KDM4A-dependent copy gains are transient and have been termed transient site-specific copy gains (TSSGs). The regions are rereplicated during S phase and the gains are lost as cells exit S phase prior to the end of G2. Thus, the copy gains are not integrated and not inherited. However, cells retain the ability to rereplicate and reproduce the gains during successive cell divisions. The transient generation of copy gains is an intriguing mechanism for cells to respond to changes in developmental or environmental conditions, which raises the possibility that this mechanism could be a biological response.
To determine whether physiological or environmental conditions could promote TSSG, cells were screened with conditions observed during development and tumorigenesis. We demonstrated that hypoxia could promote TSSG in cancer cell lines and primary cells through KDM4A protein stabilization.Citation4 Hypoxic breast and lung tumors also exhibited increased copy number of the same regions with site-specific gain in cell lines. Importantly, the hypoxia-induced copy gain was conserved at a syntenic region in zebrafish cells. Like the KDM4A-dependent TSSG, hypoxia-dependent gains were transient and resulted from rereplication during S phase. Returning cells to normoxia resulted in the rapid loss of the gains. Interestingly, hypoxia-dependent TSSG was not dependent on hypoxia inducible-factors HIF1α or HIF2α. These studies establish that changes in the cellular environment can serve as a cue to specify distinct genomic loci to undergo site-specific copy gain ().
We next sought to determine whether hypoxia-dependent TSSG resulted in increased gene expression. Upon analyzing both hypoxic breast and lung tumors from the TCGA tumor data, we identified genes with increased copy number and increased gene expression. This observation led to the identification of CKS1B, a gene commonly amplified in multiple myeloma that promotes drug resistance to multiple chemotherapeutic treatments.Citation5 We recapitulated these results in a breast cancer cell line where hypoxia promoted CKS1B copy gain and an increase in CKS1B transcripts. Upon return to normoxia, the copy number and expression of CKS1B reverted to baseline levels. Thus, TSSG in response to hypoxia could provide another mechanism for tumors to acquire copy number and transcript heterogeneity as well as drug resistance.
Finally, we demonstrated that succinate (a natural inhibitor for JmjC demethylasesCitation6) or inhibition of KDM4A with a small molecule abrogated the copy gains observed in hypoxia. These data establish that TSSG can also be regulated by metabolism (succinate generation is integral to the TCA cycle) and suggest that other metabolites could also promote or inhibit TSSG. The ability of succinate and small molecule KDM inhibitors to block hypoxia-dependent copy gain also establishes that copy number in tumors might be druggable. Since drug resistant oncogenes like CKS1B are amplified, this may provide a novel mechanism for modulating these TSSGs and provide a method for reducing 1q21-associated drug resistance.
Our results describe a mechanism and trigger for transient site-specific copy gain, which provides a pathway for generating intra-tumoral copy number heterogeneity. Cells within the tumor could experience different levels of hypoxia, different division rates, different levels of KDM4A or experience metabolic challenge, and in turn, present altered copy number within a tumor. This heterogeneity would create a diverse cell population that could allow tumors a more effective method to handle stress and/or chemotherapeutic intervention. It is important to consider that even though the gain is transient, the ability to regenerate the gain remains. Furthermore, under the right conditions, such as selective pressure from chemotherapy, the TSSG could become integrated or inherited. Thus, it will be important to continue to identify and understand all the regulators of TSSG and how we can leverage this pathway to improve cancer treatment ().
References
- Hanahan D, Weinberg RA. Cell 2011; 144:646-74; PMID:21376230; http://dx.doi.org/10.1016/j.cell.2011.02.013
- Burrell RA, et al. Nature 2013; 501:338-45; PMID:24048066; http://dx.doi.org/10.1038/nature12625
- Black JC, et al. Cell 2013; 154:541-55; PMID:23871696; http://dx.doi.org/10.1016/j.cell.2013.06.051
- Black JC, et al. Genes Dev 2015; 29:1018-31; PMID:25995187; http://dx.doi.org/10.1101/gad.259796.115
- Shi L, et al. Oncotarget 2010; 1:22-33; PMID:20930946
- Smith EH, et al. Hum Mol Genet 2007; 16:3136-48; PMID:17884808; http://dx.doi.org/10.1093/hmg/ddm275