
Transformed cells from 80 to 85% of human cancers display measureable levels of active TERT, the reverse transcriptase subunit of telomerase which is essential for growth and survival of these cells.Citation1 It is well known that the TERT promoter is transcriptionally silenced in development soon after stem cells loose stemness and differentitae. Although interesting from the point of view of regeneratrive medicine, the mechanism by which TERT promoter is shut off during differentiation remains to be understood. But perhaps more importantly, from the view point of cancer biology, the mechanism by which the TERT promoter is transcriptioanlly reactivated in many cancers is a major question to be addressed. Several labs over the last decade have tried to tackle this central question with little or no success.
Recently, 2 prevalent and mutually exclusive somatic mutations in the human TERT promoter were described, initially in over 70% of melanoms and then in a plethora of other cancers with varying frequency.Citation2 The fact that these mutations at positions −124 and −146 from the start (also reffered to as C250T and C228T), correlated with reactivation of TERT and predicted poor prognosis for the patients harboring them, spiked the intetrest of many researchers. It was believed that these mutations could somehow molecularly define how TERT promoter is turned on, at least in these select cancers. The intitial excitement in understaning the mechanism of TERT reactivation by these mutations was somewhat muted due to the realization that both these mutations create a predicted binding site for E26 (ETS) transcription factors, several sites for which exist in the wild type TERT promoter. It was not apparent how does the creation of a new ETS site endow an all-or-none shift in the activation of the TERT promoter. It is known that auto-inhibition is a rate limiting step in activation of ETS factors which need to hetero/homo dimerize with other factors to activate transcription. The signaling cascade(s) that eventually lead to the binding of one of the many ETS factors and presumably a new cancer specific factor to these mutant TERT promoters was also not defined. Furthermore, it was not clear if both the recurrent C250T and C228T mutations molecularly work in the same fashion.
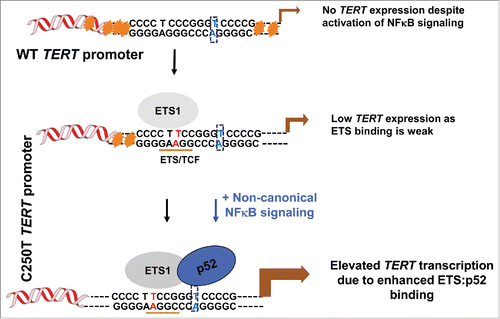
In a recent paper,Citation3 we suggest that one transcription factor that dimerizes with and activates ETS dependent transcription, only from the C250T site is the p52 NF-κB subunit. This subunit is specifically activated by non-canonical NFκB signaling, which is known to be hyperactive in gliomas. Upregulation of Fn14 receptor, which could be a key driver of non-canonical NFκB signaling is also documented in GBMs. It is also plausible that NIK kinase which is required to generate and hence activate p52, is upregulated in GBM or other such cancers with TERT promoter mutations by yet to be identified mechanims. Using glioblastomas, 83% of which display one of the 2 TERT promoter mutations, we show that p52 specifically binds the C250T (and not C228) site along with ETS1/2 in these cancers, for the first time suggesting functional differences in the operation of the 2 TERT promoter mutations.
While C250T mutant promoters require non-canonical NFκB signaling for stable ETS binding and further chromatin remodelling, the WT promoter does not seem to work in this fashion. Bioinformatic and molecular assays explain why TERT is not activated in somatic cells with WT TERT promoter with frequent activation of ETS factors or NFκB signaling which is a housekeeping signaling pathway. The juxtaposition of a ETS binding site next (created due to mutation) to a pre-existing p52 half site close to the C250T position is what turns the C250T mutant promoter. This combination of sites does not occure at the WT (or the C228T position) (). Indeed several other positions in the human genome may posses this combination of sites and could be functionally turned on by ETS and NFκB, much like what is known for ETS and RUNX factors in B cell development.
Figure 1. A model for reactivation of C250T mutant TERT promoter in cancers. Wild type TERT promoter is heavily methylated (as indicated by orange stars). The transcription start sites are denoted by a arrow. Upon mutaion at C250T position, binding site for ETS factors is created. If the cells also activate p52 via NFκB signaling, ETS and p52 stabilize each others binding on this location and cause productive transcription by gradual opening of the promoter and loss of repressive marks. The C228T mutation also creates a ETS binding site but does not have an adjacent p52 half site, as depicted by the blue residues boxed by doted lines.Citation3 Hence this ETS:p52 synergy is not seen in the context of C228T mutant TERT promoters.
These results also suggest that developing therapeuticsCitation4 which prevent non-canonical NFκB pathway activation in cells harboring mutant TERT promoter could be a strategy that will benefit cancer patients. Why do cancer cells co-opt to use p52 and ETS binding to activate C250T promoter? Unlike the canonical NFκB pathwayCitation5,6 which is rapidly activated and turned off, the non-canonical NFκB pathway is slow and persistent and is known to regulate lymphoid organogeneis, as well as bone and B cell development. Since mutant TERT promoters must remain persistently open following reactivation, cancer cells may have co-opted to use p52 bidning driven by non-canonical NFκB activity for mild but presistent expression of TERT. It is possible that once open by distinct mechanims, all TERT promoters are driven by other transcriptions factors like MYC which have been documented to activate this promoter in many contexts.Citation7
Several questions remain to be addressed. Why are TERT promoter mutations seen only in some cancers and not others? What are the functional differences between C250T and C228T mutations in terms of the upstream signaling pathways that drive them (or stabilize ETS)? It is clear that these mutations are not seen in stem cells suggesting that the mechanism by which TERT promoter remains ON during stem cell maintenance are distinct from those used to regulate its activity and levels in cancers.
References
- Low KC, Tergaonkar V. TIBS 2013; 38(9):426-34; PMID:23932019
- Horn S, et al. Science 2013; 339 (6122):959-961; PMID:23348503; http://dx.doi.org/10.1126/science.1230062
- Li Y, et al. Nat Cell Biol 2015; 17(10):1327-38; PMID:26389665; http://dx.doi.org/10.1038/ncb3240
- Li Y, Tergaonkar V. Cancer Res 2014; 74(6):1639-44; PMID:24599132; http://dx.doi.org/10.1158/0008-5472.CAN-13-3568
- Tong et al. Biosci Rep 2014; 34(3):pii:e00115
- Wong E, et al. Cell Cycle 2008; 7(23):3759-67; PMID:19029824; http://dx.doi.org/10.4161/cc.7.23.7213
- Koh CM, et al. J Clin Invest 2015; 125(5):2109-22; PMID:25893605; http://dx.doi.org/10.1172/JCI79134