
Abstract
Cisplatin (cis-diaminedichloroplatin (II), CDDP) is part of the standard therapy for a number of solid tumors including Non-Small-Cell Lung Cancer (NSCLC). The initial response observed is in most cases only transient and tumors quickly become refractory to the drug. Tumor cell resistance to CDDP relies on multiple mechanisms, some of which still remain unknown. In search for such mechanisms, we examined the impact of CDDP on mRNA translation in a sensitive and in a matched resistant NSCLC cell line. We identified a set of genes whose mRNAs are differentially translated in CDDP resistant vs. sensitive cells. The translation of the mRNA encoding the Ubiquitin-Specific Peptidase 1 (USP1), a Ubiquitin peptidase with important function in multiple DNA repair pathways, is inhibited by CDDP exposure in the sensitive cells, but not in the resistant cells. This lack of down-regulation of USP1 expression at the translational level plays a primary role in CDDP resistance since inhibition of USP1 expression or activity by siRNA or the small molecule inhibitor ML323, respectively is sufficient to re-sensitize resistant cells to CDDP. We involved the USP1 mRNA translation as a major mechanism of CDDP resistance in NSCLC cells and suggest that USP1 could be evaluated as a candidate predictive marker and as a therapeutic target to overcome CDDP resistance. More generally, our results indicate that analysis of gene expression at the level of mRNA translation is a useful approach to identify new determinants of CDDP resistance.
Introduction
CDDP is part of the standard first line therapy to several solid cancers including lung, ovarian, colorectal, head and neck, bladder and testicular cancer. With the exception of testicular germ cell cancer to which CDDP is used with a curative aim, the initial tumor response to CDDP is nearly invariably followed by the emergence of resistance to the cytotoxic effect of the drug. For the treatment of solid tumors such as the NSCLC, CDDP resistance and the ensuing disease relapse is of particular clinical relevance and the understanding of the resistance mechanisms has been a major quest ever since CDDP was introduced into the clinic.
Thus, the most advanced high throughput technologies have enabled the exploration of the cellular traits associated with increased resistance to CDDP at multiple levels. For instance, the genomic and epigenetic features associated with the loss of CDDP sensitivity have been thoroughly examined trough the search for Small Nucleotide Polymorphism (SNP) associated with CDDP resistanceCitation1-4 or by exome sequencing.Citation5 Gene expression programs associated with CDDP resistance have also been intensively scrutinized by cDNA microarrays analysis or exploring alternative levels of gene expression regulation such as those mediated by microRNAs or long non-coding RNAs.Citation6-9 Besides, functional screenings have been performed using small interfering RNA (siRNA)-mediated gene silencing or chemical library screens in order to identify cellular functions conferring resistance to CDDP.Citation10,11 Finally, some mechanism-driven, candidate-gene strategies have enabled the identification of ERCC1, whose gene product is involved in DNA repair, as a potential marker of non-small-cell lung cancer response to CDDP.Citation12 Overall, mechanisms of CDDP resistance are now better understood. Some gene expression or SNP signatures have been identified as potential predictors of CDDP sensitivity and functional screenings have led to the identification of promising candidate targets even though those findings still await clinical validation.
In order to identify new molecular events associated with CDDP resistance, we analyzed the regulation of gene expression at the mRNA level upon exposure of cells to CDDP for the following reasons. First, DNA damage such as those triggered by UV treatment lead to specific regulation of mRNA translation.Citation13-16 Second, the regulation of gene expression is perhaps best reflected by its peptide product(s) level. The proteome, however, is still far less accessible compared to the transcriptome, the genome or the epigenome. As a result, only a few studies have examined the proteome associated with resistance of NSCLC to CDDP.Citation17,18 Experimental strategies have however been devised to circumvent the technical hurdle of global protein level analysis. For instance, the ‘nascent translatome’ is experimentally accessible by resolving cellular mRNA according to their translation rate. Indeed an untranslated mRNA present as a ribonucleotide particle has a different sedimentation coefficient from polysome, i.e an mRNA associated with ribosomes.Citation13 Therefore, untranslated mRNA can be separated from the actively translated mRNA by virtue of their sedimentation properties. cDNA microarray analysis of highly translated vs untranslated mRNA can then be performed in different experimental conditions and enables the mRNA translation rates to be assessed.Citation19 We implemented this approach in order to reveal the changes in protein synthesis associated with the response to CDDP of a sensitive vs. resistant NSCLC cell lines.Citation20
Giving an initial focus to the DNA repair processes, we identified a number of mRNAs regulated specifically at the translation level in the CDDP-resistant NSCLC cell line, among which the mRNA encoding the Ubiquitin-Specific Peptidase 1 (USP1). In complex with UAF1 (USP1 associated factor 1), USP1 catalyzes the removal of a monoubiquitin from its target proteins, 2 of which, FANCD2 and PCNA, are involved in DNA repair-associated processes.Citation21,22 While the monubiquitination of FANCD2 is required for its recruitment to chromatin, the USP1/UAF1-mediated removal of the FANCD2 monoubiquitin was shown to be required for the repair process to proceed. As a result, disruption of USP1/UAF1 complex's function leads to sensitization to crosslinking agents.Citation23,24 In the case of PCNA, its deubiquitination by USP1/UAF1 is proposed to act as a switch between the translesion synthesis (TLS) DNA polymerase and the regular, more accurate DNA polymerase.Citation25-27
In this study, we identified an altered regulation of USP1 mRNA translation in CDDP-resistant cells and explored its contribution to CDDP resistance in NSCLC cells.
Results
Identification of translationally regulated mRNA involved in resistance to CDDP
In search for novel genes involved in CDDP resistance, we used a set of 2 matched human NSCLC cell lines: the relatively CDDP-sensitive A549 and an A549-derived cell clone, clone A549-R that has acquired CDDP resistance through long-term exposure to the drug.Citation20 Indeed the A549-R cells show a 3-time increase of their IC50 (90 µM) compared to the parental A549 cells (30 µM) ().Citation20
Figure 1. The CDDP-induced translational regulation of USP1 mRNA is lost in the resistant cell line (A) A549 and A549-R cells survival was measure after 48 hours exposure to CDDP using a colorimetric proliferation assay. The IC50 of CDDP determined by non-linear fitting of the absorbance is 30 µM for the A549 cell line and 90 µM for the A549-R cells. Error bars represent standard error of the mean. (B) Cell extracts from CDDP- or vehicle-treated A549 and A549-R cells were separated on a sucrose density gradient and collected into 22 fractions across the gradient. mRNA was isolated from individual fractions 8 to 22 and qPCR was performed using primers specific to USP1 mRNA. The percentage of USP1 mRNA in each fraction was plotted. Error bars represent standard error of the mean. (C) Western blot detection of USP1 protein expression in A549 and A549-R cells treated with CDDP as indicated. GADPH detection is used as a loading control. Band quantification and USP1/GAPDH ratio are shown underneath blots. (D) Western blot detection of USP1 and TP53 proteins in A549 and A549-R cells treated with cycloheximide (CHX) for the time indicated. β-Actin and GAPDH were used as loading controls. Bands were quantified and the percentage of signal relative to untreated conditions are shown underneath blots.
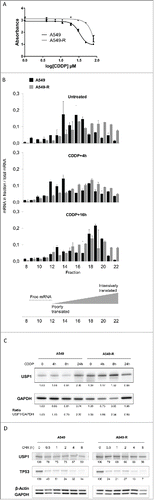
To identify mRNAs regulated at the translational level by CDDP treatment, we performed a cDNA microarray analysis of mRNAs present in polysomes. In brief, the cells were exposed to 30 µM CDDP for 4 and 16 hours. Cytoplasmic cell extracts were then prepared and fractionated into 22 fractions on a sucrose gradient. The high density fractions (fractions 19 to 22) containing the highly translated mRNA (polysomal fraction) were pooled. Extracted RNAs from the polysomal fraction as well as total RNAs (Input) from untreated and CDDP-treated cells were hybridized on GE 8X60K microarray (Agilent). We selected genes for which mRNAs were differentially present in the polysomal fraction (PF) after 4 hours or 16 hours exposure to CDDP compared to control untreated cell extracts (limma test p-value < 0.05). mRNAs regulated in a similar fashion in the input fraction were filtered out in order to exclude transcriptional regulation. shows a list of DNA repair-associated genes that fit these criteria (see also Fig. S1). Of note, several FA genes or regulators of the FA pathway are regulated at the translational level upon CDDP exposure, some of them in a cell line specific manner (FANCB, FANCF and USP1). As opposed to FANCB and USP1, FANCF translation is slightly repressed in the resistant A549-R cells ().
Table 1. Identification of DNA repair genes differentially translated after cisplatin exposure by cDNA microarray. DNA repair related gene transcripts that were differentially detected in the polysomal fraction upon CDDP treatment (limma test p-value < 0.05) and not in the input (limma test p-value > 0.05) are shown. Genes in bold are differentially regulated in a cell-line specific manner and may therefore be associated to CDDP-resistance. HRR, Homologous Recombination repair, NHEJ, Non-Homologous End-Joining, NER, Nucleotide Excision Repair, BER, Base Excision Repair.
CDDP-dependent translational regulation of USP1 mRNA in CDDP-sensitive vs. resistant cells
Among the genes identified in our microarray analysis, USP1 was of particular interest as several small molecule inhibitors of its activity have previously been developed.Citation28,29 As our results suggest a differential regulation of the USP1 mRNA translational rate in the 2 cell lines, we further examined the levels of USP1 mRNA in each fraction individually (). Strikingly, USP1 mRNA relocated from moderately translated (fractions 14 to 17) to untranslated fractions (fractions 12 to 14) after 4 hours exposure to CDDP in A549 cells. After 16 hours of CDDP exposure, the USP1 mRNA was predominantly found in the highly translated mRNA fractions (fractions 18 to 20), suggesting a biphasic regulation of USP1 mRNA translation. In the resistant A549-R cells, however, the regulation of USP1 translation is lost and the mRNA is distributed among the fractions in the same manner in CDDP-treated or untreated A549-R cells. The distribution of ABCA1 mRNA, taken as a control, was comparable in both cell lines, suggesting a specific alteration of USP1 mRNA translational regulation in the A549-R cells (Fig. S2).
To investigate whether the effect of CDDP treatment on USP1 mRNA translation rate impacted on the gene's protein product levels, we performed Western blot analysis of USP1 protein in A549 and A549-R cells treated with CDDP for various times. After a slight decrease, USP1 protein levels increased at a later time point upon CDDP exposure in the A549 cells, but not in the A549-R cells, confirming the mRNA translation analysis ().
Finally, as cellular protein level is the result of both synthesis and degradation, we ascertained that USP1 protein stability was not altered in the A549-R cells. For that purpose, we treated both cell lines with the protein synthesis inhibitor Cycloheximide (CHX) for various time in order to reveal differences in protein half-life in each cell line. TP53, a protein of notoriously short half-life, was monitored to ensure efficient protein synthesis inhibition in our experimental conditions (). As shown in , USP1 protein displays a comparable half-life in the A459 and the A549-R cell lines.
Interfering with USP1 function sensitizes cells to CDDP
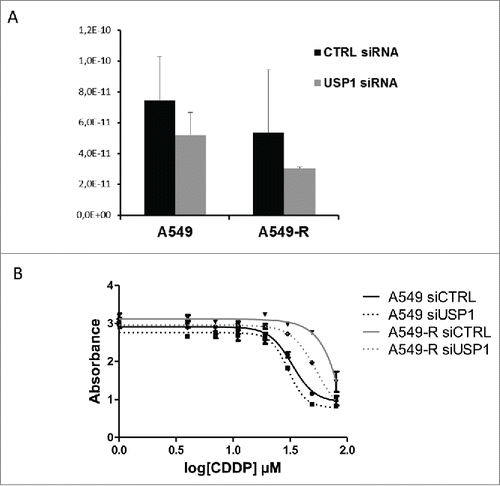
We next sought to evaluate the contribution of the regulation of USP1 expression to CDDP resistance. For that purpose, we used an siRNA targeting USP1 that lead to a modest but consistent downregulation of USP1 gene expression in both A549 and A549-R cells (). We then assessed the sensitivity of these USP1-depleted cells to CDDP. While USP1 knock-down had no effect on A549 sensitivity to CDDP, the A549-R cells were partially but clearly re-sensitized to the drug after transfection with the USP1-targeting siRNA (IC50 90 µM), but not with the control, non-targeting siRNA (IC50 50 µM, ).
Figure 2. siRNA-mediated silencing of USP1 partially re-sensitize A549-R cells to CDDP (A) RNAs from A549 and A549-R transfected with USP1-targetting or control siRNA were isolated. qPCR was performed to assess USP1 mRNA levels. Error bars represent standard error of the mean. (B) CDDP dose-response of A549 and A549-R cells transfected with control or USP1-targetting siRNA as indicated. Drug treatment was started 24 h after transfection. Cell survival was measure after 48 h exposure to CDDP using a colorimetric proliferation assay. The IC50 of CDDP was determined by non-linear fitting of the absorbance. Error bars represent standard error of the mean.
The siRNA-mediated knock-down of USP1 is only partial. We therefore tested the effect of the recently developed small molecule inhibitor of USP1 activity, ML323, on the sensitivity of A549 and A549-R cells to CDDP.Citation29 We first assessed the potency of ML323 in our cell models by examining the monoubiquitination of 2 USP1/UAF1 target proteins, PCNA and FANCD2. As previously reported, CDDP treatment induced a slight increase of the monoubiquitinated forms of the 2 target proteins ().Citation22,25-27 The ML323 treatment lead to an accumulation of the monubiquitinated forms of PCNA and FANCD2 in basal and CDDP-treated conditions, confirming that ML323 efficiently inhibits the USP1/UAF1 complex activity in our cell models ().
Figure 3. The USP1/UAF1 complex small molecule inhibitor ML33 restore CDDP sensitivity in A549-R resistant cells (A) Western blot analysis of PCNA and FANCD2 monoubiquitination. Protein extract were prepared from A549 and A549-R cells treated with a combination of CDDP (100 µM) and ML323 (100 µM) as indicated. Western blot analyses were performed with antibodies against PCNA (top panel) and FANCD2 (bottom panel). Β-tubulin detection was used as a loading control. (B) CDDP dose-response of A549 (top panel) and A549-R cells (bottom panel) co-treated with ML323 30 µM or 100 µM as indicated. Drug treatment was started 24 h after cell plating. Cell survival was measure after 48 h exposure to the drug(s) using a colorimetric proliferation assay. The IC50 of CDDP was determined by non-linear fitting of the absorbance. Error bars represent standard error of the mean.
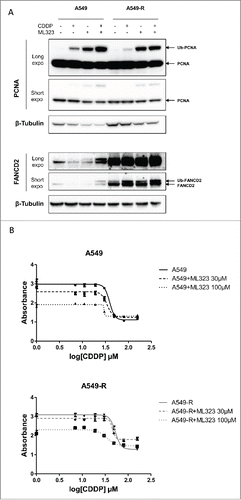
We then evaluated the impact of ML323-mediated inhibition of USP1 function on the survival to CDDP treatment. A549 and A549-R cells were exposed to CDDP in the absence or in the presence of 30 µM or 100 µM ML323. As shown in , the presence of ML323 resulted in a dose-dependent sensitization of the A549 cells to CDDP. Most importantly, while the low dose of ML323 has no effect on A549-R cells resistance to CDDP, higher concentrations of the inhibitor re-sensitized the resistant cells to levels comparable to the parental A549 cell line (IC50 34 µM with ML323 at 100 µM, ). Taken together, these results suggest that USP1 function is a major determinant of CDDP cytotoxic activity in our cell models.
Resistance to CDDP is associated with shortening of USP1 mRNA 5′ UTR
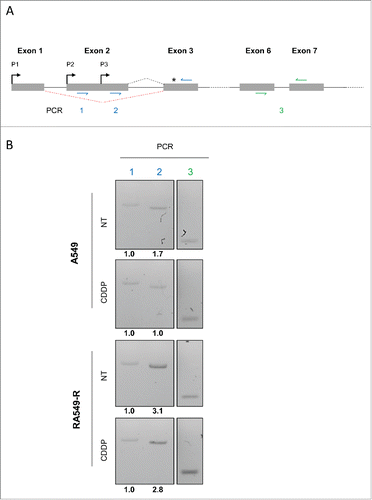
The resistance of the A549-R cells appears to be mediated in part by the deregulation of USP1 mRNA translation. The translation rate of an mRNA is mainly regulated at the initiation stage, which can be controlled, among other features, by the presence of cis-regulatory element in the 5′-untranslated region (5′ UTR) of the gene transcript(s). USP1 gene can generate transcripts with differing 5′ UTRs resulting from alternative promoter usage (see for example https://fasterdb.lyon.unicancer.fr) (). We performed an RT-PCR analysis of the 5′ UTRs of the USP1 mRNAs in A549 and A549-R cells. We could detect 2 mRNAs potentially arising from alternative promoter usage within the exon 2 (PCR 1 and 2, Fig. 4A and Fig. S3). Interestingly, the relative abundance of these mRNA variants was found to be cell line specific (). While the transcription of the USP1 gene seems to arise predominantly from the second transcription start site downstream of the second promoter (P2) in A549 cells, the A549-R cells also express a shorter mRNA transcribed from the P3 promoter. Interestingly, when analyzed by an RNA secondary structure prediction software (see for example http://mfold.rna.albany.edu), the longer USP1 mRNA 5′UTR is predicted to have a higher minimum free energy (ΔG -314.50) than the shorter 5′ UTR (ΔG -163.80) Fig. S3). The initiation of the translation of the shorter 5′ UTR may therefore be less prone to secondary structure-mediated repression. Alternatively, the USP1 mRNA bearing a shorter 5′ UTR expressed in the A549-R cells could potentially lack cis-regulatory regions responsible for the translational regulation observed in the CDDP-sensitive A549 cells. These two levels of translational regulation are proposed as candidate mechanisms for the USP1 translational deregulation associated with CDDP resistance in the A549-R cells.
Figure 4. Alternative USP1 mRNA 5′-UTR in the resistant A549-R cells (A) Schematic representation of the 5′-region of USP1 transcript. Exons are represented as boxes. Intronic sequences are represented by solid lines. Red dotted line represents alternative splicing. Putative promoters are denoted by black arrows and the translation start site by a black stars. Position of the primers used in the PCR analysis are shown (colored arrows). (B) Total RNAs from A549 and A549-R cells treated with CDDP 30 µM or with vehicle for 16 h were analyzed by semi-quantitative RT-PCR using primers as depicted in . Band quantification is shown underneath blots.
Discussion
Resistance to chemotherapeutic agents can involve various intrinsic cellular processes including drug efflux, increased resistance to apoptosis, or increased DNA repair capabilities in the case of platinum salts or other DNA damaging drugs. We chose to give an initial focus on the involvement of the DNA repair processes in resistance to CDDP. In this study we employed a high throughput approach designed to identify genes regulated at the translational level upon CDDP treatment. Performing this analysis concomitantly in a sensitive and a matched resistant cell line enabled us to identify mRNAs differentially regulated at the translation level by CDDP in sensitive vs. resistant cell lines. We specifically identified translationally-regulated mRNAs encoding DNA repair genes, among which USP1 was chosen for further investigation because previously developed small molecule inhibitors are available for pre-clinical studies as well as for potential clinical developments.Citation28,29
USP1 was shown to be involved in DNA repair as a deubiquitinating enzyme (DUB) for monoubiquitinated FANCD2.Citation22 FANCD2 monoubiquitination is crucial to initiate DNA lesion processing and removal and is catalyzed by the FA core complex upon DNA damage in an ATR-dependent process.Citation30 Interestingly, 2 FA core complex genes required for FANCD2 monoubiquitination, FANCB and FANCF, were also identified in our screen ( and Fig. S1).Citation31,32 In a subsequent step of the FA pathway activation, the USP1/UAF1-mediated removal of the ubiquitin moiety is required for FANCD2 to delocalize away from the chromatin and for the FA pathway to proceed.Citation23,24 Thus, USP1/UAF1 activity does not antagonize the FA core complex ubiquitin-ligase activity, but instead enable the FA pathway to proceed and triggers DNA lesion processing. In the sensitive A549 cell line, USP1 protein levels initially drop slightly upon CDDP exposure, potentially enabling FAND2 monoubiquitination. Later on, the USP1 protein level rises through increased mRNA translation rate. This identifies mRNA translation as a candidate mechanism for the transient and timely control of FANCD2 monoubiquitination ().
Beside the increased levels of USP1 in the resistant A549-R cells, the levels of both unmodified and monubiquitinated FANCD2 are also clearly much higher in these cells compared to the sensitive cell line. Taken together, these data point to an increase activity of the FA pathway in the resistant A549-R cells and suggest that ICL, one of the platinum-induced DNA lesions that are processed by FA, may be a crucial determinant of CDDP cytotoxic activity in this cell model.
Another crucial function of the USP1/UAF1 complex is the removal of PCNA monoubiquitination that regulates the recruitment of translesion synthesis (TLS) DNA polymerase.Citation25-27 TLS through damaged DNA can participate in cell resistance to chemotherapeutic agents and the USP1 deregulation in our resistant cell model may well impact on TSL polymerase activity, even though further investigation is needed to test this possibility.
In agreement with previous reports, we found that interfering with USP1 function using siRNA or a small molecule inhibitor was sufficient to re-sensitize the resistant cells to CDDP.Citation28,29 This opens the possibility of targeting USP1 activity using small molecule inhibitors to circumvent resistance to CDDP, even though this remains to be clinically assessed.
In this study, we report that the resistant A549-R cells express a shortened USP1 mRNA. As USP1 translational regulation is lost in these cells, we hypothesized that this short mRNA may be less prone to secondary structure-mediated translational repression or may lack regulatory sequences in its 5′-UTR. The trans-acting factor(s) that potentially fail to bind the regulatory region of USP1 mRNA remain(s) to be identified. Moreover, whether this level of regulation is commonly disrupted in CDDP-resistant NSCLC cell lines is an important question that still needs to be addressed. More generally, to our knowledge, USP1 gene expression has not been studied at the translational level and it remains to be determined whether the mechanism described in this study may be generalized across additional cancer cell lines. Nevertheless, as the total mRNA levels of USP1 are stable in our resistant and the sensitive cells lines, and as USP1 protein detection in lung tumor samples gives conflicting results, we propose the assessment of the USP1 5′-UTR as an alternative predictive marker of CDDP resistance easily amenable to routine practices.Citation33,34
Material and Methods
Cell culture, siRNA transfection, drug treatment and colorimetric proliferation assay
The A549 and A549-R NSCLC cell lines were maintained in DMEM supplemented with 10% Fetal Calf Serum, Penicillin (100 U/mL) and Streptomycin (100 µg/mL) at 37C, 10% CO2 in humidified atmosphere. The A549-R cell line was derived from the A549 cell line by chronic exposure to CDDP as previously described.Citation20
siRNA transfection was perform using Lipofectamine® RNAiMAX Reagent following manufacturer instructions. siRNA were from Santa Cruz biotechnology, INC (Control siRNA, sc-37007 and USP1 siRNA, sc-88494).
Proliferation was measured using the Cell Proliferation Reagent WST-1 (Roche) according to manufacturer′s instructions.
The USP1 inhibitor ML323 was from Axon Medchem (2309). Cycloheximide was purchased from sigma and used at a final concentration of 300µg/mL.
Polysome profiling and RT-PCR
A549 and A549-R cells were treated with vehicle or with CDDP at 30 µM for 4 h or 16 h. Cells were treated with cycloheximide at 100 µg/mL for 15 min to avoid ribosome off loading, then rinsed twice with cold PBS supplemented with 100 µg/mL cycloheximide and harvested by scrapping on ice. Every further steps were performed on ice whenever possible. Cell pellet was resuspended in 1 volume of LSB buffer (20 mM Tris pH7.5, 100 mM NaCl, 3 mM MgCl2, 1 mM DTT, 100 U/mL RNAsin® Plus RNAse inhibitor (Promega), 100 µg/mL cyclohexamide). One volume of LSB-ST buffer (LSB supplemented with 0.5 M sucrose and 2.4% TritonX100) was then added, the cell suspension was kept on ice for 30 min before centrifugation at 12,000xg at 4C for 10 min. Supernatant was collected and salt concentrations were adjusted to NaCl 150 mM and MgCl2 10mM. Samples were snap-frozen in liquid nitrogen if not processed immediately.
An aliquot of 1/10 of the cell extract was spared as the Input sample. The remaining extract was loaded on a sucrose density gradient ranging from 15% to 50% sucrose in LSB buffer. Sample separation was performed by ultracentrifugation at 38,000xg for 2 h at 4C. Samples were fractionated across the density gradient using an Isco gradient fractionation system.
For the cDNA microarray analysis, mRNA from the 4 heaviest fractions were pooled (polysomal fraction, FP) and from the total cells extract (Input) were prepared using the TRizol® RNA Isolation Reagent (Life technologies) according to manufacturer′s instructions. Quadruplicates were analyzed on a GE 8X60K (Agilent) microarray platform.
The same samples were also reversed transcribed using Mulv Reverse Transcriptase (Life Technologies) and Random hexamers and analyzed by semi-quantitative RT-PCR (GoTaq® Hota Start Polymerase, Promega) for validation of the microarray data and the analysis of the USP1 mRNA 5′-UTR using primers listed in Table S1.
For polysome profiling, mRNA was isolated using TRizol® LS Reagent (Life technologies) according to manufacturer′s instructions, reverse transcribes and analyzed by qPCR using Thermo ScientificTM LuminarisTM Color HiGreen HiRox master mix and the primers listed in Table S1.
Statistical tests
Differential expression was computed using the limma package in R that estimates the fold changes and standard errors by fitting a linear model for each gene and applying empirical Bayes smoothing to the standard errors.Citation35
Antibodies and Western blot analysis
Cells were washed in PBS, harvested by scrapping, pelleted and lysed in RIPA buffer supplemented with cOmplete (Roche) protease inhibitors cocktail for 30 min on ice. After a centrifugation at 20,000xg for 15 min at 4C, the supernatant was used as a protein extract. Protein extracts were resolved by SDS-PAGE in 10% Bis-Tris acrylamide (for USP1 and PCNA) or 3%-8% Tris-Acetate acrylamide (FANCD2) gels, transferred onto a nitrocellulose membrane and probed with the following antibodies in TBS, 5% milk, 0.05% Tween R 20: anti-USP1 (Abcam ab108104, 1/1,000), anti-PCNA (Santa Cruz biotechnology, INC, cs-56, 1/1,000), anti-FANCD2 (Santa Cruz biotechnology, INC, sc-20022, 1/1,000), TP53 (Santa Cruz biotechnology, INC, sc-47698, 1/1,000), anti-β-Tubulin (Sigma, T8328, 1/1,000), anti-β-Actin (Sigma, A5441, 1/1,000) and anti-GAPDH (Millipore, MAB 374, 1/2,000). ECL was performed using SuperSignalTMWest Pico Chemiluminescence Substrate (Life technologies) according to manufacturer′s instructions. Bands were quantified using the Image Lab 5.1-Beta software (Bio-Rad Laboratories).
Abbreviations
| 5′ UTR | = | 5′-Untranslated Region |
| ABCA1 | = | ATP-binding cassette, sub-family A, member 1 |
| CDDP | = | cis-diaminedichloroplatin (II) |
| ERCC1 | = | Exision Repair Cross-Complemention group 1 |
| FA | = | Fanconi Anemia |
| FANCB | = | Fanconi Anemia complementation group B |
| FANCD2 | = | Fanconi Anemia complementation group D2 |
| FANCF | = | Fanconi Anemia complementation group F |
| NSCLC | = | Non-Small-Cell Lung Cancer |
| PCNA | = | Proliferating Cell Nuclear Antigen |
| SNP | = | Small Nucleotide Polymorphism |
| TLS | = | Trans-Lesion Synthesis |
| UAF1 | = | USP1 associated factor 1 |
| USP1 | = | Ubiquitin-Specific Peptidase 1. |
Disclosure of Potential Conflicts of interest
No Potential Conflicts of interest were disclosed.
1120918_Supplemental_Material.zip
Download Zip (1.4 MB)Acknowledgments
We thank Guillaume Meurice from the Gustave Roussy genomic platform (Unité de Génomique Fonctionnelle) for assistance with microarray data analysis.
Funding
This work was supported by grants from the ARC Foundation (Fondation ARC pour la recherche sur le cancer), Paris Sud University (Attractivité 2012), the Fondation de France, the MMO (Médecine Moléculaire en Oncologie) program from the ANR (ANR-10-IBHU-0001) and the SIRIC-SOCRATE.(INCa-DGOS-INSERM 6043).
Related Research Data
References
- Hu L, Wu C, Zhao X, Heist R, Su L, Zhao Y, Han B, Cao S, Chu M, Dai J, et al. Genome-wide association study of prognosis in advanced non-small cell lung cancer patients receiving platinum-based chemotherapy. Clin Cancer Res 2012; 18:5507-14; PMID:22872573; http://dx.doi.org/10.1158/1078-0432.CCR-12-1202
- Lin M, Stewart DJ, Spitz MR, Hildebrandt MA, Lu C, Lin J, Gu J, Huang M, Lippman SM, Wu X. Genetic variations in the transforming growth factor-β pathway as predictors of survival in advanced non-small cell lung cancer. Carcinogenesis 2011; 32:1050-6; PMID:21515830; http://dx.doi.org/10.1093/carcin/bgr067
- Li Y, Sun Z, Cunningham JM, Aubry MC, Wampfler JA, Croghan GA, Johnson C, Wu D, Aakre JA, Molina J, et al. Genetic variations in multiple drug action pathways and survival in advanced stage non-small cell lung cancer treated with chemotherapy. Clin Cancer Res 2011; 17:3830-40; PMID:21636554; http://dx.doi.org/10.1158/1078-0432.CCR-10-2877
- Wagner KW, Ye Y, Lin J, Vaporciyan AA, Roth JA, Wu X. Genetic variations in epigenetic genes are predictors of recurrence in stage I or II non-small cell lung cancer patients. Clin Cancer Res 2012; 18:585-92; PMID:22252258; http://dx.doi.org/10.1158/1078-0432.CCR-11-2087
- Murtaza M, Dawson SJ, Tsui DW, Gale D, Forshew T, Piskorz AM, Parkinson C, Chin SF, Kingsbury Z, Wong AS, et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature 2013; 497:108-12; PMID:23563269; http://dx.doi.org/10.1038/nature12065
- Lai TC, Chow KC, Fang HY, Cho HC, Chen CY, Lin TY, Chiang IP, Ho SP. Expression of xeroderma pigmentosum complementation group C protein predicts cisplatin resistance in lung adenocarcinoma patients. Oncology Reports 2011; 25:1243-51; PMID:21327329
- Wang Q, Zhong M, Liu W, Li J, Huang J, Zheng L. Alterations of microRNAs in cisplatin-resistant human non-small cell lung cancer cells (A549/DDP). Exp Lung Res 2011; 37:427-34; PMID:21787234; http://dx.doi.org/10.3109/01902148.2011.584263
- Zhou L, Qiu T, Xu J, Wang T, Wang J, Zhou X, Huang Z, Zhu W, Shu Y, Liu P. miR-135a/b modulate cisplatin resistance of human lung cancer cell line by targeting MCL1. Pathol Oncol Res 2013; 19:677-83; PMID:23640248; http://dx.doi.org/10.1007/s12253-013-9630-4
- Yang Y, Li H, Hou S, Hu B, Liu J, Wang J. The noncoding RNA expression profile and the effect of lncRNA AK126698 on cisplatin resistance in non-small-cell lung cancer cell. PloS One 2013; 8:e65309; PMID:23741487; http://dx.doi.org/10.1371/journal.pone.0065309
- Jacquemont C, Simon JA, D'Andrea AD, Taniguchi T. Non-specific chemical inhibition of the Fanconi anemia pathway sensitizes cancer cells to cisplatin. Mol Cancer 2012; 11:26; PMID:22537224; http://dx.doi.org/10.1186/1476-4598-11-26
- Galluzzi L, Goubar A, Olaussen KA, Vitale I, Senovilla L, Michels J, Robin A, Dorvault N, Besse B, Validire P, et al. Prognostic value of LIPC in non-small cell lung carcinoma. Cell Cycle (Georgetown, Tex) 2013; 12:647-54; PMID:23343765; http://dx.doi.org/10.4161/cc.23517
- Olaussen KA, Dunant A, Fouret P, Brambilla E, Andre F, Haddad V, Taranchon E, Filipits M, Pirker R, Popper HH, et al. DNA repair by ERCC1 in non-small-cell lung cancer and cisplatin-based adjuvant chemotherapy. N Eng J Med 2006; 355:983-91; PMID:16957145; http://dx.doi.org/10.1056/NEJMoa060570
- Mazan-Mamczarz K, Kawai T, Martindale JL, Gorospe M. En masse analysis of nascent translation using microarrays. Bio Techn 2005; 39:61-2: 4: 6-7
- Powley IR, Kondrashov A, Young LA, Dobbyn HC, Hill K, Cannell IG, Stoneley M, Kong YW, Cotes JA, Smith GC, et al. Translational reprogramming following UVB irradiation is mediated by DNA-PKcs and allows selective recruitment to the polysomes of mRNAs encoding DNA repair enzymes. Genes Dev 2009; 23:1207-20; PMID:19451221; http://dx.doi.org/10.1101/gad.516509
- Cammas A, Pileur F, Bonnal S, Lewis SM, Leveque N, Holcik M, Vagner S. Cytoplasmic relocalization of heterogeneous nuclear ribonucleoprotein A1 controls translation initiation of specific mRNAs. Mol Biol Cell 2007; 18:5048-59; PMID:17898077; http://dx.doi.org/10.1091/mbc.E07-06-0603
- Dutertre M, Lambert S, Carreira A, Amor-Gueret M, Vagner S. DNA damage: RNA-binding proteins protect from near and far. Trends Biochem Sci 2014; 39:141-9; PMID:24534650; http://dx.doi.org/10.1016/j.tibs.2014.01.003
- Zeng HZ, Qu YQ, Zhang WJ, Xiu B, Deng AM, Liang AB. Proteomic analysis identified DJ-1 as a cisplatin resistant marker in non-small cell lung cancer. Int J Mol Sci 2011; 12:3489-99; PMID:21747690; http://dx.doi.org/10.3390/ijms12063489
- Huang W, Ding X, Li B, Fan M, Zhou T, Sun H, Yi Y, Zhang J. Serum biomarkers analyzed by LC-MS/MS as predictors for short outcome of non-small cell lung cancer patients treated with chemoradiotherapy. Neoplasma 2013; 60:11-8; PMID:23067211; http://dx.doi.org/10.4149/neo_2013_002
- Kumaraswamy S, Chinnaiyan P, Shankavaram UT, Lu X, Camphausen K, Tofilon PJ. Radiation-induced gene translation profiles reveal tumor type and cancer-specific components. Cancer Res 2008; 68:3819-26; PMID:18483266; http://dx.doi.org/10.1158/0008-5472.CAN-08-0016
- Michels J, Vitale I, Galluzzi L, Adam J, Olaussen KA, Kepp O, Senovilla L, Talhaoui I, Guegan J, Enot DP, et al. Cisplatin resistance associated with PARP hyperactivation. Cancer Res 2013; 73:2271-80; PMID:23554447; http://dx.doi.org/10.1158/0008-5472.CAN-12-3000
- Cohn MA, Kowal P, Yang K, Haas W, Huang TT, Gygi SP, D'Andrea AD. A UAF1-containing multisubunit protein complex regulates the Fanconi anemia pathway. Mol Cell 2007; 28:786-97; PMID:18082604; http://dx.doi.org/10.1016/j.molcel.2007.09.031
- Nijman SM, Huang TT, Dirac AM, Brummelkamp TR, Kerkhoven RM, D'Andrea AD, Bernards R. The deubiquitinating enzyme USP1 regulates the Fanconi anemia pathway. Mol Cell 2005; 17:331-9; PMID:15694335; http://dx.doi.org/10.1016/j.molcel.2005.01.008
- Oestergaard VH, Langevin F, Kuiken HJ, Pace P, Niedzwiedz W, Simpson LJ, Ohzeki M, Takata M, Sale JE, Patel KJ. Deubiquitination of FANCD2 is required for DNA crosslink repair. Mol Cell 2007; 28:798-809; PMID:18082605; http://dx.doi.org/10.1016/j.molcel.2007.09.020
- Kim JM, Parmar K, Huang M, Weinstock DM, Ruit CA, Kutok JL, D'Andrea AD. Inactivation of murine Usp1 results in genomic instability and a Fanconi anemia phenotype. Dev Cell 2009; 16:314-20; PMID:19217432; http://dx.doi.org/10.1016/j.devcel.2009.01.001
- Hussain S, Wilson JB, Medhurst AL, Hejna J, Witt E, Ananth S, Davies A, Masson JY, Moses R, West SC, et al. Direct interaction of FANCD2 with BRCA2 in DNA damage response pathways. Hum Mol Genetics 2004; 13:1241-8; PMID:15115758; http://dx.doi.org/10.1093/hmg/ddh135
- Howlett NG, Taniguchi T, Durkin SG, D'Andrea AD, Glover TW. The Fanconi anemia pathway is required for the DNA replication stress response and for the regulation of common fragile site stability. Hum Mol Genetics 2005; 14:693-701; PMID:15661754; http://dx.doi.org/10.1093/hmg/ddi065
- Huang TT, Nijman SM, Mirchandani KD, Galardy PJ, Cohn MA, Haas W, Gygi SP, Ploegh HL, Bernards R, D'Andrea AD. Regulation of monoubiquitinated PCNA by DUB autocleavage. Nat Cell Biol 2006; 8:339-47; PMID:16531995
- Chen J, Dexheimer TS, Ai Y, Liang Q, Villamil MA, Inglese J, Maloney DJ, Jadhav A, Simeonov A, Zhuang Z. Selective and cell-active inhibitors of the USP1/ UAF1 deubiquitinase complex reverse cisplatin resistance in non-small cell lung cancer cells. Chem Biol 2011; 18:1390-400; PMID:22118673; http://dx.doi.org/10.1016/j.chembiol.2011.08.014
- Liang Q, Dexheimer TS, Zhang P, Rosenthal AS, Villamil MA, You C, Zhang Q, Chen J, Ott CA, Sun H, et al. A selective USP1-UAF1 inhibitor links deubiquitination to DNA damage responses. Nat Chem Biol 2014; 10:298-304; PMID:24531842; http://dx.doi.org/10.1038/nchembio.1455
- Garcia-Higuera I, Taniguchi T, Ganesan S, Meyn MS, Timmers C, Hejna J, Grompe M, D'Andrea AD. Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol Cell 2001; 7:249-62; PMID:11239454; http://dx.doi.org/10.1016/S1097-2765(01)00173-3
- Rajendra E, Oestergaard VH, Langevin F, Wang M, Dornan GL, Patel KJ, Passmore LA. The genetic and biochemical basis of FANCD2 monoubiquitination. Mol Cell 2014; 54:858-69; PMID:24905007; http://dx.doi.org/10.1016/j.molcel.2014.05.001
- de Winter JP, van der Weel L, de Groot J, Stone S, Waisfisz Q, Arwert F, Scheper RJ, Kruyt FA, Hoatlin ME, Joenje H. The Fanconi anemia protein FANCF forms a nuclear complex with FANCA, FANCC and FANCG. Hum Mol Genetics 2000; 9:2665-74; PMID:11063725; http://dx.doi.org/10.1093/hmg/9.18.2665
- Liu Y, Luo X, Hu H, Wang R, Sun Y, Zeng R, Chen H. Integrative proteomics and tissue microarray profiling indicate the association between overexpressed serum proteins and non-small cell lung cancer. PloS One 2012; 7:e51748; PMID:23284758; http://dx.doi.org/10.1371/journal.pone.0051748
- Zhiqiang Z, Qinghui Y, Yongqiang Z, Jian Z, Xin Z, Haiying M, Yuepeng G. USP1 regulates AKT phosphorylation by modulating the stability of PHLPP1 in lung cancer cells. J Cancer Res Clin Oncol 2012; 138:1231-8; PMID:22426999; http://dx.doi.org/10.1007/s00432-012-1193-3
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 2015; 43:e47; PMID:25605792; http://dx.doi.org/10.1093/nar/gkv007