
Abstract
The transcription factor, TBX3, is critical for the formation of, among other structures, the heart, limbs and mammary glands and haploinsufficiency of the human TBX3 gene result in ulnar-mammary syndrome which is characterized by hypoplasia of these structures. On the other hand, the overexpression of TBX3 is a feature of a wide range of cancers and it has been implicated in several aspects of the oncogenic process. This includes its ability to function as an immortalizing gene and to promote proliferation through actively repressing negative cell cycle regulators. Together this suggests that TBX3 levels may need to be tightly regulated during the cell cycle. Here we demonstrate that this is indeed the case and that TBX3 mRNA and protein levels peak at S-phase and that the TBX3 protein is predominantly localized to the nucleus of S-phase cells. The increased levels of TBX3 in S-phase are shown to occur transcriptionally through activation by c-Myc at E-box motifs located at −1210 and −701 bps and post-translationally by cyclin A-CDK2 phosphorylation. Importantly, when TBX3 is depleted by shRNA the cells accumulate in S-phase. These results suggest that TBX3 is required for cells to transit through S-phase and that this function may be linked to its role as a pro-proliferative factor.
Introduction
T-box factors comprise an archaic family of transcription factors that are defined by a conserved DNA binding motif known as the T-box.Citation1,2 Members of this family are best known for their roles in development where they are expressed in a wide range of tissues and organs and their functions are integral to early cell-fate decisions and organogenesis.Citation3 The T-box factor TBX3 is crucial for the development of the limb, heart and mammary glands and mutations resulting in haplo-insufficiency of TBX3 are the genetic basis for the human ulnar-mammary syndrome.Citation4 Apart from its well defined role in development, a wealth of information has linked the overexpression of TBX3 to a growing list of cancers where it has been shown to impinge on multiple signaling pathways to promote the oncogenic process.Citation5 One such mechanism is the ability of TBX3 to override key cell cycle checkpoints to suppress senescence and promote proliferation by transcriptionally repressing the cyclin-dependent kinase inhibitors, p21WAF1/CIP1/SDII (hereafter referred to as p21) and p14ARF.Citation6,7 TBX3 also promotes proliferation of head and neck squamous carcinoma cells by directly repressing the tumor suppressor PTENCitation8 and embryonic stem cells by up-regulating Zscan4 to elongate telomeres as well as through repressing NFjBIB and p14ARF.Citation9,10 In light of these findings it appears that the role of TBX3 in cancer may, in part, be linked to its ability to regulate the cell cycle, which suggests that its levels and transcriptional activity may have to be tightly regulated during this process.
At a molecular level the cell cycle is regulated by the ordered activation of serine/threonine-specific cyclin dependent kinase (CDK) complexesCitation11 which phosphorylate numerous substrates to drive progression through the cell cycle.Citation12 The D type cyclins (cyclins D1, D2, D3), in association with CDK4/CDK6, regulate entry into G1 and cyclin E-CDK2 regulates progression from G1 into S-phase.Citation13,14 Cyclin A-CDK2 is a key promoter of S-phase progression and cyclin B-CDK1 is critical for a number of mitotic events.Citation15-17 The c-Myc transcription factor also plays an important role in cell cycle control, in part, through its ability to upregulate the G1 cyclinsCitation18-20 and through repressing levels of the CDK inhibitors, p21 and p27.Citation21-25 Whereas the former occurs through direct binding of c-Myc to consensus E-box motifs (CACGTG), p21 repression occurs by an indirect mechanism involving c-Myc interfering with MIZ-1, a p21 activator, or by sequestering and consequently inhibiting SP-1 and SP-3 binding to the p21 promoter.Citation24,25
Here we show that the expression of TBX3 is regulated during the cell cycle and that at S-phase TBX3 mRNA and protein levels peak and the protein localizes predominantly to the nucleus. Importantly, knockdown of TBX3 results in an S-phase arrest indicating that it is required for progression through S-phase. We show that TBX3 protein levels are upregulated at S-phase due to transcriptional activation by c-Myc and phosphorylation by cyclin A-CDK2 which provide additional evidence to support a role for TBX3 in S-phase.
Results
TBX3 protein levels and nuclear localization are highest in S-phase
To explore the possibility that TBX3 protein levels are regulated during the various phases of the cell cycle, we synchronized PNT1A and SW1353 cells, which express TBX3, at specific phases of the cell cycle and analyzed the protein by western blotting and confocal microscopy. Flow cytometry analysis confirmed successful synchronization in each phase of the cell cycle () and western blotting of the levels of cyclin A and cyclin B1 confirmed synchronization of cells in S and G2/M respectively (). Importantly, TBX3 protein levels increase in G1 and peak in S () and while the protein is both nuclear and cytoplasmic in G1 and G2 arrested cells, it is predominantly nuclear in S-phase cells (). These results provide evidence that TBX3 levels and nuclear localization is regulated during the cell cycle and that it may have a role in S-phase. Indeed, when TBX3 is depleted by shRNA, the majority of cells accumulates in S-phase and do not progress to G2 ().
Figure 1. TBX3 protein levels are elevated in S-phase. (A) Flow cytometry of asynchronous PNT1A and SW1353 cells or cells arrested at specific stages of the cell cycle as indicated. (B) Western blotting of cell extracts prepared from asynchronous or synchronised cells with antibodies to the indicated proteins. Anti-p38 antibody was used as a loading control.
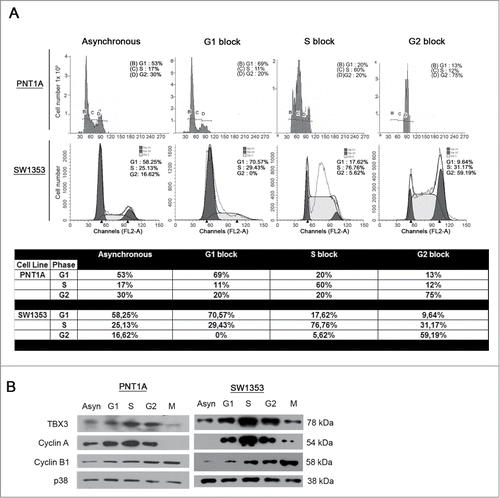
Figure 2. TBX3 protein levels and nuclear localization are highest in S-phase and it is required for progression through S-phase into G2 (A) Immunofluorescence at 40X magnification of PNT1A and SW1353 cells using a rabbit polyclonal anti-TBX3 antibody. All cells were stained with DAPI, to determine the location of the nuclei. (B) Subcellular fractionation was performed using PNT1A and SW1353 cell lysates. Nuclear and cytoplasmic extracts were subjected to western blot analyses and probed for TBX3 using anti-TBX3 antibody. GAPDH (cytoplasmic protein) and p84 (nuclear protein) expression were determined by anti-GAPDH and -p84 antibodies. (C) Upper panel: Flow cytometry of SW1353 and ATDC5 shTBX3 and shCtrl cells. Middle panel: Table showing percentages of cells in each phase of the cell cycle. Lower panel: Knockdown of TBX3 protein in SW1353 and ATDC5 cells was confirmed by western blotting using an antibody to TBX3. Anti-p38 antibody was used as a loading control.
c-Myc transcriptionally upregulates TBX3 through E-box motifs at positions −1210 and −701 bps
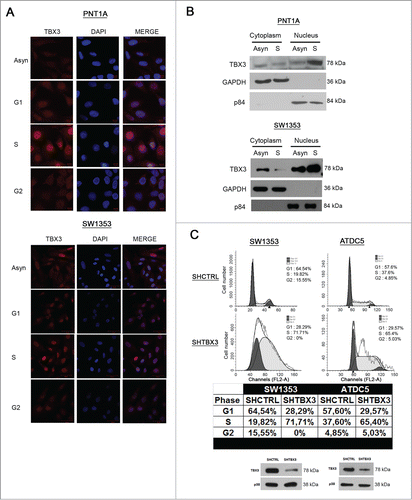
To determine the mechanism(s) responsible for upregulating TBX3 levels in S-phase, quantitative real-time PCR was performed using RNA from cells synchronized as described earlier and results show that the levels of TBX3 mRNA match the trend seen for TBX3 protein levels (compare with ). Furthermore, treatment of cells synchronized in S-phase with Actinomycin D (AD), a transcriptional inhibitor, abolished the increase in TBX3 mRNA and protein () suggesting that TBX3 levels are upregulated transcriptionally in S-phase.
Figure 3. TBX3 levels are upregulated transcriptionally and post-transcriptionally in S-phase (A) Quantitative RT-PCR of TBX3 mRNA derived from PNT1A and SW1353 cells or cells synchronised at specific stages of the cell cycle as indicated. Bars, SEM. *p < 0.05; ** p< 0.002; *** p <0.0005. (B) Quantitative RT-PCR of TBX3 mRNA or (C) Western blot analyses of TBX3 protein derived from PNT1A and SW1353 cells or cells synchronized in S-phase and treated with or without the transcriptional inhibitor, Actinomycin D (AD). The levels of TBX3 mRNA expression were analyzed using primers specific to TBX3 and normalized against GUSB. Bars, SEM. *p < 0.05; *** p< 0.0005
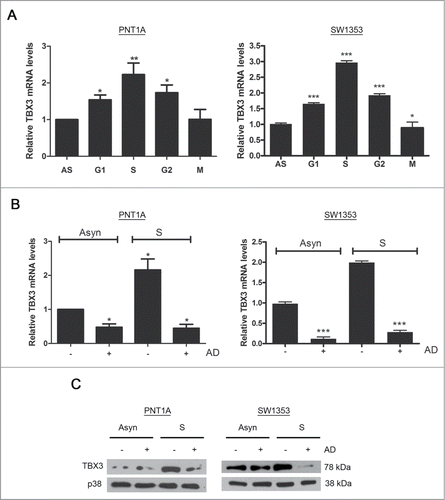
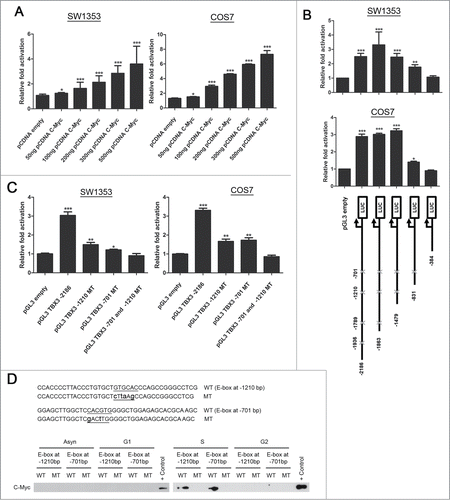
We considered the possibility that c-Myc may be responsible for the above transcriptional upregulation because of its role in S-phaseCitation26 and because the TBX3 promoter contains 4 highly conserved E-box motifs (). This was explored by performing chromatin immunoprecipitation assays to determine whether c-Myc binds these motifs in vivo in an S-phase dependent manner. Briefly, DNA bound by c-Myc was immunoprecipitated from asynchronous, G1, S, or G2 cells and subjected to quantitative real time PCR with primers spanning the E-box sites as indicated in . Compared to the IgG negative control, binding of c-Myc to the E-box sites at −1210 and −701 bps was enhanced by 600 fold and 80 fold respectively in S-phase cells and there was minimal occupancy of c-Myc at these sites in asynchronous, G1 and G2 cells (). Interestingly, no PCR product was detected with primers spanning the E-box sites at −1936 and 1789 bps, indicating that these sites are not bound by c-Myc (data not shown). In addition, silencing c-Myc expression by siRNA substantially reduced TBX3 protein levels in S-phase cells (). Together this data provide in vivo evidence that c-Myc upregulates TBX3 levels during S-phase by directly binding a region of the TBX3 promoter that harbours the E-box sites located at −1210 and −701 bps.
Figure 4. During S-phase, c-Myc directly binds a region of the TBX3 promoter containing the E-box sites located at −1210 and −701 bps and upregulates its levels. (A) TBX3 promoter contains 4 highly conserved E-box motifs and the position of each E-box is indicated. (B) Upper panel: Primers spanning the E-box sites as indicated by arrows. Lower panel: Chromatin immunoprecipitation assays were performed using DNA immunoprecipitated from asynchronous cells and cells synchronized in G1, S, or G2 with an antibody directed against c-Myc or IgG (negative control). Immunoprecipitated DNA was assayed by qRT-PCR with primers spanning the E-box sites in the TBX3 promoter as indicated in upper panel. Bars, SD. *** p< 0.0005. (C) SW1353 cells synchronized in S-phase were transiently transfected with 50nM siRNAs to c-Myc (sic-Myc#1 and sic-Myc#2) or the equivalent concentration of control siRNA (siCtrl) for 48 hours. Protein extracts were subjected to western blot analyses with antibodies to c-Myc, TBX3 and p38 (loading control).
To further explore the regulation of TBX3 by c-Myc, luciferase reporter assays were performed and the results show that c-Myc significantly activates a −2186 bp TBX3 promoter in a dose dependent manner (). This activation was significantly reduced in a −831 deletion construct which contained only the E-box motif at −701 and was abolished in a −384 deletion construct lacking an E-box motif (). Importantly, mutation of both E-boxes at −1210 and −701 led to the abrogation of TBX3 activation by c-Myc (). Furthermore, we used DNA affinity immunoblot (DAI) assays to determine whether c-Myc does indeed bind the TBX3 E-box motifs at −1210 and −701 bps in vitro. Briefly, nuclear extracts from asynchronous, G1, S and G2 cells were incubated with biotinylated DNA probes containing either wild-type or mutant E-boxes and protein-bound DNA was isolated and analyzed by western blotting. The results show that there was only an interaction between c-Myc in nuclear extracts from S-phase cells to probes with the wild type E-boxes (). These results confirmed that c-Myc directly binds and activates the TBX3 promoter at the E-boxes located at −1210 and −701 bps.
Figure 5. c-Myc transcriptionally upregulates TBX3 through E-box motifs at positions −1210 and −701 bps. Luciferase assays of SW1353 and COS7 cells transfected with (A) 500 ng of human TBX3 with 50–500 ng of c-Myc expression construct, and (B) Upper panel: transfected with 500 ng of each TBX3 promoter reporter deletion constructs (see lower panel) of 100ng c-Myc expression construct. Lower panel: Schematic representations of TBX3 promoter deletion constructs cloned upstream of a luciferase reporter. The putative E-box motifs are indicated by crosses. (C) Luciferase assays of cells transfected with 100 ng of c-Myc expression construct together with either 500ng of TBX3 promoter reporter or with TBX3 promoter reporter with mutated E-box motifs. Bars, SEM. *p < 0.03; ** p< 0.002; *** p< 0.0005. (D) Biotinylated DNA probes of the TBX3 promoter containing the homologous wild-type (WT) or mutant (MT) E-box motifs were immobilized on streptavidin beads and incubated with nuclear extracts from SW1353 cells or cells synchronized in the different phases of the cell cycle. The DNA-bound protein complexes were isolated and analyzed by western blotting using antibody to c-Myc.
Cyclin A-CDK2 binds and phosphorylates TBX3
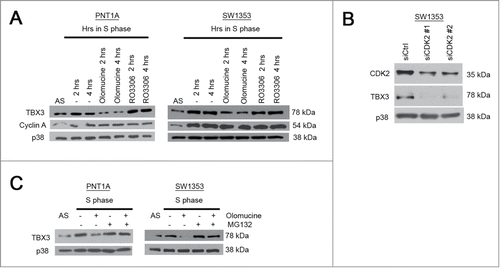
The increase in TBX3 protein levels and its exclusive nuclear localization during S-phase suggests that the structure and function of the protein may also be regulated by phosphorylation. Indeed, when cells were synchronized in S-phase and treated with olomoucine, an inhibitor of cyclin A-CDK2 and cyclin B-CDK1, the levels of TBX3 were significantly reduced (). However, treatment with R03306, which specifically inhibits the cyclin B-CDK1 complex, had no impact on TBX3 protein levels () suggesting that the results obtained with olomoucine were specifically due to the inhibition of CDK2. Indeed, when CDK2 was silenced by siRNA, TBX3 levels decreased to undetectable levels (). Furthermore, the proteosome inhibitor MG132 could rescue TBX3 protein levels in olomoucine treated cells (). These results suggest that the increase in TBX3 protein levels in S-phase is also due to phosphorylation by CDK2.
Figure 6. TBX3 protein levels are upregulated by CDK2. Western blot analyses with antibodies to the indicated proteins was performed using protein extracts from PNT1A and SW1353 cells: (A) Cells synchronized in S-phase for 2 or 4 hrs were treated with either olomoucine (an inhibitor of cyclin A-CDK2) or R03306 (inhibits the mitotic cyclinB1 complex); (B) cells transiently transfected with 50nM siRNAs to CDK2 (siCDK2#1 and siCDK2#2) or the equivalent concentration of control siRNA (siCtrl) for 48 hours; (C) cells synchronized in S-phase were treated with or without olomoucine and/or MG132 (proteasome inhibitor).
To determine if TBX3 is a substrate for the cyclin A-CDK2 complex, the ability of TBX3 to bind cyclin A in vivo was investigated by immunoprecipitation assays using cells synchronized in S-phase and an antibody to cyclin A. The rationale for this approach was based on the fact that phosphorylation by a CDK is dependent on direct interaction between its associated cyclin and its substrate.Citation12 The results show that TBX3 specifically interacts with cyclin A in S-phase (). The minimum consensus sequence recognized by CDK2 is serine-proline (SP) and the TBX3 protein sequence has 11 SP motifs (). Having established that cyclin A binds to TBX3, we next investigated whether TBX3 is phosphorylated by the CDK2 kinase in vitro. Either the wild type (WT) full-length TBX3 or WT N-terminal (1–371) TBX3 proteins () were expressed with a GST tag and used as substrates in in vitro kinase assays and GST was used as a negative control. The results indicate that both the WT full-length and N-terminal TBX3 fusion proteins were phosphorylated by CDK2 and that phosphorylation occurs within the N-terminal half of the protein.
Figure 7. Cyclin A-CDK2 binds and phosphorylates TBX3. (A) Cyclin A immunoprecipitation assays using cells synchronized in S-phase and an antibody to cyclin A. Western blot analyses with antibodies to the indicated proteins was performed using protein extracts from PNT1A and SW1353. (B) Schematic representation of the wild type full length (TBX3 FL) protein used as a substrate in kinase assays. Eleven putative serine proline (SP) motifs was identified. (C) Upper panel: In vitro CDK2 kinase assays were performed using purified GST-TBX3 fusion proteins as substrates in the presence of the recombinant activated CDK2 kinase and [γ−32P] ATP. Kinase assay using the indicated TBX3 proteins is shown in the upper panel after SDS-PAGE and autoradiography. Lower panel: a Coomassie Blue stained gel indicating that comparable amounts of protein were used in the kinase assay.
![Figure 7. Cyclin A-CDK2 binds and phosphorylates TBX3. (A) Cyclin A immunoprecipitation assays using cells synchronized in S-phase and an antibody to cyclin A. Western blot analyses with antibodies to the indicated proteins was performed using protein extracts from PNT1A and SW1353. (B) Schematic representation of the wild type full length (TBX3 FL) protein used as a substrate in kinase assays. Eleven putative serine proline (SP) motifs was identified. (C) Upper panel: In vitro CDK2 kinase assays were performed using purified GST-TBX3 fusion proteins as substrates in the presence of the recombinant activated CDK2 kinase and [γ−32P] ATP. Kinase assay using the indicated TBX3 proteins is shown in the upper panel after SDS-PAGE and autoradiography. Lower panel: a Coomassie Blue stained gel indicating that comparable amounts of protein were used in the kinase assay.](/cms/asset/e1ec92bb-075c-41c7-bc41-911609485410/kccy_a_1080398_f0007_b.gif)
Discussion
Several lines of evidence suggest that, during embryonic development and cancer, the T-box transcription factor TBX3 represses key cell cycle regulators such as p21, NFjBIB and p14ARF to impact on proliferation.Citation6-10,27-30 This suggests that TBX3 levels may need to be tightly regulated during the various phases of the cell cycle. Here we show that this is indeed the case and that TBX3 mRNA and protein levels are highest in S-phase which is due to direct transcriptional activation by c-Myc and phosphorylation of the TBX3 protein by cyclin A-CDK2. Furthermore, the subcellular localization of TBX3 also appears to be regulated during the cell cycle with the protein being localized predominantly to the nucleus in S-phase. Importantly, the depletion of TBX3 by shRNA results in cells arresting in S-phase. Together our data suggest a role for TBX3 in S-phase and/or in the transition from S into G2.
TBX2 the homologous partner of TBX3, also has an established role as an anti-senescence and pro-proliferative factor and like TBX3 it represses p21 and p14ARF. This may suggest that TBX2 and TBX3 levels are similarly regulated and have overlapping roles during the cell cycle. However, contrary to our findings that TBX3 levels peak in S-phase, Tbx2 levels were shown to peak in G2.Citation31 Furthermore, unlike our current data showing that depleting TBX3 leads to an S-phase arrest, we have previously shown that knocking down TBX2 in breast cancer and melanoma cells compromises an S-phase arrest in response to cisplatin induced DNA damage.Citation32 Together our data provide additional support that, while highly homologous, TBX2 and TBX3 have distinct biological roles during the cell cycle.
This study identifies TBX3 as a novel target of c-Myc during S-phase of the cell cycle. We demonstrate using in vitro and in vivo assays that c-Myc can directly bind the TBX3 promoter at the E-boxes located at −1210 and −701 bps which is sufficient to activate TBX3 transcription. While the E-box situated at −1210 bps is not a canonical c-Myc recognition site, a recent study by Selmi et al.Citation33 also demonstrated that c-Myc directly binds and activates the TWIST1 promoter through this site. In this regard it is worth noting that chromatin context is now recognized to be an important determinant of c-Myc binding to E-boxes i.e. it appears to preferentially bind E-boxes in “active chromatin.”Citation34-36 Furthermore, it has been established that cells in S-phase have genetically active open chromatin, and hence our data showing that c-Myc preferentially binds the TBX3 promoter in S-phase supports this possibility.Citation37
We show that TBX3 is required for progression through S-phase which begs the question as to the link between c-Myc-TBX3 and S-phase. c-Myc has been heavily implicated in regulating S-phase entry by, in part, activating CDC25A and Cyclin E and in DNA replication by for example activating CDT1 and the DNA metabolic enzyme, thymidine kinase.Citation21,38-41 Interestingly, the role of c-Myc in regulating S-phase entry and DNA replication appears to both involve its ability to repress p21, a known TBX3 direct target gene.Citation7,42 c-Myc was reported to repress p21 by preventing its activation by Sp1/Sp3 and MIZ-1 to allow cells to bypass the G1 checkpoint and progress into S-phase.Citation24,25 In vitro, TBX3 can directly bind and repress a T-element in the p21 promoter and it is tempting to speculate that to facilitate entry and progression through S-phase c-Myc prevents activation of p21 through multiple mechanisms including activation of TBX3.
Here we identify TBX3 as a substrate for the cyclin A-CDK2 complex, another important regulator of entry and progression through S-phase. We show that phosphorylation of TBX3 by this complex leads to an increase in protein stability. Indeed, treatment of cells with olomoucine led to a dramatic decrease in TBX3 protein levels in S phase and this could be rescued when the proteasome was inhibited with MG132. We further show that TBX3 is predominantly nuclear in S-phase cells, and it will be interesting to explore whether its translocation to the nucleus is regulated by cyclin A-CDK2 phosphorylation. This possibility is consistent with the nuclear localization of TBX2 being regulated by phosphorylation in response to cellular stress.Citation43 Given the importance of cyclin A-CDK2 in S-phase, these results provide additional support that TBX3 must play an important role during this stage of the cell cycle. Cyclin A-CDK2 promotes S-phase progression by either directly phosphorylating substrates such as Cdh1, Rb, p21 and p27 or indirectly by phosphorylating substrates that are able to regulate these factors.Citation44-47 It is tempting to speculate that TBX3 may be an example of the latter to ensure repression of p21. This would be important to ensure a negative feedback loop because p21 can inhibit cyclin A-CDK2 activity.Citation46
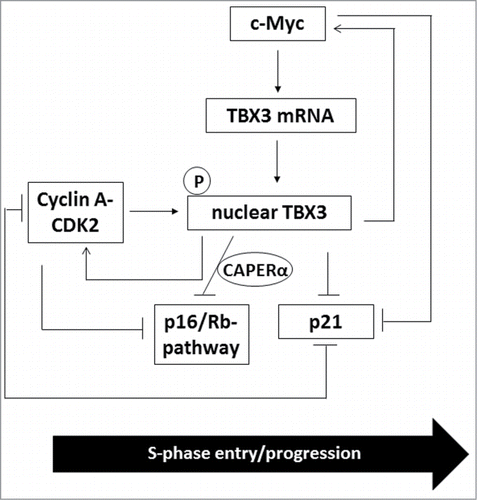
Cyclin A-CDK2 also maintains S-phase progression by phosphorylating and inactivating Rb to ensure the release of E2F.Citation45 Interestingly, recent reports have demonstrated that TBX3 can bypass senescence by forming a co-repressor complex with CAPERα to inhibit the Rb pathway, in part, through repressing CDKN2A-p16INK transcription and destabilizing p16INK mRNA.Citation48 It would therefore be important to explore whether phosphorylation of TBX3 by cyclin A-CDK2 also plays a role in inhibiting the Rb pathway during S-phase progression. Furthermore, Kumar et al.Citation48 also showed that TBX3−/− mouse embryonic fibroblasts have decreased CDK2 and c-Myc levels. This data together with our data suggest a positive feedback loop between TBX3, c-Myc and cyclin A-CDK2 in S-phase progression (see for proposed model). In conclusion, the current study provides a molecular basis for the regulation of TBX3 in S-phase which may begin to provide insight into its role in the cell cycle and in cancer.
Figure 8. A proposed model for regulation and the role of TBX3 during S-phase. During S-phase, TBX3 is transcriptionally activated by c-Myc which results in an increase in nuclear TBX3 protein levels which are stabilized through phosphorylation by cyclin A-CDK2. This results in an additional mechanism through which p21 is repressed by c-Myc and cyclin A-CDK2 and prevents p21 mediated inhibition of cyclin A-CDK2. c-Myc and CDK2 levels are also positively regulated by TBX3 through a feedback loop. Finally, cyclin A-CDK2 and TBX3/CAPERα pathways converge to negatively regulate the Rb pathway, thus leading to the release of E2F to activate S-phase target genes and ultimately S-phase progression.
Materials and Methods
Plasmids and mutagenesis
The human pGEX-TBX3 full length (FL), pGEX TBX3 N- terminal and the human TBX3 promoter luciferase reporter constructs were generated as previously described.Citation49,50 The −2186–base pair TBX3-luciferase plasmid was modified to introduce point mutations by site-directed mutagenesis using the Stratagene QuikChange system. Successful introduction of the mutation was confirmed by sequencing. Flag tagged c-Myc expression construct was kindly provided by Professor Luscher from Institut für Biochemie, Germany.
Cell culture and transfections
PNT1A normal human prostate epithelium (provided by Professor Nicolas Novitzky, Division of Haematology, University of Cape Town), COS7 (fibroblast-like cell line derived from monkey kidney tissue), ATDC5 mouse chondrosarcoma and SW1353 human chondrosarcoma (ATCC HTB-94) cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 200 units/ml penicillin and 100 μg/ml streptomycin. Cells were maintained in a 37°C incubator (95% air/5% CO2, 65% humidity). Knockdown of CDK2 and c-Myc expression was achieved by transfecting cells with siRNAs that specifically target CDK2 (si-CDK2#1 (Sigma, USA) or siCDK2#2 (Qiagen, USA)) or c-Myc (sic-Myc#1 (Dharmacon, Lafayette, CO, USA) or sic-Myc#2 (Qiagen, USA)) or a control (non-silencing) siRNA (Qiagen, USA), using Hiperfect (Qiagen, USA) according to the manufacturer's instructions. For luciferase assays, cells were transfected with 500 ng of the TBX3 luciferase reporter plasmid plus 50 ng, 100 ng, 200 ng, 300 ng and 500 ng of c-Myc expression vector or empty vector plasmid. The vector pRL-TK, containing the thymidine kinase promoter which drives the expression of a renilla reporter, was used as an internal control for transfection efficiency (40 ng per transfection). Cells were cultured for 30 hr and extracts were then assayed for firefly and renilla luciferase activity using the dual luciferase assay system (Promega, Madison, WI, USA) according to the manufacturer's instructions. Luciferase activities were measured using the Luminoskan Ascent luminometer (Thermo Labsystems, Franklin, MA, USA).
Cell cycle synchronization and Flow cytometry
Cells were synchronized in specific phases of the cell cycle by a double thymidine (Sigma, USA) block (15 hr 2 mM thymidine, 9 hr release, 15 hr thymidine) to obtain a G1/S population, or were released into fresh medium for 4 hours to obtain S-phase cells. G2 cells were obtained by a single thymidine block followed by 16 hr treatment with 0.1 μg/ml nocodazole (Sigma, USA), at the end of which mitotic cells were separated by shake-off. The cell cycle phase distribution was assessed by measuring DNA content by flow cytometry as described previously.Citation51
Western blot analyses
Cells were harvested and solubilized at 4°C in RIPA buffer (150 mM NaCl, 1% Triton X-100, 0.1% SDS, 20 mM Tris pH 7.5, 1% deoxycholate and a cocktail of protease inhibitors), centrifuged at 12,000g for 20 min at 4°C and the supernatants recovered. Protein were resolved by SDS-PAGE and transferred to Hybond C (Amersham, United Kingdom). Membranes were probed with appropriate primary antibodies followed by peroxidase-conjugated anti-rabbit antibody and visualized by enhanced chemiluminescence (Pierce). The primary antibodies used were as follows: rabbit polyclonal anti-TBX3 antibody (1:500 dilution) (Zymed 42–4800; Zymed Laboratories), rabbit polyclonal anti-p38 antibody (1:5000 dilution) (Sigma, Missouri, USA), rabbit polyclonal anti-cyclin A (1:1000 dilution) and anti-cyclin B1 (1:500 dilution) all from Santa Cruz Biotechnology, CA, USA.
qRT-PCR
Total RNA was extracted from cells using the RNeasy Plus Mini kit (Qiagen). Reverse transcription of RNA (1μg) was performed according to the manufacturer's instructions using the InProm-IITM reverse transcription system (A3800; Promega, Madison, WI, USA). Using 2 μl of cDNA, quantitative real time PCR was conducted on an Applied Biosystems StepOne Plus thermal cycler or LightCycler Version 3 (Roche, Basle, Switzerland) using 2x SYBR green master mix (Applied Biosystems, Carlsbad, CA, USA), or SensiMix Lite Kit (QT 405–05; Quantace, Taunton, MA) respectively, according to the manufacturer's protocols. PCR cycle parameters were: denaturation for 15 min at 95ºC, combined annealing and extension for 35 cycles at 60ºC for 1min. Each DNA sample was quantified in triplicate and a negative control without cDNA template was run with every assay to assess the overall specificity. Melting curve analyses was carried out to ensure product specificity. Relative mRNA expression levels were normalized to glucuronidase β (GUSB) using the 2-ΔΔCt method. Primers used to amplify the human TBX3 (QT00022484) and GUSB (QT00046046) were purchased from Qiagen.
Treatments
To inhibit the kinase activity of cyclin A/CDK2 and/or cyclin B1/CDK1, cells were synchronized in G1 phase with a double thymidine block and released into S-phase in the presence of 50 μM (PNT1A) or 100 μM (SW1353) olomoucine (Sigma, USA) and 20 μM (PNT1A) or 40 μM R03306 (Alexis Biochemicals, Heidelberg, Germany) for 4 hr for western blot analyses. For inhibition of de novo transcription, cells were treated with 5 μg/ml (PNT1A) or 4 μM (SW1353) Actinomycin D (Sigma, USA) for 4 hr after release into S-phase. For inhibition of ubiquitin mediated protein degradation, cells were pre-treated with 10 μM MG132 (PNT1A) or 25 μM (SW1353) (Calbiochem, USA) for 30 minutes prior to addition of olomoucine.
Immunofluorescence
Cells grown on glass coverslips, were fixed in ice cold absolute methanol at −20°C for 5 min and permeabilized in 0.2% Triton X-100 in PBS for 10 min before blocking for 1hr in 5% swine serum in PBS. After overnight incubation at 4ºC with the rabbit polyclonal anti-TBX3 antibody (1:25 dilution) (Zymed 42–4800; Zymed Laboratories), cells were incubated with the appropriate fluor-conjugated secondary Cy3 donkey anti-rabbit IgG (Jackson ImmunoResearch Laboratories, Inc., USA). For DNA staining, cells were incubated with 1 μg/ml DAPI (Sigma, Germany) diluted in PBS for 10 min in the dark. Coverslips were mounted on slides with Mowial mounting medium (Hoechst, Germany) containing Anti-Fade (Sigma, USA) and fluorescent cells were viewed using standard FITC and DAPI filters on an Axiovert confocal microscope (Zeiss, USA).
Protein expression and purification
All glutathione S-transferase (GST) fusion proteins were expressed in Escherichia coli strain pLysS. Isopropyl-1-thio-D-galactopyranoside (IPTG) induction was performed for 4 hr at 37ºC. GST fusion proteins were purified using Glutathione Sepharose 4B beads according to the manufacturer's instructions (Amersham Biosciences, USA). The GST-fusion proteins were transformed into the Escherichia coli (E. coli) strain pLysS and protein expression induced for 4 hrs with 0.5 M isopropyl-1-thio-Dgalactopyranoside IPTG) (Promega, USA) at 37ºC with shaking. Bacterial cells were collected by centrifugation at 3000 rpm for 30 min at room temperature and the resulting pellet resuspended in 1 ml PBSTi buffer and lysed by sonication. Cellular debris was removed by centrifugation at 13000 rpm for 30 min at 4ºC. Protein lysates were incubated with 140 μl PBSTi-equilibrated Glutathione Sepharose 4B beads (Amersham Biosciences, USA) for 1 hr at 4ºC with constant rolling. The bead-bound complexes were washed 4 times in 1 ml PBSTi buffer. Following washes, the purified proteins were resuspended in 200 μl PBSTi and stored at 4ºC. Quantification was accomplished by resolving equal volumes of GST and GST-fusion protein on a 10% SDS PAGE followed by Coomassie staining for 1 hr and destaining overnight.
Protein kinase assays
Kinase assays were performed using 0.2 units/μl recombinant cyclin A-CDK2 (New England BioLabs, UK) in a 40 μl reaction volume (20 mM MOPS pH 7.2, 25 mM β- glycerophosphate, 5 mM EGTA, 1 mM sodium orthovanadate, 1 mM dithiothreitol). The kinase was added to TBX3-recombinant protein, in the presence of [γ−32P]ATP (10 μM Ci diluted with 9μl of 400 μM unlabelled ATP, 75 mM MgCl2) and incubated for 30 min at 30°C. Following the kinase reaction, beads were washed 5 times with 1 ml of reaction buffer, and 20 μl of protein denaturing buffer were added. Proteins were resolved on a 10% SDS-PAGE, stained with Coomassie and destained before being dried for autoradiography.
Immunoprecipitation assays
PNTIA and SW1353 cells synchronized in S-phase were lysed in 150 mM RIPA buffer [0.05 M Tris-HCl (pH 8), 0.15 M NaCl, 0.1% NP-40, 0.1% sodium deoxycholate, 0.1% SDS, 5 mM EDTA, 1 mM DTT, 10 mM NaF, 0.01 mM sodium orthovanadate, 2 complete proteinase inhibitor tablets (Roche, Switzerland)]. Cellular debris was removed by centrifugation at 12 000 rpm for 20 min at 4°C. The supernatant of each sample was added to 40 µl of protein A/G Sepharose beads (Santa Cruz Biotechnology) and incubated for 30 min at 4°C with rotation. Rabbit polyclonal anti-cyclin A (5 µg) and a non-specific IgG (Santa Cruz Biotechnology) antibody were added to separate lysates and incubated at 4°C with rotation overnight. The immune complexes were immunoprecipitated with 20 μl of protein A/G Sepharose beads at 4°C with rotation for 4 hr, washed in cold PBS, boiled in 2X SDS loading buffer and protein samples were analyzed by SDS-PAGE and western blotting using antibodies to TBX3 (1:500) and cyclin A (1:1000).
DNA affinity immunoblot assay
Biotinylated DNA probes spanning E-Boxes present at −1210 and −701 relative to the TBX3 transcriptional start site were designed and purchased from IDT (Integrated DNA technologies, USA) (forward, 5′-CACTCGACCTGTGAAAACCAC-3′; reverse, 5′-Bio-AGAGCTCCTCGCCACCCT-3′). Subconfluent SW1353 cells were blocked in S-phase and nuclear extracts isolated. For each DNA-binding reaction, 40 μg of nuclear extract was incubated with 1 μg of biotinylated DNA probe in 200 μl of binding buffer (20 mM Tris-HCl pH 7.6, 50 mM NaCl, 1 mM MgCl2, 0.2 mM EDTA, 0.5 mM dithiothreitol, 5% glycerol, and 10 ng/μl poly(dI-dC)). The beads were extensively washed with binding buffer and boiled in 25 μl of 2× protein loading buffer (125 mM Tris-HCl, pH 6.5, 0.4% SDS, 10% β-mercaptoethanol, and 20% glycerol). Proteins bound to the biotinylated probes were analyzed by SDS–PAGE, followed by immunoblotting using rabbit polyclonal anti-c-Myc (Cat# N262, Santa Cruz Biotechnology, Santa Cruz, CA).
Chromatin immunoprecipitation assays
Chromatin immunoprecipitation assays were carried out as previously described.Citation52 Briefly, SW1353 cells blocked in G1-, S- or G2- phases of the cell cycle were fixed in 1% formaldehyde and the chromatin extracted, sonicated, and immunoprecipitated using antibodies against c-Myc (sc-N262) or IgG (negative control; Santa Cruz Biotechnology). DNA precipitated was analyzed by qRT-PCR using the primer pairs: TBX3 E-box −1210 (forward, 5′- GAG AAG ATA CCA GGC TGG C-3′; reverse, 5′ CAT ATT CCA CCT GGA TGT GGG3′); E-box −701 (forward, 5′- GAG ACC AGC ACC GAG ACA C-3′; reverse, 5′ GGC CAC TCG GTT CTA CAA AAG-3′) and GAPDH (nonspecific promoter region control) (forward, 5′-GAAGGCTGGGGCTCATTT-3′; reverse, 5′-CAGGAGGCATTGCTGATGAT-3'). Crossing values (Ct) of E-box −1210 and E-box −701 precipitated DNA were normalized against the Ct values of IgG. Fold enrichment was determined using the ΔΔ Ct method: fold enrichment = 2−(ΔCt1 − ΔCt2), where ΔCt1 is the ChIP of interest and ΔCt2 is the IgG.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We would like to thank Yoonus Abrahams for general assistance for the luciferase assays.
Funding
This work was supported by grants from the SA Medical Research Council, the National Research Foundation (NRF), Cancer Association of South Africa (CANSA), Cancer Research Initiative of South Africa (CARISA), Hasso Plattner Foundation funding via the University of Cape Town. The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies.
References
- Papaioannou V, Silver LM. The T-box gene family. Bioessays 1998; 20(1):9-19; PMID:9504043; http://dx.doi.org/10.1002/(SICI)1521-1878(199801)20:1%3c9::AID-BIES4%3e3.0.CO;2-Q.
- Smith J. T-box genes: What they do and how they do it. Trends Genet 1999; 15(4):154-8; PMID:10203826; http://dx.doi.org/10.1016/S0168-9525(99)01693-5
- Naiche LA, Harrelson Z, Kelly RG, Papaioannou VE. T-box genes in vertebrate development. Ann Rev Genet 2005; 39:219-39; PMID:16285859; http://dx.doi.org/10.1146/annurev.genet.39.073003.105925.
- Packham EA, Brook JD. T-box genes in human disorders. Hum Mol Genet 2003; 12:R37-44; PMID:12668595; http://dx.doi.org/10.1093/hmg/ddg077.
- Wansleben S, Peres J, Hare S, Goding CR, Prince S. T-box transcription factors in cancer biology. BBA–Rev on Cancer 2014; 1846(2):380-91.
- Lingbeek ME, Jacobs JJL, van Lohuizen M. The T-box repressors TBX2 and TBX3 specifically regulate the tumor suppressor gene p14ARF via a variant T-site in the initiator. J Biol Chem 2002; 277:26120-7; PMID:12000749; http://dx.doi.org/10.1074/jbc.M200403200
- Hoogaars WM, Barnett P, Rodriguez M, Clout DE, Moorman AF, Goding CR, Christoffels VM. TBX3 and its splice variant TBX3 + exon 2a are functionally similar. Pigment Cell Melanoma Res 2008; 21(3):379-87; PMID:18444963; http://dx.doi.org/10.1111/j.1755-148X.2008.00461.x
- Burgucu D, Guney K, Sahinturk D, Ozbudak IH, Ozel D, Ozbilim G, Yavuzer U. Tbx3 represses PTEN and is over-expressed in head and neck squamous cell carcinoma. BMC Cancer 2012; 12:481; PMID:23082988; http://dx.doi.org/10.1186/1471-2407-12-481.
- Dan J, Li M, Yang J, Li J, Okuka M, Ye X, Liua L. Roles for Tbx3 in regulation of two-cell state and telomere elongation in mouse ES cells. Sci Rep 2013; 3:3492; PMID:24336466.
- Esmailpour T, Huang T. TBX3 promotes human embryonic stem cell proliferation and neuroepithelial differentiation in a differentiation stage-dependent manner. Stem Cells 2012; 30(10):2152-63; PMID:22865636; http://dx.doi.org/10.1002/stem.1187.
- Morgan DO. Principles of CDK regulation. Nature 1995; 374:131-4; PMID:7877684; http://dx.doi.org/10.1038/374131a0.
- Lim S, Kaldis P. Cdks, cyclins, and CKIs: roles beyond cell cycle regulation. Development 2013; 140:3079-93; PMID:23861057; http://dx.doi.org/10.1242/dev.091744.
- Sherr CJ. The ins and outs of RB: coupling gene expression to the cell cycle clock. Trends Cell Biol 1994; 4(1):15-8; PMID:14731824; http://dx.doi.org/10.1016/0962-8924(94)90033-7.
- Ohtsubo M, Theodoras AM, Schumacher J, Roberts JM, Pagano M. Human cyclin E, a nuclear protein essential for the G1-to-S phase transition. Mol Cell Biol 1995; 15:2612-24; PMID:7739542.
- Heichman KA, Roberts JM. Rules to replicate by. Cell 1994; 79:557-62; PMID:7954822; http://dx.doi.org/10.1016/0092-8674(94)90541-X.
- King RW, Jackson PK, Kirschner MW. Mitosis in transition. Cell 1994; 79:563-71; PMID:7954823; http://dx.doi.org/10.1016/0092-8674(94)90542-8.
- Arellano M, Moreno S. Regulation of CDK/cyclin complexes during the cell cycle. Int J Biochem Cell Biol 1997; 29:559-73; PMID:9363633; http://dx.doi.org/10.1016/S1357-2725(96)00178-1.
- Wade M, Wahl GM. c-myc, genome instability, and tumorigenesis: the devil is in the details. Curr Top Microbiol Immunol 2006; 302:169-203; PMID:16620029.
- Leone G, DeGregori J, Sears R, Jakoi L, Nevins JR. Myc and Ras collaborate in inducing accumulation of active cyclin E/Cdk2 and E2F. Nature 1997; 387:422-6; PMID:9163430; http://dx.doi.org/10.1038/387422a0.
- Rudolph B, Saffrich R, Zwicker J, Henglein B, Muller R, Ansorge W, Eilers M. Activation of cyclin-dependent kinases by Myc mediates induction of cyclin A, but not apoptosis. EMBO J 1996; 15:3065-76; PMID:8670807.
- Evan G, Wyllie A, Gilbert C, Littlewood T, Land H, Brooks M, Waters C, Penn L, Hancock D. Induction of apoptosis in fibroblasts by c-myc protein. Cell 1992; 63:119-25; http://dx.doi.org/10.1016/0092-8674(92)90123-T.
- Perez-Roger I, Solomon DLC, Sewing A, Land H. Myc activation of cyclin E/Cdk2 kinase involves induction of cyclin E gene transcription and inhibition of p27Kip1 binding to newly formed complexes. Oncogene 1997; 14:2373-81; PMID:9188852; http://dx.doi.org/10.1038/sj.onc.1201197.
- Vlach J, Hennecke S, Alevizopoulos K, Conti D, Amati B. Growth arrest by the cyclin-dependent kinase inhibitor p27Kip1 is abrogated by c-Myc. EMBO J 1996; 15:6595-604; PMID:8978686.
- Wu S, Cetinkaya C, Munoz-Alonso M.J, von der Lehr N, Bahram N, Beuger V, Eilers M, Leon J, Larsson L. Myc represses differentiation-induced p21CIP1 expression via Miz-1-dependent interaction with the p21 core promoter. Oncogene 2003; 22:351-60; PMID:12545156; http://dx.doi.org/10.1038/sj.onc.1206145.
- Gartel AL, Ye X, Goufman E, Shianov P, Hay N, Najmabadi F, Tyner AL. Myc represses the p21(WAF1/CIP1) promoter and interacts with Sp1/Sp3. Proc Natl Acad Sci USA 2001; 98(8):4510-5; PMID:11274368; http://dx.doi.org/10.1073/pnas.081074898.
- Pusch O, Soucek T, Hengstschläger-Ottnad E, Bernaschek G, Hengstschläger M. Cellular targets for activation by c-Myc include the DNA metabolism enzyme thymidine kinase. DNA Cell Biol 1997; 16(6):737-47; PMID:9212167.
- Hanson KD, Shichiri M, Follansbee MR, Sedivy JM. Effects of c-Myc expression on cell cycle progression. Mol Cell Biol 1994; 14(9):5748-55; PMID:8065309; http://dx.doi.org/10.1128/MCB.14.9.5748.
- Suzuki A, Sekiya S, Büscher D, Izpisúa Belmonte JC, Taniguchi H. Tbx3 controls the fate of hepatic progenitor cells in liver development by suppressing p19ARF expression. Development 2008 May; 135(9):1589-95; http://dx.doi.org/10.1242/dev.016634.
- Platonova N, Scotti M, Babich P, Bertoli G, Mento E, Meneghini V, Egeo A, Zucchi I, Merlo GR. TBX3, the gene mutated in ulnar-mammary syndrome, promotes growth of mammary epithelial cells via repression of p19ARF, independently of p53. Cell Tissue Res 2007; 328:301-16; PMID:17265068; http://dx.doi.org/10.1007/s00441-006-0364-4.
- Govoni KE, Linares GR, Chen ST, Pourteymoor S, Mohan S. T-box 3 negatively regulates osteoblast differentiation by inhibiting expression of osterix and runx2. J Cell Biochem 2009 Feb 15; 106(3):482-90; http://dx.doi.org/10.1002/jcb.22035.
- Bilican B, Goding CR. Cell cycle regulation of the T-box transcription factor tbx2. Exp Cell Res 2006; 312:2358-66; PMID:16730707; http://dx.doi.org/10.1016/j.yexcr.2006.03.033.
- Wansleben S, Davis E, Peres J, Prince S. A novel role for the anti-senescence factor TBX2 in DNA repair and cisplatin resistance. Cell Death Dis 2013 Oct 10; 4:e846; http://dx.doi.org/10.1038/cddis.2013.365.
- Selmi A, de Saint-Jean M, Jallas AC, Garin E, Hogarty MD, Bénard J, Puisieux A, Marabelle A, Valsesia-Wittmann S. TWIST1 is a direct transcriptional target of MYCN and MYC in neuroblastoma. Cancer Lett 2015 Feb 1; 357(1):412-8; http://dx.doi.org/10.1016/j.canlet.2014.11.056.
- Guo J, Li T, Schipper J, Nilson KA, Fordjour FK, Cooper JJ, Gordân R, Price DH. Sequence specificity incompletely defines the genome-wide occupancy of Myc. Genome Biol 2014; 15(10):482; PMID:25287278; http://dx.doi.org/10.1186/s13059-014-0482-3.
- Sab∫ A, Amati B. Genome recognition by MYC. Cold Spring Harb Perspect Med 2014 Feb 1; 4(2): pii: a014191.
- Thomas LR, Wang Q, Grieb BC, Phan J, Foshage AM, Sun Q, Olejniczak ET, Clark T, Dey S, Lorey S, et al. Interaction with WDR5 Promotes Target Gene Recognition and Tumorigenesis by MYC. Mol Cell 2015 May 7; 58(3):4 40-52.
- Barton MC, Crowe AJ. Chromatin alteration, transcription and replication: What's the opening line to the story? Oncogene 2001 May 28; 20(24):3094-9; http://dx.doi.org/10.1038/sj.onc.1204334.
- Leone G, DeGregori J, Sears R, Jakoi L, Nevins JR. Myc and Ras collaborate in inducing accumulation of active cyclin E/Cdk2 and E2F. Nature 1997; 387:422-6; PMID:9163430; http://dx.doi.org/10.1038/387422a0.
- Pusch O, Soucek T, Hengstschläger-Ottnad E, Bernaschek G, Hengstschläger M. Cellular targets for activation by c-Myc include the DNA metabolism enzyme thymidine kinase. DNA Cell Biol 1997; 16(6):737-47; PMID:9212167.
- Valovka T, Schönfeld M, Raffeiner P, Breuker K, Dunzendorfer-Matt T, Hartl M, Bister K. Transcriptional control of DNA replication licensing by Myc. Sci Rep 2013 Dec 6; 3:3444.
- Alevizopoulos K, Vlach J, Hennecke S, Amati B. Cyclin E and c-Myc promote cell proliferation in the presence of p16INK4a and of hypophosphorylated retinoblastoma family proteins. EMBO J 1997 Sep 1; 16(17):5322-33; http://dx.doi.org/10.1093/emboj/16.17.5322.
- Ogryzko VV, Wong P, Howard BH. WAF1 retards S-phase progression primarily by inhibition of cyclin-dependent kinases. Mol Cell Biol 1997 Aug; 17(8):4877-82.
- Abrahams A, Mowla S, Parker MI, Goding CR, Prince S.UV-mediated regulation of the anti-senescence factor Tbx2. J Biol Chem 2008 Jan 25; 283(4):2223-30; http://dx.doi.org/10.1074/jbc.M705651200.
- Oakes V, Wang W, Harrington B, Lee WJ, Beamish H, Chia KM, Pinder A, Goto H, Inagaki M, Pavey S, Gabrielli B. Cyclin A/Cdk2 regulates Cdh1 and claspin during late S/G2 phase of the cell cycle. Cell Cycle 2014; 13(20):3302-11; PMID:25485510; http://dx.doi.org/10.4161/15384101.2014.949111.
- Akiyama T, Ohuchi T, Sumida S, Matsumotot K, Toyoshima K. Phosphorylation of the retinoblastoma protein by cdk2. Proc Natl Acad Sci USA 1992; 89:7900-4; PMID:1518810; http://dx.doi.org/10.1073/pnas.89.17.7900.
- Jaumot M, Estañol JM, Casanovas O, Graña X, Agell N, Bachs O. The cell cycle inhibitor p21CIP is phosphorylated by cyclin A-CDK2 complexes. Biochem Biophys Res Commun 1997 Dec 18; 241(2):434-8; http://dx.doi.org/10.1006/bbrc.1997.7787.
- Dai L, Liu Y, Liu J, Wen X, Xu Z, Wang Z, Sun H, Tang S, Maguire AR, Quan J, Zhang H, Ye T. A novel cyclinE/cyclinA-CDK inhibitor targets p27(Kip1) degradation, cell cycle progression and cell survival: implications in cancer therapy. Cancer Lett 2013 Jun 1; 333(1):103-12; http://dx.doi.org/10.1016/j.canlet.2013.01.025.
- Kumar PP, Emechebe U, Smith R, Franklin S, Moore B, Yandell M, Lessnick SL, Moon AM. Coordinated control of senescence by lncRNA and a novel T-box3 co-repressor complex. Elife 2014 May 29; 3.
- Mowla S, Pinnock R, Leaner VD, Goding CR, Prince S. PMA-induced up-regulation of TBX3 is mediated by AP-1 and contributes to breast cancer cell migration. Biochem J 2011 Jan 1; 433(1):145-53; http://dx.doi.org/10.1042/BJ20100886.
- Peres J, Mowla S, Prince S. The T-box transcription factor, TBX3, is a key substrate of AKT3 in melanomagenesis. Oncotarget 2015 Jan 30; 6(3):1821-33.
- Davis E, Teng H, Bilican B, Parker MI, Liu B, Carriera S, Goding CR, Prince S. Ectopic Tbx2 expression results in polyploidy and cisplatin resistance. Oncogene 2008 Feb 7; 27(7):976-84; http://dx.doi.org/10.1038/sj.onc.1210701.
- Prince S, Carreira S, Vance KW, Abrahams A, Goding CR. Tbx2 directly represses the expression of the p21(WAF1) cyclin-dependent kinase inhibitor. Cancer Res 2004 Mar 1; 64(5):1669-74; http://dx.doi.org/10.1158/0008-5472.CAN-03-3286.