
RIPK1 is a key molecule determining cellular fate downstream of several innate immune receptors. It is a serine/threonine kinase consisting of an N-terminal kinase domain linked by a largely unstructured intermediate domain to a C-terminal death domain (). In the TNF signaling pathway, RIPK1 paradoxically promotes cell survival as well as cell death.Citation1,2 These opposite cellular functions are mediated by 2 distinct faces of RIPK1. Upon binding of TNF to TNFR1, RIPK1 is recruited to the receptor complex I where it acts as a scaffold protein promoting cell survival, in part, by activating the canonical NF-κB pathway. Specific conditions can however activate RIPK1, and its kinase activity then regulates assembly of 2 possible cytosolic death-inducing complexes, namely complex IIb (RIPK1-FADD-Caspase-8) and the necrosome (RIPK1-RIPK3-MLKL). These complexes respectively drive TNF-mediated apoptosis or necroptosis, a regulated form of necrosis. The precise molecular mechanism(s) controlling RIPK1 activation is (are) currently unknown. Similarly, how RIPK1 kinase activity contributes to both cell death processes still remains unclear. Despite this lack of understanding, it is evident that RIPK1 can play a dual role downstream of TNFR1 and that its kinase activity needs tight repression to avoid unnecessary damage to the organism.
Figure 1. The domain structure of human RIPK1 with all reported phosphorylation sites annotated. The figure is based on and updated from data collected on the PhosphoSitePlus® website, www.phosphositeplus.org.Citation7
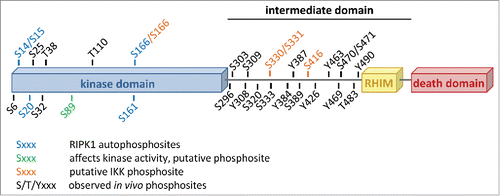
The conjugation of ubiquitin chains to RIPK1 has been reported to play an important role in the activation of the NF-κB pathway and in the repression of RIPK1's cell death potential.Citation2 In contrast, very little is known on the contribution of phosphorylation to the regulation of RIPK1's cellular functions, despite accumulating evidence indicating that RIPK1 is phosphorylated on various serine, threonine and tyrosine residues (). In 2008, Degterev and colleagues identified Ser14/15, Ser20, Ser161 and Ser166 as RIPK1 autophosphorylation sites.Citation3 Apart from Ser161 for which contrasting results have been reported, phosphorylation on none of these residues appeared to have a major influence on cell death induction.Citation3,4 Mass spectrometric analysis and large-scale proteomics studies have revealed existence of many additional residues of RIPK1 being phosphorylated in cells, but the kinases responsible for their respective phosphorylation and the functional consequences of these phosphorylation events are currently unknown (). Using site-directed mutagenesis to examine the effects of putative phosphoacceptor sites on RIPK1, the group of Francis Chan made the interesting observation that the inactivating Ser89Ala substitution in the kinase domain of RIPK1 greatly increased its enzymatic activity while the phosphomimetic Ser89Asp substitution repressed it.Citation4 These results therefore indicate that RIPK1 kinase-dependent cellular functions, such as cell death induction, can be modulated through phosphorylation by (other) kinases. It is however important to mention that, although greatly conserved during evolution, there is currently no physiological evidence that RIPK1 is actually phosphorylated on Ser89. Further experimental work is therefore needed to better characterize the potential regulatory mechanism associated to the phosphorylation of this residue. Taken together, these studies demonstrate that RIPK1 contains multiple phosphorylation sites and that phosphorylation presumably regulates certain aspects of RIPK1 functions. However, a clear link between RIPK1 phosphorylation and RIPK1-mediated function has been lacking.
In a study recently published in Molecular Cell, we revealed an unexpected new role of IKKα and IKKβ in the active repression of RIPK1 pro-death function through phosphorylation.Citation5 We demonstrated that RIPK1 is a bona fide substrate of IKKα and IKKβ, and that IKKα/IKKβ-mediated phosphorylation of RIPK1 in TNFR1 complex I protects cells from RIPK1 kinase-dependent death. Mass spectrometry analysis of kinase assays performed with recombinant proteins allowed us to identify Ser166, Ser331, and Ser416 as highly conserved RIPK1 residues phosphorylated by IKKα and IKKβ (). Unfortunately, we were, at that stage, unable to test the physiological relevance of these phosphorylation events due to abnormal behavior of RIPK1 deficient cells reconstituted with WT RIPK1, used as control cells. A future challenge is therefore to understand how IKKα/IKKβ-dependent RIPK1 phosphorylation prevents RIPK1-dependent death. We demonstrated that IKKα/IKKβ inhibition resulted in death-inducing complex assembly following TNF stimulation, but different scenarios can explain our findings. Indeed, IKKα/IKKβ may directly repress RIPK1 kinase activity by addition of an inhibitory phosphate group on RIPK1. Alternatively, IKKα/IKKβ-dependent phosphorylation may directly affect binding to the death-inducing complex components. Another possibility is that IKKα/IKKβ retain RIPK1 in complex I thereby reducing the cytosolic RIPK1 pool available for cell death induction. Interestingly, another NF-κB-related kinase, NIK, has recently been reported to phosphorylate RIPK1 in vitro and to be required for TNF-mediated RIPK1-dependent apoptosis in conditions leading to NIK stabilization (cIAP1/2 depletion).Citation6 Under these conditions, NIK deficiency was shown to prevent assembly of the apoptosis-inducing complex IIb. Together, these results indicate that RIPK1 phosphorylation can positively as well as negatively influence cell death triggered by TNF.
In conclusion, these new studies add an extra layer of complexity to the biology of RIPK1, and open the doors to exciting future work on the role of phosphorylation as crucial mechanism regulating RIPK1-dependent signaling.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Related Research Data
References
- Pasparakis M, Vandenabeele P. Nature 2015; 517(7534):311-20; PMID:25592536; http://dx.doi.org/10.1038/nature14191
- Weinlich R, Green DR. Mol Cell 2014; 56(4):469-80; PMID:25459879; http://dx.doi.org/10.1016/j.molcel.2014.11.001
- Degterev A, et al. Nat Chem Biol 2008; 4(5):313-21; PMID:18408713; http://dx.doi.org/10.1038/nchembio.83
- McQuade T, et al. Biochem J 2013; 456(3):409-15; PMID:24059293; http://dx.doi.org/10.1042/BJ20130860
- Dondelinger Y, et al. Mol Cell 2015; 60(1):63-76; PMID:26344099
- Boutaffala L, et al. Cell Death Differ 2015; PMID:26045047
- Hornbeck PV, et al. Nucleic Acids Res 2012; 40(Database issue):D261-270; PMID:22135298; http://dx.doi.org/10.1093/nar/gkr1122