
Abstract
Sarcomas are rare cancers and the current treatments in inoperable or metastatic disease have not been shown to prolong survival. In order to develop novel targeted therapies, we tested the efficacy of polo-like kinase 1 (PLK-1) inhibitor (TAK-960) in sarcoma. All the sarcoma cell lines were sensitive to TAK-960 with IC50s in the low nanomolar range. We chose MPNST, CHP100 and LS141 for our studies and of which MPNST cells exclusively underwent polyploidy after a delay in mitosis for about 18 hours; CHP100 cells, after a 24h mitotic delay, died of apoptosis; LS141, after a delay in mitosis stayed at 4N with mild apoptosis. Apoptosis induced by TAK-960 in CHP100 was associated with down-regulation of Mcl-1 and the effect was recapitulated by down-regulating PLK1 by siRNA, confirming that the effect of TAK-960 on Mcl-1 expression is target specific. With suppression of Mcl-1 by siRNA, TAK-960 induced apoptosis in MPNST cells as well. These effects were confirmed in vivo, such that TAK-960 more effectively inhibited CHP100 than MPNST xenografts. In the setting of PLK-1 inhibition, Mcl-1 down regulation is shown to be an important determinant of apoptosis. Collectively, the net effect of this is to drive cells to apoptosis, resulting in a greater anti-tumor effect in vivo. Therefore, targeting PLK-1 should have a greater impact in treating sarcomas provided there is concomitant suppression of Mcl-1. These results further indicate that Mcl-1 could be an important biomarker to predict sensitivity to the induction of apoptosis by PLK-1 targeted therapy in sarcoma.
Keywords:
Introduction
The human Polo-like kinase 1 (PLK1) expression has been correlated with cell proliferation.Citation1 The use of small molecule inhibitors has revealed roles of PLK1 in cell cycle regulation, mainly during mitosis.Citation2 Consistent with these functions, PLK1 localization is also dynamic during cell cycle progression, moving from centrosomes to spindle poles, kinetochores and midbodies.Citation3 Elevated PLK1 activity, often observed in cancer cells, supports increased proliferation, compromising genomic integrity which could be due to altered transcriptional programs.Citation4 The importance of PLK1 in cell proliferation and its overexpression in various human cancers appear to be sufficient to override cellular checkpoints and induce genetic instability, promoting tumorigenesis.Citation5-7 Also the level of PLK1 positively correlates with aggressiveness of tumor progression, and patients with high PLK1 expression in their tumors exhibit a significantly poorer rate of survival than those with low PLK1 expression.Citation8-11 Induction of apoptosis occurs in most tumor cells but not in normal cells upon interfering with PLK1 function,Citation12-14 and tumor regressions in mouse xenograft models has also been observed.Citation15,16
Based on these and other related studies, PLK1 has become a key target in cancer therapy and small-molecule inhibitors of PLK1 have become attractive candidates for anticancer drug development. Among several inhibitors targeting PLKs, some are at different stages of clinical development. TAK-960 is an investigational PLK1 inhibitor that binds to the ATP-binding pocket of PLK1.Citation17,18 It is an orally bioavailable, potent and selective PLK1 inhibitor. It has been shown to exhibit single-agent antitumor activity in a variety of tumor cell lines, xenograft models of solid tumors and hematologic malignancies.Citation18
Sarcoma is rare cancer in adults (1%) but there are many subtypes of sarcomas each of which are genetically and biologically distinct such as; liposarcoma, the most common type of soft tissue sarcoma, accounting for approximately 20% of the tumors in adults, is characterized by amplifications of CDK4 and MDM2, malignant peripheral nerve sheath tumors (MPNST), highly malignant tumors of the Schwann cell lineage that have loss of Neurofibromatosis 1 (NF1), Ewing sarcoma (ES), a malignancy of the bone and soft tissue, is characterized by translocations of EWS/FLI1.Citation19-21 Since surgery, radiation and chemotherapy have a limited ability to control inoperable or metastatic disease, there is an extreme need to develop novel targeted therapies and strategies.
The present study is an attempt to utilize a small molecule inhibitor of PLK1, TAK-960 in the treatment of sarcomas. TAK-960 is shown to induce growth suppression in the cell lines and in xenograft models tested through different cellular fate. Of the different mechanisms by which TAK-960 suppresses growth, our finding suggests apoptosis, rather than polyploidy, as the preferred biological effect of this drug. Also, our results show that inhibition of PLK1 induces apoptosis through increased proteasomal degradation of Mcl-1. This leads to the release of cytochrome c followed by caspase 3 cascade activation and apoptosis. The role of Mcl-1 in the apoptotic process was further confirmed by testing the apoptotic effect upon siRNA down regulation or transient overexpression of Mcl-1, stabilization of Mcl-1 using proteasome inhibitor, MG132 with the inhibition of PLK-1. We further show that inhibition of PLK-1 along with Mcl-1 down regulation can push polyploid cells to apoptosis.
Results
TAK-960, a selective PLK1 inhibitor, effective in inducing growth inhibition in sarcoma cell lines
TAK-960, a selective inhibitor of PLK1, was tested for its sensitivity in a panel of sarcoma cell lines. shows the IC50 values of TAK-960 in 15 different sarcoma cell lines in the low nanomolar range determined by the cell death assay. summarizes the p53 status of the cell lines along with the cell cycle effect of TAK-960 upon the cell lines, as determined by flow cytometry with 36 hours of 50 nM TAK-960 exposure. depicts the cell cycle distribution of cells exposed to TAK 960 for 24 hours. TAK-960 induced apoptotic (sub-G1) cells in the CHP100 and SaOs2 cell lines (all null for p53 expression) and A673 (undetectable p53 by western blot), as well as in DSCRT, HSSY, SYO and SK-UT1B cell lines (all wild type for p53 expression), indicating the apoptotic effect induced by TAK-960 is p53 independent. Though both LS141 and DDLS (wild type for p53 expression) underwent mild to negligible apoptosis, the predominant effect was induction of a G2 cell cycle arrest. Another notable effect was the induction of polyploidy (cells with DNA content >4N), preferably in cells expressing mutant p53 ().
Figure 1. PLK1 inhibitor TAK-960 induces growth suppression by target specific inhibition. (A) (i) IC-50 values of TAK-960 in 15 different Sarcoma cell lines. IC-50s were determined by cell proliferation assay by using the Dojindo Cell Counting Kit done in 6 replicates. (B) Sarcoma cell lines were treated with TAK-960 (50 nM) for 48 hours and % of cells showing polyploidy (>4N) or apoptosis (<2N) were determined and quantitated by flow cytometry analysis. (C) Sarcoma cell lines were treated with TAK-960 (50 nM) for 48 hours and distribution of cell cycle phases sub G1 (apoptosis or <2N), G1 (2N), G2 (4N) and polyploidy (>4N). (% of cells showing polyploidy (>4N) or apoptosis (<2N) were determined by flow cytometry analysis. (D) Sarcoma cell lines mentioned were treated with TAK-960 (25 nM) for 24 hours and the total protein lysates were probed with antibodies, as indicated. Tubulin was used to confirm equal loading of protein. Results shown are representative or the mean of 3 or more independent experiments.
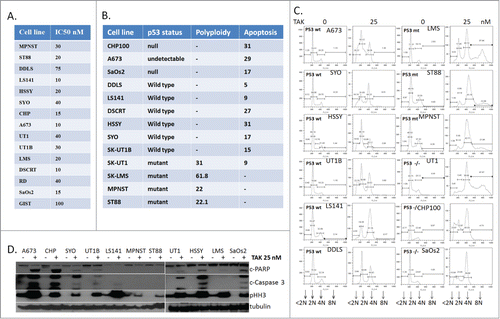
The relationship between apoptosis and p53 was further explored by protein gel blot (). Though TAK-960 induced phospho Histone H3 (S10), a marker of mitosis, in all the cell lines, apoptosis, as evidenced by induction of cleaved PARP and cleaved caspase 3, was seen in selective lines upon exposure to 25 nM TAK-960 for 24 hours. This effect was independent of p53 and generally correlated with the flow cytometry results.
PLK1 inhibition leads to accumulation of cells in mitosis
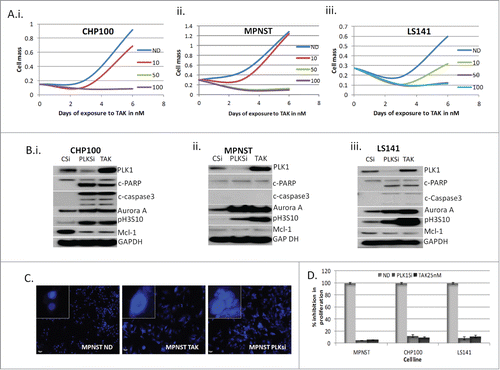
From the cell lines tested 3 representative cell lines, with different cellular effects induced by TAK-960, MPNST (polyploidy), LS141 (G2 arrest and apoptosis) and CHP100 (apoptosis), were chosen for further study. show the dose effect of TAK-960 in CHP100, MPNST, and LS141 with complete inhibition in growth at concentrations as low as 50 nM. Again, all 3 cell lines showed a significant induction of mitotic markers, pHH3 (S10) as well as Aurora Kinase A (). Following prolonged drug exposure (48 hours), both the CHP100 and the LS141 cell lines underwent apoptosis as measured by induction in cleaved PARP and cleaved caspase 3 (). In contrast, even with prolonged drug exposure and with higher dose, MPNST cells still showed no apoptotic effects (), but did show evidence of polyploidy with formation of giant multi-nucleated cells (). As shown in , these particular TAK-960 effects were recapitulated by down regulating PLK1 by siRNA in all the cell lines (PLKsi), indicating that these effects were target specific. Similar to the effect of TAK-960, though the induction of apoptosis with PLK1 siRNA was only observed in the LS141 and CHP100 cells, the induction of Aurora A and pH3 was seen in all 3 cell lines, consistent with the mitotic effect induced by PLK1 inhibition. Even though the degree of growth inhibition is comparable across all 3 cell lines with PLK1 inhibition (either by drug or siRNA), the mechanisms by which the growth suppression occurred appears different. Interestingly, upon inhibition of PLK-1, in CHP100 and LS-141 cells, downregulation of Mcl-1 protein occurred along with the induction of apoptosis which was not evident in the MPNST cells and which show no evidence of either PARP cleavage or caspase 3 activation. Complete inhibition of cell growth in all 3 cell lines upon inhibiting PLK1 was achieved either by down regulation (siRNA) or by exposure to 25 nM TAK-960 (p values LS141: ND vs PLKsi = 0.0018 and vs. TAK = 0.002); MPNST: ND vs PLKsi = 0.0007 and vs. TAK = 0.00068; CHP100: ND vs. PLKsi 0.004 and vs. TAK = 0.004)) ().
Figure 2. Inhibition of PLK1 induces mitotic arrest due to failure of chromosome alignment. (A) Dose- time dependency of TAK-960 in CHP100, MPNST and LS141 as a measure of proliferation determined using Dojindo Cell Counting Kit done in 6 duplicates. (B) MPNST, CHP100 and LS141 cells were transfected with either specific siRNA for PLK1 (PLKsi) for 48 hours or control siRNACitation36 for 24 hours followed by TAK-960 (25 nM) for 48 hours and the total protein lysates were probed with antibodies, as indicated. GAPDH was used to confirm equal loading of protein. (C) MPNST cells were treated with 50 nM TAK-960 or down regulated PLK1 using specific siRNA for 48 hours and DAPI (4′, 6-diamidino-2-phenylindole) staining shows enlarged multinucleated polyploid nuclei. (D) Down regulation of PLK1 by siRNA recapitulates the TAK-960 proliferation inhibitory effect as determined by measuring cell proliferation using the Dojindo Cell Counting Kit, indicating the percentage of growth/proliferation inhibition as compared with control. All the results are representative of 3–4 independent experiments.
PLK1 inhibition induces polyploidy in MPNST and apoptosis in CHP100
We next elected to examine the cell cycle distributions of MPNST, LS141 and CHP100 cells under either treated or untreated with 25 nM TAK-960 for 48 hours or transfected with control siRNA (48 hours) or siRNA to PLK1 (48 hours). As shown in , inhibition of PLK1 by either siRNA or TAK-960 induces mitotic accumulation (as measured by phospho-MPM2 staining) with induction of apoptosis (sub-G1 peak) to 23% with TAK-960 and to 20% with PLK1 siRNA) in CHP100 cells, mitotic accumulation with polyploidy, (>4N 19% with TAK-960 and 17% with siRNA) but without apoptosis in MPNST cells, and mitotic accumulation but with induction of apoptosis (5% with TAK-960 and 3% with PLK1 siRNA) in LS141 cells.
Figure 3. Inhibition of PLK1 by siRNA recapitulates the effect of TAK-960. (A) Cell cycle distribution and mitotic effect as determined by FACScan analysis in MPNST, CHP100 and LS141 cells treated with either 25 nM TAK-960 for 24 hours or control siRNA or PLK1 siRNA for 36 hours. (B) % of cells in G1 (2N), G2 (4N), mitosis (phospho MPM2), apoptosis (<2N) and polyploidy (>4N) population determined by flow cytometry analysis after probing for phospho MPM2, a mitotic marker, in all the 3 cell lines. (B) MPNST (i) and CHP100Citation15 and LS141 (iii) cells were exposed to 50 nM TAK-960, harvested at 0, 6, 18, 24, and 36 h and analyzed for its DNA content after staining with Propidium Iodide and mitotic population after phospho MPM2 staining by flow cytometry analysis. Results given are representative of 3 independent experiments.
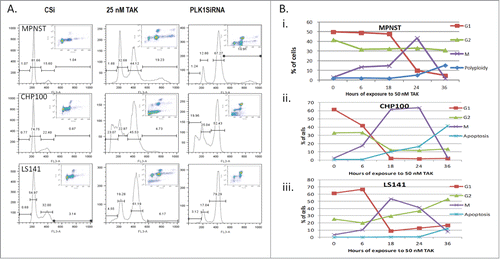
The time dependent changes in cell cycle distribution upon exposure to TAK-960 were next monitored by measuring DNA content and p-MPM2 changes over time (). In MPNST cells after 6-18 hours 10% of the cells in G2 progressed to mitosis and by 24 hours all the cells in G1 had entered 4N and remained predominantly in mitosis (). By 36 hours MPNST cells arrested in mitosis exited and underwent endoreduplication, resulting in 4N and 8N polyploid cells but with no apoptosis. However, in CHP100 cells by 6-18 hours, cells in G1 and G2 decreased with a corresponding increase in the mitotic population. By 36 hours cells had exited from mitosis and underwent apoptosis without endoreduplication (). In LS141 after 18-24 hours, there was a decrease in the G1 population with a concomitant increase in the G2 and mitotic cell populations. Upon exit from mitosis, cells seemed to stay longer in 4N with a modest increase in apoptosis by 36 hours (). Therefore, though all the cell lines exhibit an increase in the mitotic cell population with TAK-960 treatment, MPNST cells undergo polyploidy, whereas CHP100 and LS141 undergo apoptosis.
PLK1 inhibition leads to apoptosis through downregulation of Mcl-1
In order to study the mechanism by which TAK-960 and PLK1 suppression induces apoptosis we focused initially on CHP100 cells, which is devoid of p53 and therefore must undergo apoptosis through a p53 independent mechanism. Pro-survival factor Mcl-1,Citation22 has been shown to be a crucial regulator of apoptosis triggered by antitubulin chemotherapeutics.Citation23 For these studies, PLK1 was inhibited in CHP100 cells either by 24 hour exposure to TAK-960 or by downregulating its expression using specific siRNA. As shown in , apoptosis was evident with an increase in cleaved PARP and cleaved caspase 3 along with a pronounced Mcl-1 downregulation upon PLK1 inhibition either by TAK-960 or by target specific downregulation using siRNA. Mitochondrial fractionation showed cytochrome c release to the cytosol upon inhibition of PLK1 (). Therefore, in CHP100 in a p53 negative background, PLK1 inhibition induces apoptosis in association with Mcl-1 down regulation and cytochrome c release. To further study the role of Mcl-1 in the TAK-960 induced growth inhibitory effect, cell viability was compared in CHP100 cells under normal Mcl-1 or Mcl-1 overexpressed conditions. shows a moderate revival of cells in the Mcl-1 overexpressed cells despite increasing concentrations of TAK-960. The moderate effect could be attributed to the low transfection efficiency of CHP100 cells.
Figure 4. Mcl-1 mediates PLK-1 induced apoptosis. (A) (i) CHP100 cells were transfected with either specific siRNA for PLK1 (PLKsi) for 48 hours or control siRNA,Citation36 for 24 hours followed by TAK-960 (25 nM) for 24 hours and the total protein lysates were probed with antibodies for PLK1, cleaved caspase3 and PARP, Mcl-1 and GAPDH as loading control.Citation15 Treated cells were harvested for mitochondrial fractionation and probed for cytochrome c and Bax. (iii) Role of Mcl-1 overexpression on the TAK-960 induced growth suppression as determined by using the Dojindo Cell Counting Kit, indicating the percentage of growth/proliferation inhibition as compared with control. (B) Role of Mcl-1 on the apoptotic effect of TAK-960 (i). % of apoptosis as determined by FACScan analysis of cells treated with 25 nM TAK-960 under control siRNA transfected,Citation36 Mcl-1 siRNA (Mclsi) transfected or Mcl-1 over expressing (Mcl-OE) cells for 36 hours followed by staining with Propidium Iodide.Citation15 Comparison of CHP100 cells overexpressing Mcl-1 upon treatment with 25 nM TAK-960 for 36 hours as determined by protein gel blot analysis of the cell lysates. (iii) MG132 pretreated or siRNA downregulated for Mcl-1 cells treated with 25 nM TAK-960 for 36 hours, blotted for p-Mcl-1, Mcl-1, cleaved PARP and caspase 3 and GAPDH as loading control. (iv) a. Cells treated with TAK-960 (25 nM) or GSK β inhibitor (1 μM) alone or in combination for 36 hours, blotted for Mcl-1, p-Mcl-1, cleaved caspase 3 and tubulin as loading control. b. Lysates from cells treated with TAK-960 (25 nM) for 18 hours, immunoprecipitated with FBW7 antibody and followed by western blot for Mcl-1. Inputs were blotted for p-Mcl-1, Mcl-1 and tubulin. (C) Effect Mcl-1 in sarcoma cells. (i) siRNA down regulated for Mcl-1 in ST88, SKLMS, SK-UT1B, A673, TC71, SK-UT1 and SYO cells and tested for the growth inhibitory effect of TAK-960 (25 nM) by protein gel blot analysis by blotting for cleaved PARP, Mcl-1and GAPDH as control.Citation15 MPNST cells were downregulated for Mcl-1 and tested for apoptotic effect after treatment with 50 nM TAK-960 for 48 hours by western blot analysis for c-PARP, Mcl-1, c-caspase 3 and GAPDH as control. (iii) Quantitative expression of the % of cells in different cell cycle phases after exposing to 50 nM TAK-960 in MPNST transfected with control or Mcl-1 siRNA.
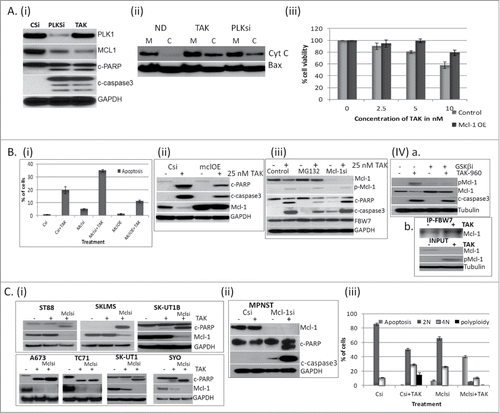
To further study the mechanism by which the change in growth inhibitory effect occurred, FACScan analysis of the CHP100 cells treated with TAK-960 under Mcl-1 overexpressed or downregulated conditions. When compared to TAK-960 in the presence of siRNA control for Mcl-1, apoptosis was further increased from 17% to 34% upon down regulating Mcl-1 in the presence of TAK-960 and reduced to 12% upon overexpressing Mcl-1 in the presence of TAK-960 (). This finding was further confirmed by a reduction in cleaved PARP and activated caspase 3 in the Mcl-1 overexpressing cells (MclOE, ) in the presence of 25 nM TAK-960. Treatment with MG132, a proteasome inhibitor, showed stabilization of Mcl-1 protein level in (Supplement 1). A reduction in cleaved PARP and activated caspase 3 was observed when Mcl-1 was stabilized in cells pretreated with MG132 in the presence of 25 nM of TAK-960 (). In contrast, there was an increase in these apoptotic markers when Mcl-1 was down-regulated with siRNA and treated with TAK-960 (). An increase in phosphorylation of Mcl-1 (p-Mcl-1) was observed upon treatment with TAK-960 (). Upon inhibiting the phosphorylation of Mcl-1 by concomitant treatment with the GSK β inhibitor (SB216763) in combination with TAK-960, stabilization of Mcl-1 protein and a partial reversal in cleaved caspase 3 was observed () consistent with the previous finding that Mcl-1 protein degradation occurs through GSK β mediated phosphorylation at S159. Marginal reversal of apoptosis is probably due to either the incomplete inhibition of phosphorylation or the existence of alternative pathway of regulation. The level of the FBW7 (F-box and WD repeat domain-containing 7) ubiquitin ligase, which has been shown to bind to and regulate Mcl-1 expression,Citation23 Citation24 did not change upon treatment with TAK-960 (). However an increased binding of Mcl-1 with FBW7 was observed using a coimmunoprecipitation assay (), suggesting that the degradation of Mcl-1 by the proteasome pathway is mediated by increased binding to FBW7 in line with previous finding.Citation25
To test whether Mcl-1 plays significant role in cell lines other than CHP100 in induction of apoptosis by PLK-1 inhibition, Mcl-1 expression level in a panel of 10 cell lines was tested with TAK-960 treatment. As shown in A673, TC71, SK-UT1 and SYO cells underwent some degree of PARP cleavage in the presence of TAK-960 with down regulation of Mcl-1. This could be further enhanced by siRNA downregulation of Mcl-1 in these cell lines. In contrast, ST88, SKLMS and SK-UT1B cells in general did not undergo PARP cleavage with TAK-960 alone and under these conditions there was no downregulation of Mcl-1. However, when Mcl-1 was suppressed with siRNA and then treated with TAK-960, apoptosis was observed, confirming the significant role Mcl-1 plays in the induction of apoptosis when PLK-1 is inhibited.
This was further tested in MPNST cells which showed a predominant polyploid effect without apoptosis upon inhibition of PLK-1. With the downregulation of Mcl-1, TAK-960 induced apoptosis in MPNST rather than polyploidy as determined by the increase in cleaved PARP and caspase 3 (). shows the quantitation of the cell cycle effect in MPNST cells with Mcl-1 down regulation along with TAK-960 treatment. TAK-960 alone induced an increase in 4N (10% to 30 %) and 8N (18%) cells in MPNST. Upon downregulating Mcl-1 by siRNA, TAK-960 induced 8N cells disappeared and the 4N cell population was reduced to 10% with a 40% induction in apoptosis. This implies the importance of targeting Mcl-1 along with inhibition of PLK-1 in achieving maximum apoptotic effect.
Polyploidy or Apoptotic in vivo
Polyploidy, apoptosis and cell cycle arrest are all shown to be efficient in tumor suppression. As TAK-960 in different cell lines shows different, we chose to determine. In order to test whether cellular fate would have an impact on in vivo growth inhibition, we elected to compare the effect of TAK-960 in vivo on the growth of MPNST cells that in vitro undergo polyploidy without suppression of Mcl-1 upon mitotic exit, whereas CHP100 cells undergo Mcl-1 suppression with apoptosis. TAK-960 (10 and 20 mg/kg) was tested in MPNST xenografts and the effect on tumor growth, as well as the biologic effects including mitotic accumulation and apoptosis, were measured. As shown in , growth suppression was not as efficient in MPNST xenograft. The MPNST tumor lysates from TAK-960 treated animals showed no evidence of apoptosis (lacking cleaved PARP or caspase 3) and no change in Mcl-1 expression (.Citation15 H&E staining was performed and, as shown in MPNST cells show enlarged nuclei, when compared to vehicle treated control at the same magnification, indicating polyploidy in vivo. By immunohistochemistry, mitotic arrest was not apparent (no induction of phospho histone H3 (Ser10). There was also no significant change in Ki67 staining and no evidence of apoptosis with no cleaved caspase 3 or TUNEL staining, consistent with the western blot analysis as well as the in vitro results (Supplement 2A).
Figure 5. Effect of TAK-960 on MPNST and CHP100 xenografts. (A) TAK-960 induces tumor suppression in MPNST in vivo. Athymic female nude mice were implanted with MPNST tumors and mice (n = 7) were treated with TAK-960 (10 mg/kg) or vehicle as described in the Materials and Methods. (A) (i) Tumor volume was measured every 2 to 3 days and the mean tumor volume was plotted against time in days. 24 hours after the final treatment, tumors were excised and the xenograft lysates were probed for apoptotic marker, cleaved PARP, PLK1, Mcl-1 and GAPDH by protein gel blot analysis. (iii) 24 hours after the final treatment, tumors were excised and analyzed by immunohistochemistry H&E. (B) TAK-960 induces tumor suppression in CHP100 in vivo. Athymic female nude mice were implanted with CHP100 tumors and mice (n = 7) were treated with TAK-960 (10 mg/kg) or vehicle as described in the Materials and Methods. (i) Tumor volume was measured every 2 to 3 days and the mean tumor volume was plotted against time in days.Citation15 24 hours after the final treatment, tumors were excised and analyzed by immunohistochemistry for phospho-histone H3 (Ser10), Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) and cleaved caspase 3. (iii) TUNEL-positive cells, phospho-histone H3 (Ser10) and cleaved caspase 3 were counted from 3 different fields and the numbers plotted as percentages. (iv) 24 hours after the final treatment, tumors were excised and the xenograft lysates were probed for apoptotic markers, cleaved PARP and caspase 3, PLK1, Mcl-1 and GAPDH by western blot analysis. All the results are representative of 3–4 independent experiments.
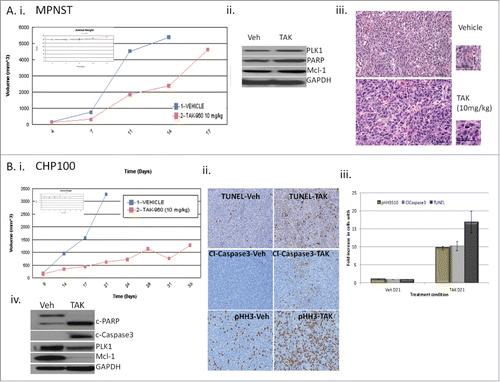
As shown in , 10 mg/kg of TAK-960 efficiently suppressed the tumor growth of CHP100 for the 21 days of therapy. depicts the immunohistochemistry of the CHP100 xenografts showing statistically significant increases in mitosis with increased p-Histone H3 (S10) and apoptosis with cleaved caspase 3 and TUNEL (p value, TAK-vs. vehicle for cleaved caspase 3: 0.0002, TUNEL: 0.002 and pHH3: 0.00025) in the post treatment samples as compared to the vehicle treated control. H&E staining showed no change in nuclear size, unlike the MPNST treated xenografts. However, there was a decrease in Ki67 staining and increase in the mitotic marker α/β tubulin with TAK-960 therapy (Supplement 2B). Similar to the in vitro effects, tumor lysates collected at the completion of the study indicated that in CHP100 treated cells TAK-960 suppressed Mcl-1 and induced apoptosis ().
Discussion
Anti-mitotic agents have been clinically validated in sarcoma but the benefits of these drugs are limited, both by low levels of clinical activity and cumulative toxicities. Thus, there is a rationale for identifying new anti-cancer agents that target mitosis. Similar to aurora kinases, PLK-1 has also been shown to be important in cell proliferation and its overexpression in various human cancers appears to be sufficient to override cellular checkpoints and induce genetic instability, promoting tumorigenesis.Citation5-7 The first small molecule inhibitor of PLK-1 was scytoneminCitation26 followed by BI2536,Citation27 and ON01910,Citation28 which has been tested in phase I and II clinical studies. The present study is an attempt to determine whether inhibition of PLK-1 by TAK-960 could represent an effective new therapeutic strategy in the treatment of sarcoma and to allow us to further understand how the inhibition of PLK-1 leads to cell death.
Previous studies have shown that TAK-960 is active against a wide range of cell lineages including colorectal, ovarian and lymphoma cell lines.Citation17 Our results now broaden these finding and indicate that TAK-960 is an extremely potent inhibitor of sarcoma cell growth. We further show with siRNA that the antiproliferative effect of TAK-960 is a result of target specific inhibition of PLK-1. The effects of of TAK-960 on morphology and growth can be recapitulated with siRNA confirming its specificity.
A closer examination of the cell cycle effect showed inhibition of cytokinesis with accumulation of cells in mitosis in all the cell lines. However, different cell lines exhibited different cellular fates upon inhibition of PLK-1 including apoptosis or polyploidy. In MPNST cells TAK-960 induced endoreduplication resulting in polyploidy upon prolonged drug exposure (48h) but without evidence of apoptosis (). Furthermore, the apoptotic effect is shown to be independent of p53 with a suppression of the pro-apoptotic molecule Mcl-1 following PLK-1 inhibition. Under normal conditions, Mcl-1 blocks apoptosis by binding and sequestering the pro apoptotic proteins such as Bak and Bax which are capable of making pores in mitochondreal membrane allowing realease of cytochrome c into the cytoplasm. Cytochrome c induces the activation of caspases lead to the macromolecular degradation of apoptotic process.Citation29 PKC-Β inhibitor is shown to be effective in combination with PLK-1 inhibitors in the absence of p53.Citation37 Based on the previous findings that anti-tubulin chemotherapeutics,Citation23 and vinca alkaloids in combination with PLK-1 inhibition,Citation38 induce prolonged mitotic arrest followed by apoptosis through down regulation of Mcl-1, we have tested the role of Mcl-1 in apoptosis induced by inhibition PLK-1. Since the predominant effect of TAK-960 was mitotic, we have tested the previous finding on the mechanism of downregulation of Mcl-1. Our finding of downregulation of Mcl-1 upon inhibition of PLK-1 with cytochrome c release and caspase 3 activation leading to apoptosis is consistant with the known effect of Mcl-1 (). The mechanism by which the down regulation occurs appears to be through proteosomal degradation of Mcl-1 as determined by the reversal of apoptosis upon pre-treatment with MG132, a proteosomal inhibitor. An increased binding of FBW7 to Mcl-1 was observed upon inhibition of PLK-1 ().Citation24 Also, pretreatment,Citation24 with GSK3B inhibitor marginally rescued the cells from TAK-960 induced apoptosis (() further cofirming the previous finding of destabilization of Mcl-1 upon phosphorylation by GSK3B. Therefore, our data suggest that PLK-1 regulates the phosphorylation of Mcl-1 through GSK3B such that blocking PLK-1 activates GSK3B to phosphorylate Mcl-1 and result in increased FBW7 binding and proteasomal degradation. The reduced Mcl-1 protein level in turn activates the caspase cascade leading to apoptosis in the setting of PLK-1 inhibition.
In order to further understand the impact of apoptosis vs. polyploidy after TAK-960 treatment, we further compared the effect of TAK-960 on 2 xenograft models MPNST and CHP100. We selected these 2 tumor models to allow us to evaluate whether apoptosis in CHP100 cells with Mcl-1 suppression or polyploidy in MPNST cells without Mcl-1 suppression would result in a greater anti-tumor effect with TAK-960 therapy. Consistent with our in vitro studies, apoptosis was detected in vivo only in the CHP100 xenografts which showed the greater growth suppression. On the other hand, polyploid cells (enlarged nuclei or multi-nucleated cells) were observed in the MPNST xenografts as expected even at a higher dose. Still, the greater growth inhibition for prolonged period in the CHP100 cells as compared to MPNST would indicate that the apoptosis induced by TAK-960 results in a more favorable biological effect and this translates into better tumor suppression in vivo, especially when there is suppression of Mcl-1. Overall these, results indicate that TAK-960 is active both in vitro and in vivo against sarcoma cell lines such that apoptosis can be achieved with substantial anti-tumor effects and this appears to be mediated in part by suppression of Mcl-1. In fact, from these results it may be possible to examine Mcl-1 suppression as a marker of improved efficacy following TAK-960 therapy.
In view of the lack of active agents for patients with sarcoma, the identification of novel targets and putative biomarkers could provide these patients new opportunities for targeted drug therapies. Our results show a prominent role for Mcl-1 in inducing apoptosis in the setting of PLK-1 inhibition. In fact one could suggest that a future direction in drug development would be combining PLK inhibitors with drugs that block Mcl-1 to either induce or further enhance the PLK inhibitory effects. In view of the lack of active agents for patients with sarcoma, the identification of novel targets could provide these patient population new opportunities for targeted drug therapies. These results indicate that PLK1 is a new target for drug development in this disease and TAK-960, as a potent PLK-1 inhibitor, merits testing as a single agent in this patient population especially in the subset of patients where suppression of Mcl-1 can be observed with the drug alone.
Materials and Methods
Cell culture
Sarcoma cell lines; A673 and CHP100, null for p53,Citation30 (Ewing), MPNST and ST88 (malignant peripheral nerve sheath, mutant for p53),Citation31 LS141 and DDLS (dedifferentiated liposarcoma, wild type for p53),Citation32 WDD (liposarcoma), SK-UT1, SK-UT1B and SK-LMS (uterine leiomyosarcoma), HSSY and SYO (synovial), SaOs2 (osteosarcoma), DSRCT (desmoplastic small round cell tumor), RD (rhabdomyosarcoma), GIST (gastrointestinal stromal tumor) were maintained in RPMI with 50 U/ml each of penicillin and streptomycin, and 10% heat-inactivated fetal bovine serum, and incubated at 37 °C in 5% CO2.
Reagents
TAK-960-001, a small molecule inhibitor of Polo like Kinase 1, was provided by Millennium (The Takeda Oncology Company, 40 Landsdowne Street, Cambridge, MA 02139). Proteasome inhibitor, MG132 and GSK β inhibitor, SB 216763 were purchased from Sigma-Aldrich (St. Louis, MO 63103).
Colorimetric cell proliferation assay
The assay was done as per the manufacturer's protocol (Dojindo Molecular Technologies, Inc..) with minor changes as follows. Briefly, 2,000 cells were plated in 100 µL volume per well of a 96-well plate, siRNA transfections were done at the time of plating and treatments were done 24 h after plating. After the incubation period of 3 days with TAK960 (0-100 nM), the media was replaced with MEM without phenol red with 10% serum and 10% CCK-8 solution, which were further incubated at 37°C for 1 to 4 h. In this assay the amount of formazan dye generated by the activity of dehydrogenases in the cells is quantified and which is directly proportional to the number of living cells. Then the optical density at 450 nm to determine the cell viability was measured using Spectra Max 340 PC (Molecular Devices Corp).
Flow cytometry
For flow cytometry,Citation33 the cells were washed and fixed in 75% ice-cold ethanol before staining with propidium iodide (50 µg/mL) containing RNase (5 µg/mL) for the measurement of DNA content. To measure the mitotic population fixed cells were labeled with the phospho MPM-2 monoclonal antibody (Millipore), which recognizes specific phosphorylated epitopes present only in mitosis and followed by Alexa Flour 488 antimouse secondary antibody (Invitrogen, Oregon, USA). Cells were then treated with RNase and propidium iodide. Samples were analyzed on a FACScan (Becton Dickinson) for cell cycle distribution and mitotic index (fraction of cells positive for phospho MPM-2) using the Cell Quest software. 10,000 events were examined per sample.
siRNA transfection
Cells were plated on 60-mm plates, and transfections using lipofectamine RNAiMAX (Invitrogen) were performed according to the manufacturer's protocol. The siRNA sequences for PLK1 and Mcl-1 were purchased from Santa Cruz Biotechnology, Ambion Inc.. (Life Technologies, 3175 Staley Road, Grand Island, NY14072, USA) and Dharmacon (Lafayette, CO, USA). Cells were harvested 48 hours after transfection for Western Blot analysis or FACScan analysis.
Mcl-1 Overexpression
Cells were transiently transfected with an MCL-1 expression plasmid as previously described.Citation34 The MCL-l expression plasmid was a gift from Dr. Hannah Rabinowich and Dr. Leslie Goldstein (University of Pittsburgh School of Medicine, Pittsburgh, PA).Citation35
Cell lysate extraction, immunoblotting and immunoprecipitation
Cell lysates were prepared by lysis of both floating and adherent cells in whole-cell lysis buffer (50 mmol/L Tris, pH 7.4, 150 mmol/L NaCl, 1% NP-40, 1 mmol/L EDTA, 0.25% sodium deoxy cholate, 0.1 mmol/L Na3VO4, with protease inhibitor cocktail tablet (Roche)), allowed to lyse on ice for 10 min, syringed and cleared by centrifugation in a microcentrifuge at 13,000 rpm for 10 min at 4°C. Thirty micrograms of protein were fractionated by SDS-PAGE and transferred onto Immobilon membranes (Millipore). After blocking with 5% nonfat milk, membranes were probed with primary antibodies. The following antibodies were used in this study: mouse monoclonal to cleaved poly (ADP-ribose) polymerase (PARP), rabbit polyclonal to PLK1, rabbit polyclonal to cleaved caspase 3, rabbit polyclonal to phospho Histone H3 (S10), rabbit polyclonal to phospho and Aurora A Kinase, rabbit polyclonal to phospho-Mcl-1 and Mcl-1, rabbit polyclonal to Bax and rabbit polyclonal to GAPDH were purchased from Cell Signaling (Danvers, MA 01923, USA) and mouse monoclonal to Rb protein and cytochrome c were purchased from BD PharMingen (San Jose, CA 95131, USA). Bound primary antibodies were detected with horseradish peroxidase-conjugated secondary antibodies and visualized by enhanced chemiluminescence reagent (both from GE Healthcare UK Limited).
Immunoprecipitation was performed by using 500 μg of soluble protein. Lysates were incubated with 2 μg of the indicated primary antibody overnight at 4°C followed by incubation with 50 μl of protein A-agarose beads (Upstate Biotechnology). Immunocomplexes were washed 5 times in lysis buffer and fractionated by SDS-PAGE. Western blot analysis was performed as previously described.
Mitochondrial extraction
Mitochondrial extraction was done as per manufacturer's protocol (Thermo Scientific, Rockford, IL, USA) with the following changes. 5 million cells were resuspended in 100 μL reagent A, after 2 minutes of incubation on ice, 2.5 μL of Reagent B was added and incubated at room temperature for 5 minutes with vortex every minute. After adding 100 μL of Reagent C, the manufacturer's protocol was strictly followed.
Quantitative fluorescent microscopy (QFM)
Cells were collected after drug treatment and fixed in 3% paraformaldehyde. The nuclear morphology of cells was examined using fluorescence microscopy after staining with 4′, 6-diamidino-2-phenylindole (DAPI) at a concentration of 25 µg/ mL. Number of cells with decondensed, fragmented chromatin was taken as a measure for apoptosis. A minimum of 400 cells were counted for each sample and taken as a percentage of untreated cells.
Xenograft studies
Athymic mice bearing MPNST or CHP100 tumors (7 mice/cohort) of 150 mm3 diameters were either treated with vehicle control, 10 mg/kg of TAK-960 p.o. once daily 5× for 3 weeks. Twenty-4 hours after the treatment on day 12 one animal from each cohort was sacrificed and the tumors examined for H&E, Ki67, cleaved caspase 3, Phospho Histone H3 (S10), α/β tubulin and terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL). Tumors were measured every 2 to 3 d with calipers, and tumor volumes were calculated by the formula 4/3 × r3 ((r = (larger diameter + smaller diameter)/4)). The percentage of tumor regression was calculated as the percentage ratio of difference between baseline and final tumor volume to the baseline volume. Toxicity was monitored by weight loss. These studies were done in accordance with the Principles of Laboratory Animal Care (NIH Publication No. 85-23, released 1985).
Histopathology
For immunohistochemistry analysis, representative sections were deparaffinized, rehydrated in graded alcohols, and subjected to antigen retrieval by microwave oven treatment using standard procedures. H&E staining was carried out using Gill's hematoxylin (Poly Scientific R&D Corp.) for 10 min as per the manufacturer's protocol followed by counterstaining with eosin (Poly Scientific R&D Corp.) for 4 min. The immunohistochemistry was performed at Molecular Cytology Core Facility of Memorial Sloan Kettering Cancer Center using Discovery XT processor (Ventana Medical Systems). TUNEL staining was done by proteinase K treatment 20 ug/ml followed by TdT-biotin-dUTP labeling mix (Roche) for 1 hour. The percentage of TUNEL-positive cells from tumor sections was determined by counting at least 100 cells each from at least 3 randomly selected fields. For Ki-67 (Vector labs) and PHH3S10 (5 μg/mL, Cell Signaling Technologies INC.), cleaved caspase 3 (Asp175) (0.1 μg/mL, Cell Signaling Technologies INC.) were used followed by biotinylated goat anti-rabbit IgG Streptavidin- HRP and DAB detection kit (Ventana Medical Systems) were used according to the manufacturer instructions. The immunofluorescence detection of α/β tubulin was performed using a rabbit polyclonal anti-α/β tubulin (Cell Signaling Technologies) was used in 1:50 dilution. The tissue sections were blocked for 30 min in 10% normal goat serum, 2% BSA in PBS. The incubation with the primary antibody was done for 6 hours followed by 60 minutes incubation with biotinylated goat anti-rabbit IgG. Blocker D, Streptavidin-HRP D (Ventana Medical Systems), followed by incubation with Tyramide-Alexa Fluor 488.
Statistical Analysis
All in vitro experiments were carried out at least 3 times. The statistical significance of the experimental results was determined by the 2-sided t test. We chose P = 0.05 as statistically significant in individual comparisons. For in vivo studies, the 2-sided t test was used as a summary measure for each mouse. Tumor volume was compared between groups of mice. P values were calculated using the Wilcoxon Rank Sum test.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
1078033_supplemental_files.zip
Download Zip (1.2 MB)References
- Golsteyn RM, Schultz SJ, Bartek J, Ziemiecki A, Ried T, Nigg EA. Cell cycle analysis and chromosomal localization of human Plk1, a putative homologue of the mitotic kinases Drosophila polo and Saccharomyces cerevisiae Cdc5. J Cell Sci 1994; 107 (Pt 6):1509-17; PMID:7962193
- Lenart P, Petronczki M, Steegmaier M, Di Fiore B, Lipp JJ, Hoffmann M, Rettig WJ, Kraut N, Peters JM. The small-molecule inhibitor BI 2536 reveals novel insights into mitotic roles of polo-like kinase 1. Curr Biol 2007; 17(4):304-15; PMID:17291761; http://dx.doi.org/10.1016/j.cub.2006.12.046
- Strebhardt K. Multifaceted polo-like kinases: drug targets and antitargets for cancer therapy. Nature Rev Drug Discovery 2010; 9(8):643-60; http://dx.doi.org/10.1038/nrd3184
- Laoukili J, Kooistra MR, Bras A, Kauw J, Kerkhoven RM, Morrison A, Clevers H, Medema RH. FoxM1 is required for execution of the mitotic programme and chromosome stability. Nat Cell Biol 2005; 7(2):126-36; PMID:15654331; http://dx.doi.org/10.1038/ncb1217
- Eckerdt F, Yuan J, Strebhardt K. Polo-like kinases and oncogenesis. Oncogene 2005; 24(2):267-76; PMID:15640842; http://dx.doi.org/10.1038/sj.onc.1208273
- Strebhardt K, Ullrich A. Targeting polo-like kinase 1 for cancer therapy. Nat Rev Cancer 2006; 6(4):321-30; PMID:16557283; http://dx.doi.org/10.1038/nrc1841
- Takai N, Hamanaka R, Yoshimatsu J, Miyakawa I. Polo-like kinases (Plks) and cancer. Oncogene 2005; 24(2):287-91; PMID:15640844; http://dx.doi.org/10.1038/sj.onc.1208272
- Knecht R, Elez R, Oechler M, Solbach C, von Ilberg C, Strebhardt K. Prognostic significance of polo-like kinase (PLK) expression in squamous cell carcinomas of the head and neck. Cancer Res 1999; 59(12):2794-7; PMID:10383133
- Knecht R, Oberhauser C, Strebhardt K. PLK (polo-like kinase), a new prognostic marker for oropharyngeal carcinomas. Int J Cancer 2000; 89(6):535-6; PMID:11102900; http://dx.doi.org/10.1002/1097-0215(20001120)89:6%3c535::AID-IJC12%3e3.0.CO;2-E
- Kneisel L, Strebhardt K, Bernd A, Wolter M, Binder A, Kaufmann R. Expression of polo-like kinase (PLK1) in thin melanomas: a novel marker of metastatic disease. J Cutaneous Pathol 2002; 29(6):354-8; http://dx.doi.org/10.1034/j.1600-0560.2002.290605.x
- Strebhardt K, Kneisel L, Linhart C, Bernd A, Kaufmann R. Prognostic value of pololike kinase expression in melanomas. JAMA 2000; 283(4):479-80; http://dx.doi.org/10.1001/jama.283.4.479
- Liu X, Lei M, Erikson RL. Normal cells, but not cancer cells, survive severe Plk1 depletion. Mol Cell Biol 2006; 26(6):2093-108; PMID:16507989; http://dx.doi.org/10.1128/MCB.26.6.2093-2108.2006
- Luo J, Emanuele MJ, Li D, Creighton CJ, Schlabach MR, Westbrook TF, Wong KK, Elledge SJ. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell 2009; 137(5):835-48; PMID:19490893; http://dx.doi.org/10.1016/j.cell.2009.05.006
- Sur S, Pagliarini R, Bunz F, Rago C, Diaz LA Jr, Kinzler KW, Vogelstein B, Papadopoulos N. A panel of isogenic human cancer cells suggests a therapeutic approach for cancers with inactivated p53. Proc Natl Acad Sci USA 2009; 106(10):3964-9; PMID:19225112; http://dx.doi.org/10.1073/pnas.0813333106
- Elez R, Piiper A, Kronenberger B, Kock M, Brendel M, Hermann E, Pliquett U, Neumann E, Zeuzem S. Tumor regression by combination antisense therapy against Plk1 and Bcl-2. Oncogene 2003; 22(1):69-80; PMID:12527909; http://dx.doi.org/10.1038/sj.onc.1206038
- Steegmaier M, Hoffmann M, Baum A, Lénárt P, Petronczki M, Krssák M, Gürtler U, Garin-Chesa P, Lieb S, Quant J, et al. BI 2536, a potent and selective inhibitor of polo-like kinase 1, inhibits tumor growth in vivo. Curr Biol 2007; 17(4):316-22; PMID:17291758; http://dx.doi.org/10.1016/j.cub.2006.12.037
- Nie Z, Feher V, Natala S, McBride C, Kiryanov A, Jones B, Lam B, Liu Y, Kaldor S, Stafford J, et al. Discovery of TAK-960: an orally available small molecule inhibitor of polo-like kinase 1 (PLK1). Bioorg Med Chem Lett 2013; 23(12):3662-6; PMID:23664874; http://dx.doi.org/10.1016/j.bmcl.2013.02.083
- Hikichi Y, Honda K, Hikami K, Miyashita H, Kaieda I, Murai S, Uchiyama N, Hasegawa M, Kawamoto T, Sato T, et al. TAK-960, a novel, orally available, selective inhibitor of polo-like kinase 1, shows broad-spectrum preclinical antitumor activity in multiple dosing regimens. Mol Cancer Ther 2012; 11(3):700-9; PMID:22188812; http://dx.doi.org/10.1158/1535-7163.MCT-11-0762
- O'Neill A, Shah N, Zitomersky N, Ladanyi M, Shukla N, Uren A, Loeb D, Toretsky J. Insulin-like growth factor 1 receptor as a therapeutic target in ewing sarcoma: lack of consistent upregulation or recurrent mutation and a review of the clinical trial literature. Sarcoma 2013; 2013:450478; PMID:23431249; http://dx.doi.org/10.1155/2013/450478
- Singer S, Millis K, Souza K, Fletcher C. Correlation of lipid content and composition with liposarcoma histology and grade. Ann Surg Oncol 1997; 4(7):557-63; PMID:9367021; http://dx.doi.org/10.1007/BF02305536
- Kar M, Deo SV, Shukla NK, Malik A, DattaGupta S, Mohanti BK, Thulkar S. Malignant peripheral nerve sheath tumors (MPNST)–clinicopathological study and treatment outcome of twenty-four cases. World J Surg Oncol 2006; 4:55
- Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev 2008; 9(1):47-59; PMID:18097445; http://dx.doi.org/10.1038/nrm2308
- Wertz IE, Kusam S, Lam C, Okamoto T, Sandoval W, Anderson DJ, Helgason E, Ernst JA, Eby M, Liu J, et al. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature 2011; 471(7336):110-4; PMID:21368834; http://dx.doi.org/10.1038/nature09779
- Zhong Q, Gao W, Du F, Wang X. Mule/ARF-BP1, a BH3-only E3 ubiquitin ligase, catalyzes the polyubiquitination of Mcl-1 and regulates apoptosis. Cell 2005; 121(7):1085-95; PMID:15989957; http://dx.doi.org/10.1016/j.cell.2005.06.009
- Inuzuka H, Fukushima H, Shaik S, Liu P, Lau AW, Wei W. Mcl-1 ubiquitination and destruction. Oncotarget 2011; 2(3):239-44; PMID:21608150
- Stevenson CS, Capper EA, Roshak AK, Marquez B, Eichman C, Jackson JR, Mattern M, Gerwick WH, Jacobs RS. The identification and characterization of the marine natural product scytonemin as a novel antiproliferative pharmacophore. J Pharmacol Ex Ther 2002; 303(2):858-66; PMID:12388673; http://dx.doi.org/10.1124/jpet.102.036350
- Hofheinz RD, Al-Batran SE, Hochhaus A, Jäger E, Reichardt VL, Fritsch H, Trommeshauser D, Munzert G. An open-label, phase I study of the polo-like kinase-1 inhibitor, BI 2536, in patients with advanced solid tumors. Clin Cancer Res 2010; 16(18):4666-74; PMID:20682708; http://dx.doi.org/10.1158/1078-0432.CCR-10-0318
- Jimeno A, Li J, Messersmith WA, Laheru D, Rudek MA, Maniar M, Hidalgo M, Baker SD, Donehower RC. Phase I study of ON 01910.Na, a novel modulator of the Polo-like kinase 1 pathway, in adult patients with solid tumors. J Clin Oncol 2008; 26(34):5504-10; PMID:18955447
- Thomas LW, Lam C, Edwards SW. Mcl-1; the molecular regulation of protein function. FEBS Letters 2010; 584(14):2981-9; PMID:20540941; http://dx.doi.org/10.1016/j.febslet.2010.05.061
- Moll UM, Ostermeyer AG, Haladay R, Winkfield B, Frazier M, Zambetti G. Cytoplasmic sequestration of wild-type p53 protein impairs the G1 checkpoint after DNA damage. Mol Cell Biol 1996; 16(3):1126-37
- Subramanian S, Thayanithy V, West RB, et al. Genome-wide transcriptome analyses reveal p53 inactivation mediated loss of miR-34a expression in malignant peripheral nerve sheath tumours. J Pathol 2010; 220(1):58-70; PMID:19890883; http://dx.doi.org/10.1002/path.2633
- Singer S, Socci ND, Ambrosini G, Lee CH, Beck AH, Zhu S, Downs-Kelly E, Montgomery K, Goldblum JR, Hogendoorn PC, et al. Gene expression profiling of liposarcoma identifies distinct biological types/subtypes and potential therapeutic targets in well-differentiated and dedifferentiated liposarcoma. Cancer Res 2007; 67(14):6626-36; PMID:17638873; http://dx.doi.org/10.1158/0008-5472.CAN-07-0584
- Motwani M, Delohery TM, Schwartz GK. Sequential dependent enhancement of caspase activation and apoptosis by flavopiridol on paclitaxel-treated human gastric and breast cancer cells. Clin Cancer Res 1999; 5(7):1876-83; PMID:10430095
- Nair JS, de Stanchina E, Schwartz GK. The topoisomerase I poison CPT-11 enhances the effect of the aurora B kinase inhibitor AZD1152 both in vitro and in vivo. Clin Cancer Res 2009; 15(6):2022-30; PMID:19276280; http://dx.doi.org/10.1158/1078-0432.CCR-08-1826
- Ho AL, Musi E, Ambrosini G, Nair JS, Deraje Vasudeva S, de Stanchina E, Schwartz GK. Impact of combined mTOR and MEK inhibition in uveal melanoma is driven by tumor genotype. PloS One 2012; 7(7):e40439; PMID:22808163; http://dx.doi.org/10.1371/journal.pone.0040439
- Kirschmann DA, Seftor EA, Fong SF, Nieva DR, Sullivan CM, Edwards EM, Sommer P, Csiszar K, Hendrix MJ. A molecular role for lysyl oxidase in breast cancer invasion. Cancer Res 2002; 62(15):4478-83; PMID:12154058
- Lange L, Keppner-Witter S, Grigat J, Spänkuch B. Combinatorial inhibition of Plk1 and PKCβ in cancer cells with different p53 status. Oncotarget 2014; 5:2263-75; PMID:24810255
- Czaplinski S, Hugle M, Stiehl V, Fulda S. Polo-like kinase 1 inhibition sensitizes neuroblastoma cells for vinca alkaloid-induced apoptosis. Oncotarget 2015; 5; PMID:26046302