
Abstract
The spindle assembly checkpoint (SAC) acts as a guardian against cellular threats that may lead to chromosomal missegregation and aneuploidy. Mad2, an anaphase-promoting complex/cyclosome-Cdc20 (APC/CCdc20) inhibitor, has an additional homolog in mammals known as Mad2B, Mad2L2 or Rev7. Apart from its role in Polζ-mediated translesion DNA synthesis and double-strand break repair, Rev7 is also believed to inhibit APC/C by negatively regulating Cdh1. Here we report yet another function of Rev7 in cultured human cells. Rev7, as predicted earlier, is involved in the formation of a functional spindle and maintenance of chromosome segregation. In the absence of Rev7, cells tend to arrest in G2/M-phase and display increased monoastral and abnormal spindles with misaligned chromosomes. Furthermore, Rev7-depleted cells show Mad2 localization at the kinetochores of metaphase cells, an indicator of activated SAC, coupled with increased levels of Cyclin B1, an APCCdc20 substrate. Surprisingly unlike Mad2, depletion of Rev7 in several cultured human cell lines did not compromise SAC activity. Our data therefore suggest that besides its role in APC/CCdh1 inhibition, Rev7 is also required for mitotic spindle organization and faithful chromosome segregation most probably through its physical interaction with RAN.
Introduction
A faithful distribution of chromosomes between the daughter cells is indispensable to prevent aneuploidy, a condition associated with many cancers.Citation1 Following the assembly of a functional bipolar mitotic spindle with all the chromosomes properly attached and aligned at the metaphase plate, the spindle assembly checkpoint (SAC) becomes satisfied, allowing metaphase cells to enter anaphase.Citation2 In the event of a misaligned chromosome(s) or defective mitotic spindle, SAC-mediated mitotic arrest is enforced through the sequestration of Cdc20, an anaphase-promoting complex/cyclosome (APC/C) activator. This in turn prevents Cyclin B1 degradation, thereby preventing the cells from entering anaphase.Citation3 Through another pathway, the premature exit from metaphase is prevented by the regulatory effect of nucleocytoplasmic transport factors Rae1 and Nup98 on APC/CCdh1, thereby inhibiting the untimely degradation of securin.Citation4,5 The SAC-mediated safeguard is considered vital for faithful segregation of chromosomes and hence has attracted tremendous attention in the past to understand the molecular mechanism especially in reference to cancer.Citation1,6-8 It is now widely accepted that activated SAC inhibits APC/CCdc20 through a coordinated effort of several proteins including Mad1, Mad2, BubR1, Bub1, Bub3 and Mps1, in which a closed form of Mad2 (c-Mad2) and BubR1 bind to Cdc20 thereby preventing its association with APC/C.Citation7 However, some reports also suggest that BubR1 is the main player with Mad2 acting as a facilitator for the BubR1-Cdc20 association.Citation9-11
The human REV7 gene was originally identified based on its sequence homology with the budding yeast REV7.Citation12 Rev7 was subsequently found to interact with the Rev3 catalytic subunit of DNA polymerase ζ (Polζ) as well as Y-family polymerase Rev1.Citation12-14 Due to 48% sequence similarity to Mad2, Rev7 is also known as mitotic arrest deficient protein 2 B (Mad2B) or Mad2L2.Citation15 This molecular similarity was further supported through in vitro functional studies more than a decade ago showing that Mad2B inhibits APC/C by binding to Cdc20 and Cdh1Citation16,17 and more recently in an in vivo study showing that Mad2B specifically blocks Cdh1.Citation18 In addition to APC/C inhibition and the well-established translesion DNA synthesis (TLS) activity of Polζ, several recent reports have shown that Rev7 interacts with an array of diverse proteins unrelated to TLS or APC/C, including Ets-like transcription factor (ELK-1), hepatocellular carcinoma-associated gene 2 (HCCA2), Ras-related nuclear protein (RAN), T cell factor 4 (TCF4), clathrin light chain A (CLTA) and single-minded 2 (Sim2),Citation19-24 suggesting that Rev7 plays multiple roles either in response to DNA damageCitation19 or during cell cycle progression that are not necessarily related to APC/C inhibition. Its interaction with RAN throughout the cell cycle, especially during metaphase Citation19 is of particular interest since GTP-bound RAN plays a key role in spindle assembly.Citation25-27 RAN is a member of the Ras superfamily, which when active (RAN-GTP), forms a concentration gradient with higher levels of RAN-GTP in the nucleus during interphase and around the metaphase plate during metaphase to assist in nucleocytoplasmic transport and spindle assembly, respectively.Citation25,27 However the biological relevance of the Rev7-RAN interaction with respect to the mitotic functions remains to be illustrated.
Here we report that Rev7 plays a role in spindle assembly, mostly probably through its association with RAN. Loss of Rev7 induces Mad2-mediated mitotic arrest with increased frequency of abnormal spindles, misaligned chromosomes and accumulation of Cyclin B1. Furthermore, unlike Mad2, Rev7 depletion does not affect SAC activity and its long-term depletion results in aneuploidy. Experimental depletion of RAN shows mitotic phenotypes comparable to Rev7 depletion, and Rev7 depletion appears to affect RAN distribution during mitosis. These data indicate that Rev7 has a mitotic function distinct from APC/C inhibition.
Results
Subcellular localization of Rev7
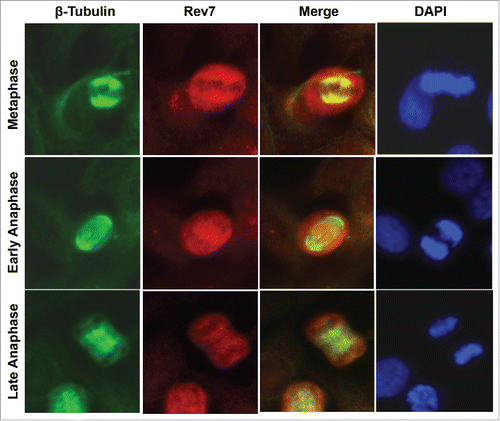
We previously observed an increased expression and distinct subcellular localization of Rev7 during metaphase of the cell cycle.Citation28 This led us to suspect that Rev7 plays a role in mitosis distinct from its TLS function. This is in agreement with a previous report that Rev7 interacts with a RAN GTPase,Citation21 an essential component of a bipolar spindle formation,Citation25,26 thereby having a possible role in the spindle formation. To first investigate the precise localization of Rev7 in mitotic cells, we performed double immunostaining with rabbit anti-β-tubulin and mouse anti-Rev7 antibodies in HeLa cells. As shown in , Rev7 is highly concentrated around the metaphase plate and co-localized with mitotic spindles. Although this localization is in agreement with a previous report,Citation21 our data indicate that Rev7 is actually around instead of being confined to the spindle structure. The Rev7 and RAN interaction was confirmed in a pull-down assay (See Fig. S3A), consistent with an early report.Citation21 The specificity of the Rev7 antibody used in this study was validated by various means as described previously.Citation28 The subcellular distribution of Rev7 was also confirmed in HCT116 cells and by using a previously-validated rabbit anti-Rev7 antibody Citation29 (data not shown).
Figure 1. Subcellular localization of Rev7. Co-immunostaining images of HeLa cells showing the subcellular localization of Rev7 during metaphase and anaphase stages of the cell cycle with respect to the spindle. During metaphase Rev7 looks to be concentrated around the metaphase plate as detected with a mouse anti-Rev7 antibody, and co-localizes with the spindle as detected with rabbit anti-β-tubulin (top panel). During early anaphase there is an increased Rev7 staining between the anaphase plates (middle panel) and in late anaphase Rev7 staining is reduced and more uniform (bottom panel).
Depletion of Rev7 causes G2/M arrest whereas Mad2 or RAN depletion causes cell death
In order to understand the cellular functions of Rev7 that are independent of TLS, we established a highly-efficient Rev7 depletion protocol (Fig. S1A). Following siRev7 treatment for 48 hrs, there was an apparent increase in the number of round, presumably metaphase cells, which became more obvious after 72 hrs of treatment. In contrast, the percentage of round cells in the mock- and control-treated cells remained constant (). To determine the biological nature of the round cells, we performed flow cytometeric analysis, which confirmed that the round cells are in fact metaphase cells but not dead cells. The percentage of G2/M-phase cells was significantly higher in the siRev7-treated compared to the control-treated cells (). The G2/M arrest after siRev7 treatment was further confirmed in NF1604 and HCT116 cells albeit at a different frequency (Fig. S2). In sharp contrast, cells treated with siRev3 did not show such a G2/M arrest (), indicating that Rev7-mediated G2/M activity is distinct from its TLS function.
Figure 2. Rev7 depletion causes mitotic arrest in HeLa cells. (A) Representative images showing increased number of round cells (red arrowheads) in siRev7-treated cells after 72 hrs of treatment. (B) Quantitative analysis of the percentage of round cells in mock-, siCTRL- and siRev7-treated cells 72 hrs post-transfection. Results are the average of 3 experiments with approximately 500 cells in each experiment. Round cells were considered to be mitotic cells and the rest to be other phases of the cell cycle. (C) Cell-cycle distribution as determined by flow cytometry after 72 hrs of treatment with indicated siRNAs. siMad2 cells were analyzed after 48 hrs of treatment. (D) Percentage distribution of cells in different phases of the cell cycle averaged from 3 experiments as analyzed from the flow cytometry data in (C). (E) Representative images of DAPI-stained cells with Rev7, RAN or Mad2 depletion showing different types of nuclear abnormalities such as nuclear constrictions (siRAN), nuclear fragmentation (siMad2), interphase bridge and micronuclei formation (siMad2). siRAN images were captured at 36 hrs post-transfection, siMad2 at 48 hrs post-transfection and siRev7 at 72 hrs post-transfection. (F) Quantitative analysis of various nuclear abnormalities observed after the siRNA treatment. I-bridges (Interphase bridges), N-fragmentation (Nuclear fragmentation). Error bars represent standard deviation. *p < 0 .05, **p < 0 .005, ***p < 0 .0005 vs. siCTRL.
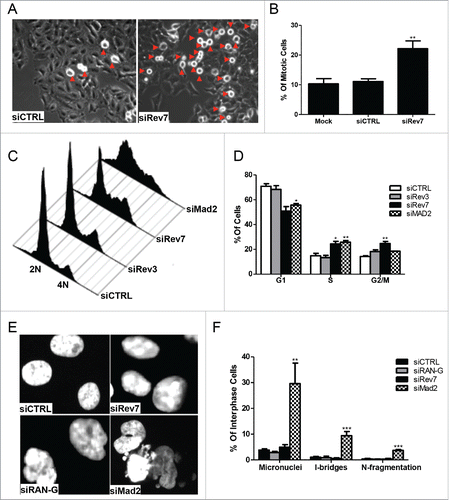
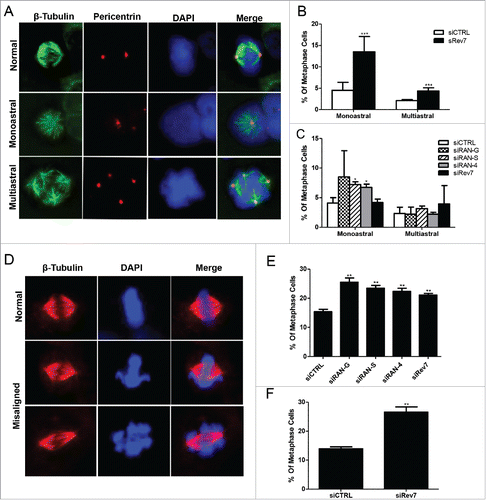
Figure 3. Rev7 and RAN depletion causes abnormal mitotic spindle formation and chromosome misalignment in HeLa cells. (A) Representative images of β-tubulin and pericentrin co-stained cells showing monoastral and multiastral spindles. (B) Quantitative analysis showing the percentage of metaphase cells carrying monoastral and multiastral spindles in 72-h siCTRL- and siRev7-treated samples. (C) Quantitative analysis of the percentage of metaphase cells carrying monoastral and multiastral spindles after 36-h treatment of siRev7 or siRAN. (D) Representative images of β-tubulin-stained cells showing normally-aligned (top panel), mildly-misaligned (middle panel) and severely-misaligned (bottom panel) chromosomes at the metaphase plate. (E) Quantitative analysis of the percentage of metaphase cells carrying misaligned chromosomes after 36 hrs of siRNA treatment. (F) Quantitative analysis of the percentage of metaphase cells carrying misaligned chromosomes after 72 hrs of siRev7 treatment. Error bars represent standard deviation from 3 independent experiments for all figures except B, which were calculated from 6 experiments. *p < 0 .05, **p < 0 .005, ***p < 0 .0005 vs. siCTRL.
In this study, we also monitored cellular phenotypes after Mad2 or RAN depletion. While depletion of Mad2 was rather effective (Fig. S1B), we were unable to achieve a highly-efficient RAN depletion despite using 3 different siRNAs at varying concentrations and in different cells lines. The specificity of Mad2 antibodies used in this study was verified by overexpressing pCDNA-Mad2 vector in HCT116 cells (Fig. S1C). All three RAN siRNAs resulted in approximately 50% depletion (Fig. S1D). Nevertheless, treatment of HeLa cells with siMad2 or siRAN for 48 hrs resulted in extensive cell death. The apparent difference between Rev7 and RAN depletion was that cells died 48 hrs after siRAN treatment but no immediate cell death was seen in siRev7-treated cells even after 72-hr treatment (). The siMad2-associated cell death was distinct from siRAN, as loss of Mad2 caused multiple cellular defects including micronuclei formation, nuclear fragmentation and interphase bridges () but siRAN resulted in a deformed nucleus.()
Both Rev7 and RAN are essential for proper spindle formation
To further look into possible causes for the observed G2/M arrest, we examined the mitotic spindle for possible defects based on our observations and previous reports.Citation21,24 As shown in , depletion of Rev7 results in a substantial increase in the number of monoastral spindles with duplicate centrosomes and some increase in the number of multiastral and abnormal spindles. The most significant increase in monoastral (threefold) and multiastral (twofold) spindles was observed 72 hrs after siRev7 transfection (). Other than the reported physical interaction between Rev7 and RAN,Citation21 the functional importance of this interaction remains unknown. To address functional relevance between Rev7 and RAN, we depleted RAN from HeLa cells using 3 different siRANs and compared the phenotypes with Rev7-depleted cells. Since all our RAN siRNAs caused cell death at 48 hrs post-transfection, we were unable to look for a similar phenotype at the 72-hr time point. Instead, we observed some comparable phenotypes 36 hrs after siRNA transfection when there was no apparent cell death and the knockdown efficiency was approximately 50%. Interestingly, treatment with all 3 siRANs resulted in some increase in monoastral phenotype but no change in multiastral spindles (). Since an abnormal spindle can cause chromosomal misalignment at the metaphase plate, we next wished to quantify chromosome misalignment in siRev7-treated cells and compare it with siRAN-treated cells. As shown in , there was a significant increase in the number of misaligned metaphases ranging from minor to severely misaligned in both siRev7- and siRAN-treated cells, suggesting that Rev7 and RAN may function in the same pathway during mitosis.
Although Rev7 indeed physically interacts with RAN in vivo (Fig. S3A), depletion of Rev7 did not alter total levels of cellular RAN (Fig. S3B). However, subcellular distribution of RAN in metaphase cells appeared to be affected. Interestingly, almost all metaphase cells with misaligned chromosomes showed abnormal RAN distribution around the chromatin in comparison to normal metaphases (Fig. S3C-D). Unfortunately, due to technical difficulties we were unable to observe the reported RAN-GTP concentration gradient in the metaphase cells Citation25 and hence cannot conclude whether and how Rev7 functions to maintain such a gradient.
Rev7 depletion leads to Mad2 activation
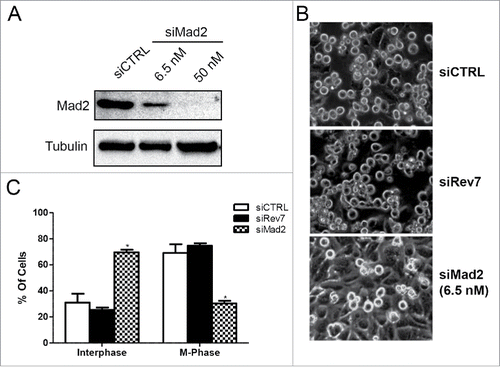
A recent study using U2OS cells indicates that Rev7 specifically sequesters Cdh1, thereby preventing early activation of APC/CCdh1.Citation18 In this case, the Rev7-deficient cells exit mitosis faster than control cells after release from nocodazole arrest. In contrast we observed an apparent G2/M arrest in HeLa cells after Rev7 depletion (), a phenomenon that was also observed in HCT116 and lung fibroblast NF1604 cells (Fig. S2). Our choice of HeLa cells was based on their extensive use in SAC-related studies and a report showing that these cells have a robust SAC.Citation30 We have previously shown that the Rev7 siRNA used in this study did not cause any change in the Mad2 protein levels.Citation28 To further rule out any SAC-related activity like its Mad2 homolog, we examined the effect of siRev7 on nocodazole-induced cell cycle arrest. Since complete depletion of Mad2 results in cell death, we managed a partial and transient depletion of Mad2 by using siRNA at a low concentration (6.5 nM) () to mimic haplo-insufficient Mad2 cells known to fail to arrest after nocodazole treatment.Citation31 Indeed the partial depletion of Mad2 was sufficient to interfere with its SAC activity in response to the nocodazole treatment (). In comparison, HeLa cells depleted of Rev7 and treated with nocodazole for 24 and 48 hrs arrested normally like siCTRL-treated cells (). We also examined nocodazole-induced G2/M arrest in HCT116 cells and did not notice alteration of mitotic index in Rev7-depleted cells (Fig. S4).
Figure 4. HeLa cells depleted of Rev7 or Mad2 respond differently to nocodazole-induced G2/M cell cycle arrest. (A) Western blots showing partial and complete depletion of Mad2 using different concentrations of siMad2. β-tubulin was used as an internal control. (B) Representative images taken from cells treated with siRev7 or 6.5 nM siMad2 for 48 hrs followed by treatment with 200 ng/ml nocodazole for an additional 24 hrs before taking photographs. (C) Quantitative analysis of the percentage of interphase (flat) and metaphase (round) cells in different treatment groups averaged from 2 experiments with approximately 300 cells in each experiment.
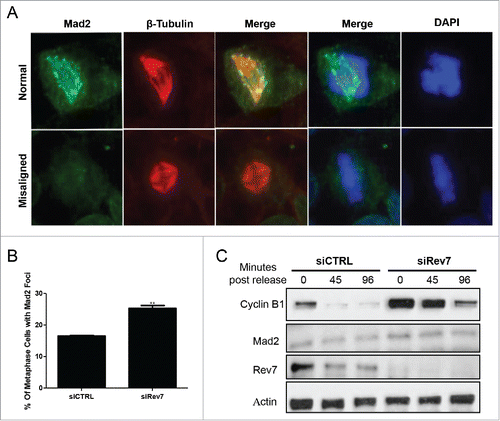
Although Rev7 depletion causes increased mitotic cells with monoastral spindle that may lead to mitotic arrest, the defective bipolar spindles with misaligned chromosomes may cause SAC activation that may also lead to mitotic arrest. During SAC activation, Mad2 localizes at the unattached kinetochores during metaphase.Citation32 We performed immunostaining using a rabbit anti-Mad2 antibody to look for Mad2 focus formation. Metaphases with misaligned chromosomes showed Mad2 foci with significantly higher frequency in siRev7-treated cells than in siCTRL cells (), indicating that loss of Rev7 in turn results in the activation of SAC. It has been previously reported that activated SAC causes an increase in the Cyclin B1 levels through APC/CCdc20 inhibition.Citation3 As expected, Rev7 depletion resulted in Cyclin B1 accumulation at selected time points in mitotic shake-off cells collected after release from RO-0303 (Cdk1 inhibitor) block (). The above observations collectively indicate that the mitotic arrest caused by Rev7 depletion is at least partially due to the activation of Mad2 checkpoint.
Figure 5. Rev7 depletion induces Mad2 focus formation and Cyclin B1 accumulation in HeLa cells. (A) Representative images of Mad2- and β-tubulin-immunostained cells showing a normal metaphase cell with no Mad2 foci (left panel) and a misaligned metaphase cell with Mad2 foci and an abnormal spindle (right panel). (B) Quantitative analysis of metaphase cells positive for Mad2 foci 72 hrs after siRev7 treatment. (C) Western blots showing increased levels of Cyclin B1 after Rev7 depletion at 0 hr post-release from RO-3306 block and delayed degradation at 45 and 96 minutes post-release when compared to siCTRL treated cells. Mad2 levels remained unchanged. Actin was used as an internal control. Error bars represent standard deviation from 3 experiments. **p < 0.005 vs. siCTRL.
Loss of Rev7 causes aneuploidy but not premature segregation
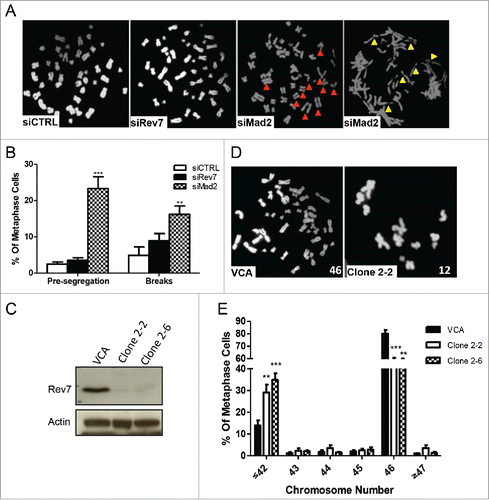
In order to further delineate Mad2 and Rev7 functions, we looked at the premature sister chromatid segregation defect in Rev7-depleted cells, a well-recognized phenotype of Mad2-depletion.Citation31 As shown in , a significantly higher percentage of metaphases had premature segregation and DNA double-strand breaks in the Mad2-depleted HCT116 cells than expected; however, no such an increase was observed in the Rev7-depleted cells when compared to the siCTRL cells. Since Rev7-depleted cells display an apparent misalignment () and lagging chromosome defects,Citation28 an obvious prediction is that loss of Rev7 could cause aneuploidy. To address this possibility, we first examined transiently-transfected cells using siRNAs and did not find an apparent difference in aneuploidy between the treatment groups (data not shown). Next, we counted metaphase chromosomes in 2 stable Rev7-depleted MSU1.1 cell lines (Clone 2.2 and 2.6) () Citation29 to see if a long-term depletion causes aneuploidy. Surprisingly, both clones showed a significant increase in the number of metaphases with ≤42 chromosomes as compared to vector control cells (). Consistently, in a freshly-generated stable Rev7-depleted HCT116 cell line (shRev7-C4), an increase in the number of metaphases with fewer than normal number of chromosomes was observed in comparison to the vector-transfected cells after 16 passages post-selection (Fig. S5).
Figure 6. Mad2 depletion causes premature segregation and DNA damage whereas long-term depletion of Rev7 causes aneuploidy. (A) Representative images of HCT116 metaphase spread showing a normal metaphase in siCTRL- and siRev7-treated cells but premature sister chromatid segregation (red arrow heads) and chromosome breaks (yellow arrow heads) in siMad2-treated cells 48 hrs post-transfection. (B) Quantitative analysis of premature segregation (Pre-segregation) and chromosomal breaks in images taken from (A). Error bars represent standard deviations from 3 experiments. **p < 0 .005, ***p<0.0005 vs. siCTRL in respective groups. (C) Western blots showing stable depletion of Rev7 in 2 selected clones of MSU1.1 cells. (D) Representative metaphase spreads from vector control MSU1.1 cells (VCA) and Rev7-depleted MSU1.1 cell clone 2–2 showing aneuploidy. (E) Quantitative analysis of metaphase spreads from Rev7-depleted MSU1.1 cell clones 2–2 and 2–6 showing the percentage of metaphase cells with ≤42 chromosomes. **p < 0 .005, ***p < 0 .0005 vs. VCA.
Discussion
The functional complexity of Rev7/Mad2B in human cells has just stared to unravel. Besides its role in TLS, Rev7 was initially believed to be a SAC protein like Mad2Citation15; however, it was later described as an APC/C inhibitor but surprisingly not a SAC protein.Citation16-18 Further adding to its complex nature, several recent reports have suggested additional functions such as upregulation of Elk-1 target genes in response to DNA damage,Citation19 epithelial-mesenchymal transdifferentiation mediated by TCF4,Citation22 central nervous system development,Citation23 DNA repair at telomeres,Citation33 DNA double-strand break resection,Citation34 cell cycle regulation Citation20 and more importantly regulation during metaphase.Citation21,24 In this study we attempted to define the mitotic function(s) of Rev7.
Previous reports suggest that Rev7 plays its mitotic role(s) through RAN and/or Clathrin. First, Rev7 is known to physically interact with RAN,Citation21 an important component of bipolar spindle formation. Second, it localizes to the spindle structure during metaphase. Unlike the previous report Citation21 that Rev7 is exclusive to the mitotic spindle, we found that Rev7 localizes in and around the metaphase spindle. It is unlikely that the discrepancy was due to our experimental variations as we used 2 independent anti-Rev7 antibodies and 3 different cell lines. Third, experimental depletion of either Rev7 or RAN results in increased monoastral cells and cells with abnormal spindles and misaligned chromosomes, indicating a defect in the spindle assembly. Assembly of a bipolar spindle can happen in the absence of a centrosome but requires the RAN-GTP gradient.Citation35 The unique Rev7 localization pattern in mitotic cells and its physical interaction with RAN led us to hypothesize that Rev7 is involved in the maintenance of the RAN-GTP gradient. Although experimental limitations did not allow us to test the RAN-GTP gradient around the metaphase plate directly, depletion of Rev7 appears to cause redistribution of RAN around the chromatin throughout metaphase without affecting the total cellular level of RAN. Based on these data we envisage that when there is no nuclear membrane to maintain the RAN-GTP gradient in metaphase cells, Rev7 play a critical role in the recruitment and/or retention of RAN-GTP toward the metaphase plate. In this aspect, Rev7 can be considered as an accessory protein of RAN-GTP.
A role of Rev7 in mediating RAN and spindle assembly does not rule out its possible involvement in the mitotic spindle checkpoint, particularly given its sequence homology with the key SAC protein Mad2. Several previous reports assign Rev7 as an APC/C inhibitor similar to Mad2 and Emi1.Citation15-17,36 Mad2 interacts exclusively with APC/CCdc20 whereas Rev7 interacts preferentially with APCCdh1 but also with APC/CCd20.Citation16-18,37-41 Most of these studies were conducted in in-vitro systems and hence their physiological relevance remains unclear. Furthermore, the subcellular localization of Rev7 during metaphase appears to be distinct from Cdh1.Citation42 Despite the fact that Mad2 is a well-characterized SAC protein, there is no direct evidence supporting a similar role for Rev7 in SAC. Several observations in the present study indicate that Rev7 cannot be a SAC protein. First, unlike Mad2, depletion of Rev7 causes a moderate mitotic arrest, suggesting that Rev7 is required for mitotic progression instead of being a checkpoint. Second, in response to nocodazole treatment, partially-depleted Mad2 cells progress as if there is no spindle poisoning. In contrast, siRev7-treated cells are arrested in response to nocodazole, suggesting an intact SAC. This appears to be consistent with a previous report, showing that the bacterial protein IpaB associates with Rev7 to cause G2/M arrest in the host cells.Citation40 Third, loss of Rev7 actually activates Mad2, indicating that these 2 proteins function in different pathways. The activated Mad2 following Rev7 depletion is further supported by the accumulation and delayed degradation of Cyclin B1.
The major difference between Rev7 and Mad2 is that while complete depletion of Mad2 caused cell death with premature sister chromatid separation, nuclear fragmentation, micronuclei formation, nuclear bridges, and chromosome breaks that are consistent with previous reports,Citation31,43,44 no such defect was observed in Rev7-depleted cells. While both depletion and overexpression of Mad2 causes genomic instability especially aneuploidy,Citation31,43-45 Rev7 depletion or overexpression does not cause aneuploidy early on; however, MSU1.1 and HCT116 cells analyzed after multiple generations with persistent Rev7 depletion did show a significant increase in aneuploidy. Even though this is a similar phenotype to the Mad2 haplo-insufficient HCT116 cells,Citation19 our data clearly indicate different underlying mechanisms. In Rev7-depleted cells the chromosome misalignment defect appears to be a major cause of aneuploidy, whereas premature sister chromatid separation and failed sister chromatid separation are thought to be the predominant reasons for aneuploidy in Mad2-depleted and overexpressed cells, respectively.Citation43,44
In conclusion, our experimental data provide compelling evidence to support an essential role for Rev7 in association with RAN to assemble a functional mitotic spindle. Furthermore, our data argue against Rev7 being a Mad2 functional ortholog despite their sequence homology.
Materials and Methods
Cell culture
HCT116, HeLa, NF1604 and MSU1.1 cells were cultured in DMEM (Sigma) containing 15 mM HEPES (Sigma), 25 mM NaHCO3 (Sigma), 1x antibiotics/antimycotics (Gibco), and 10% FBS (Gibco). For G2 cell-cycle arrest, cells were treated with 100 ng/ml nocodazole.
Depletion of target gene products
For transient depletion of a gene product, a protocol was followed similar to that described previously.Citation28 The following siRNAs were used: siGenome SMARTpool human MAD2 (M-003271–01–005), siGenome SMARTpool human RAN (M-010353–00–0005), ON-TARGETplus SMARTpool human RAN (L-010353–00–0005), all purchased from Dharmacon. Additionally, a previously-published siRNA sequence 5’-AGAAGAAUCUUCAGUACUA-3’ Citation46 was used to deplete RAN products (siRAN-4). For stable depletion, cells were transfected with a pSilencer vector either alone or expressing short hairpin against hRev7 (shRev7) Citation29 by using lipofectamine 2000 (Invitrogen) following the manufacturer's protocol. The cells were selected with 1 µg/ml puromycin and selected clones were tested for the depletion efficiency using Western blotting.
Immunocytochemistry (ICC)
Cells cultured on poly-lysine-coated cover slips were quickly washed with cold PBS followed by fixation in 4% paraformaldehyde solution for 15 min at room temperature. After three washes with PBS, cells were permeabilized with PBS containing 0.25% Triton X-100 (PBST) for 10 min followed by 3 more washes with 1xPBST. Cells were then blocked in 5% bovine serum albumin (BSA) (Sigma) for 30 min followed by incubation with selected antibodies overnight at 4 °C. To assess the spindle structure or the spindles associated with Mad2 foci, cells seeded onto poly-lysine-coated coverslips were pre-extracted in a buffer containing 80 mM PIPES pH6.8, 1 mM MgCl2, 4 mM EGTA, 0.32 M sucrose (added fresh) and 0.5% Triton X-100 at room temperature for 30 seconds. Cells were fixed in cold methanol for 3 min at -20 °C followed by rehydration by 3 washes with 1xPBST for 5 min each at room temperature. Following rehydration, cells were blocked for 30 min in 5% BSA in PBS (blocking reagent) after which they were incubated overnight at 4 °C with selected primary antibodies in 1% BSA; anti-β-Tubulin (1:500, SantaCruz, sc-166729), anti-hMAD2 (1:300, Covance Inc.., PRB-452C), or anti-Pericentrin (1:1000, Abcam, ab4448). Cells were washed 3 times the following day with 1xPBST for 10 min each and incubated with secondary antibodies (Molecular Probes) in 1% blocking reagent (1:2000 Alexa 546 donkey anti-mouse; or 1:2000 Alexa 546 goat anti-rabbit) for 1 hr at room temperature, Cells were again washed 3 times with 1xPBST for 10 min each and mounted with Prolong Gold anti-fade reagent (Invitrogen) containing 1.5 µg/ml DAPI.
Flow cytometry analysis
A procedure similar to that described previously Citation28 was followed. Briefly, around 1x106 cells were collected and the DNA was labeled in 1 ml propidium iodide solution (1% Triton X-100, 200 µg/ml DNase-free RNase A, 20 ng/ml propidium iodide). Cells were passed through a fine pore mesh before running on a Beckman Coulter Epics XL Flow Cytometer. The data were analyzed using FlowJo v9.1 software.
Metaphase spread preparation
After cells were arrested in metaphase by the addition of 0.1 µg/ml colcemid (Sigma) for 3 hrs, they were trypsinized and washed with PBS. Cells were treated with a hypotonic solution (0.56% KCl) at 37 °C for 6 min followed by the addition of 3 drops of freshly-prepared ice-cold methanol/acetic acid (3:1) to stop the reaction. After centrifugation, cells were resuspended in methanol/acetic acid fixative drop-wise while vortexing at a low speed. This step was repeated one more time before storing the samples at -20°C for further analysis. To obtain metaphase spreads, cell were dropped onto a pre-cleaned glass slide and dried overnight. Metaphases were then stained with DAPI and visualized under a microscope.
Cell synchronization and protein extraction
Following Rev7 knockdown using siRNA for 48 hrs, HeLa cells were treated with 2.5 µM Cdk1 inhibitor RO-3306 (217699, Calbiochm) for 16 to 18 hrs to synchronize them in G2 phase. The G2 block was then confirmed under microscope by the appearance of very few round cell. Cells were then washed with fresh media 3 times to remove the inhibitor. Samples were collected at 0, 45, and 90 minutes post-release and total proteins were extracted in an NP-40 buffer with a protease inhibitor cocktail (Roche). Total protein concentration was determined by the Bradford method using a commercial reagent from Bio-Rad, and the Cyclin B1 level was detected through western blotting.
Western blotting
Heat-denatured protein extracts prepared in a RIPA buffer (50 mM Tris-HCl pH7.4, 150 mM NaCl, 2 mM EDTA, 1% NP-40, 0.1% SDS, 2x protease inhibitor cocktail and 1 mM PMSF) were resolved on a 10% SDS-PAGE gel and transferred to a polyvinylidene fluoride (PVDF) membrane. Blots were incubated with selected primary antibodies in 5% blocking buffer overnight at 4 °C followed by incubation with HRP-conjugated secondary antibody for 1 hr at room temperature. Immunoreactivity was detected using HRP-conjugated secondary antibodies (1:10,000 goat anti-mouse Upstate 12–349; 1:5000 goat anti-rabbit Santa Cruz sc-2004) and developed using Western LightningTM Chemiluminescence Reagent Plus (Perkin Elmer NEL104). The relative protein levels were quantified using ImageJ software. The following antibodies were used: Mouse anti-Mad2 1:5000 (Santa Cruz sc-28261), mouse anti-Rev7 1:3000 (BD Biosciences), rat anti-HA 1:5000 (Roche Inc., 1867423), mouse anti-V5 1:5000 (Santa Cruz sc-271944), mouse anti-RAN 1:5000 (BD Transduction, 610340), mouse anti-Cyclin B1 1:1000 (sc-245, Santa Cruz) and rabbit anti-tubulin (Santa Cruz, sc-9104).
Statistical analysis
Prism v4 software was used for graphs and statistical analysis. The two-tail Students t-test was used to calculate the significance value. A p value of <0 .05 was considered statistically significant.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental material
Supplemental data for this article can be accessed on the publisher's website.
Supplemental_Material.zip
Download Zip (949.8 KB)Acknowledgments
We thank Michelle Hanna for proofreading the manuscript and other Xiao laboratory members for helpful discussion.
Funding
This study was supported by the Natural Sciences and Engineering Research Council of Canada Discovery Grant No. RGPIN-2014–04580 and Chinese National 973 Project [2013CB911003] to WX. AB is a recipient of the Saskatchewan Health Research Foundation Postdoctoral Fellowship.
Related Research Data
References
- Rajagopalan H, Lengauer C. Aneuploidy and cancer. Nature 2004; 432:338-41; PMID:15549096; http://dx.doi.org/10.1038/nature03099
- Farr KA, Cohen-Fix O. The metaphase to anaphase transition: a case of productive destruction. European J Biochem / FEBS 1999; 263:14-19; http://dx.doi.org/10.1046/j.1432-1327.1999.00510.x
- Musacchio A, Salmon ED. The spindle-assembly checkpoint in space and time. Nat Rev Mol Cell Biol 2007; 8:379-93; PMID:17426725; http://dx.doi.org/10.1038/nrm2163
- Jeganathan KB, Baker DJ, van Deursen JM. Securin associates with APCCdh1 in prometaphase but its destruction is delayed by Rae1 and Nup98 until the metaphase/anaphase transition. Cell Cycle 2006; 5:366-70; PMID:16479161; http://dx.doi.org/10.4161/cc.5.4.2483
- Jeganathan KB, Malureanu L, van Deursen JM. The Rae1-Nup98 complex prevents aneuploidy by inhibiting securin degradation. Nature 2005; 438:1036-9; PMID:16355229; http://dx.doi.org/10.1038/nature04221
- Kops GJ, Weaver BA, Cleveland DW. On the road to cancer: aneuploidy and the mitotic checkpoint. Nat Rev Cancer 2005; 5:773-85; PMID:16195750; http://dx.doi.org/10.1038/nrc1714
- Lara-Gonzalez P, Westhorpe FG, Taylor SS. The spindle assembly checkpoint. Curr Biol 2012; 22:R966-980; PMID:23174302; http://dx.doi.org/10.1016/j.cub.2012.10.006
- Foley EA, Kapoor TM. Microtubule attachment and spindle assembly checkpoint signalling at the kinetochore. Nat Rev Mol Cell Biol 2013; 14:25-37; PMID:23258294; http://dx.doi.org/10.1038/nrm3494
- Herzog F, Primorac I, Dube P, Lenart P, Sander B, Mechtler K, Stark H, Peters JM. Structure of the anaphase-promoting complex/cyclosome interacting with a mitotic checkpoint complex. Science 2009; 323:1477-81; PMID:19286556; http://dx.doi.org/10.1126/science.1163300
- Nilsson J, Yekezare M, Minshull J, Pines J. The APC/C maintains the spindle assembly checkpoint by targeting Cdc20 for destruction. Nat Cell Biol 2008; 10:1411-20; PMID:18997788; http://dx.doi.org/10.1038/ncb1799
- Kulukian A, Han JS, Cleveland DW. Unattached kinetochores catalyze production of an anaphase inhibitor that requires a Mad2 template to prime Cdc20 for BubR1 binding. Dev Cell 2009; 16:105-17; PMID:19154722; http://dx.doi.org/10.1016/j.devcel.2008.11.005
- Murakumo Y, Roth T, Ishii H, Rasio D, Numata S, Croce CM, Fishel R. A human REV7 homolog that interacts with the polymerase zeta catalytic subunit hREV3 and the spindle assembly checkpoint protein hMAD2. J Biol Chem 2000; 275:4391-7; PMID:10660610; http://dx.doi.org/10.1074/jbc.275.6.4391
- Murakumo Y, Roth T, Ishii H, Rasio D, Numata S, Croce CM, Fishel R. Interactions in the error-prone postreplication repair proteins hREV1, hREV3, and hREV7. J Biol Chem 2001; 276:35644-51; PMID:11485998; http://dx.doi.org/10.1074/jbc.M102051200
- Masuda Y, Ohmae M, Masuda K, Kamiya K. Structure and enzymatic properties of a stable complex of the human REV1 and REV7 proteins. J Biol Chem 2003; 278:12356-60; PMID:12529368; http://dx.doi.org/10.1074/jbc.M211765200
- Cahill DP, da Costa LT, Carson-Walter EB, Kinzler KW, Vogelstein B, Lengauer C. Characterization of MAD2B and other mitotic spindle checkpoint genes. Genomics 1999; 58:181-7; PMID:10366450; http://dx.doi.org/10.1006/geno.1999.5831
- Chen J, Fang G. MAD2B is an inhibitor of the anaphase-promoting complex. Genes Dev 2001; 15:1765-70; PMID:11459826; http://dx.doi.org/10.1101/gad.898701
- Pfleger CM, Salic A, Lee E, Kirschner MW. Inhibition of Cdh1-APC by the MAD2-related protein MAD2L2: a novel mechanism for regulating Cdh1. Genes Dev 2001; 15:1759-64; PMID:11459825; http://dx.doi.org/10.1101/gad.897901
- Listovsky T, Sale JE. Sequestration of CDH1 by MAD2L2 prevents premature APC/C activation prior to anaphase onset. J Cell Biol 2013; 203:87-100; PMID:24100295; http://dx.doi.org/10.1083/jcb.201302060
- Zhang L, Yang SH, Sharrocks AD. Rev7/MAD2B links c-Jun N-terminal protein kinase pathway signaling to activation of the transcription factor Elk-1. Mol Cell Biol 2007; 27:2861-9; PMID:17296730; http://dx.doi.org/10.1128/MCB.02276-06
- Li L, Shi Y, Wu H, Wan B, Li P, Zhou L, Shi H, Huo K.. Hepatocellular carcinoma-associated gene 2 interacts with MAD2L2. Mol Cell Biochem 2007; 304:297-304; PMID:17541814; http://dx.doi.org/10.1007/s11010-007-9512-8
- Medendorp K, van Groningen JJ, Vreede L, Hetterschijt L, van den Hurk WH, de Bruijn DR, Brugmans L, van Kessel AG. The mitotic arrest deficient protein MAD2B interacts with the small GTPase RAN throughout the cell cycle. PLoS One 2009; 4:e7020; PMID:19753112; http://dx.doi.org/10.1371/journal.pone.0007020
- Hong CF, Chou YT, Lin YS, Wu CW. MAD2B, a novel TCF4-binding protein, modulates TCF4-mediated epithelial-mesenchymal transdifferentiation. J Biol Chem 2009; 284:19613-22; PMID:19443654; http://dx.doi.org/10.1074/jbc.M109.005017
- Meng X, Tian X, Wang X, Gao P, Zhang C. A novel binding protein of single-minded 2: the mitotic arrest-deficient protein MAD2B. Neurogenetics 2012; 13:251-60; PMID:22660985; http://dx.doi.org/10.1007/s10048-012-0333-x
- Medendorp K, Vreede L, van Groningen JJ, Hetterschijt L, Brugmans L, Jansen PA, van den Hurk WH, de Bruijn DR, van Kessel AG. The mitotic arrest deficient protein MAD2B interacts with the clathrin light chain A during mitosis. PLoS One 2010; 5:e15128; PMID:21152103; http://dx.doi.org/10.1371/journal.pone.0015128
- Gruss OJ, Vernos I. The mechanism of spindle assembly: functions of Ran and its target TPX2. J Cell Biol 2004; 166:949-55; PMID:15452138; http://dx.doi.org/10.1083/jcb.200312112
- Royle SJ, Bright NA, Lagnado L. Clathrin is required for the function of the mitotic spindle. Nature 2005; 434:1152-7; PMID:15858577; http://dx.doi.org/10.1038/nature03502
- Sanderson HS, Clarke PR. Cell biology: Ran, mitosis and the cancer connection. Curr Biol 2006; 16:R466-468; PMID:16782004; http://dx.doi.org/10.1016/j.cub.2006.05.032
- Bhat A, Andersen PL, Qin Z, Xiao W. Rev3, the catalytic subunit of Polzeta, is required for maintaining fragile site stability in human cells. Nucleic Acids Res 2013; 41:2328-39; PMID:23303771; http://dx.doi.org/10.1093/nar/gks1442
- McNally K, Neal JA, McManus TP, McCormick JJ, Maher VM. hRev7, putative subunit of hPolzeta, plays a critical role in survival, induction of mutations, and progression through S-phase, of UV((254nm))-irradiated human fibroblasts. DNA Repair (Amst) 2008; 7:597-604; PMID:18295554; http://dx.doi.org/10.1016/j.dnarep.2007.12.013
- Andreassen PR, Skoufias DA, Margolis RL. Analysis of the spindle-assembly checkpoint in HeLa cells. Meth Mol Biol 2004; 281:213-25; PMID:15220532
- Michel LS, Liberal V, Chatterjee A, Kirchwegger R, Pasche B, Gerald W, Dobles M, Sorger PK, Murty VV, Benezra R. MAD2 haplo-insufficiency causes premature anaphase and chromosome instability in mammalian cells. Nature 2001; 409:355-9; PMID:11201745; http://dx.doi.org/10.1038/35053094
- May KM, Hardwick KG. The spindle checkpoint. J Cell Sci 2006; 119:4139-42; PMID:17038540; http://dx.doi.org/10.1242/jcs.03165
- Boersma V, Moatti N, Segura-Bayona S, Peuscher MH, van der Torre J, Wevers BA, Orthwein A, Durocher D, Jacobs JJ. MAD2L2 controls DNA repair at telomeres and DNA breaks by inhibiting 5′ end resection. Nature 2015; 521:537-40; PMID:25799990; http://dx.doi.org/10.1038/nature14216
- Xu G, Chapman JR, Brandsma I, Yuan J, Mistrik M, Bouwman P, Bartkova J, Gogola E, Warmerdam D, Barazas M, et al. REV7 counteracts DNA double-strand break resection and affects PARP inhibition. Nature 2015; 521:541-4; PMID:25799992; http://dx.doi.org/10.1038/nature14328
- Heald, R, Tournebize R, Blank T, Sandaltzopoulos R, Becker P, Hyman A, Karsenti E. Self-organization of microtubules into bipolar spindles around artificial chromosomes in Xenopus egg extracts. Nature 1996; 382:420-5; PMID:8684481; http://dx.doi.org/10.1038/382420a0
- Reimann JD, Freed E, Hsu JY, Kramer ER, Peters JM, Jackson PK. Emi1 is a mitotic regulator that interacts with Cdc20 and inhibits the anaphase promoting complex. Cell 2001; 105:645-55; PMID:11389834; http://dx.doi.org/10.1016/S0092-8674(01)00361-0
- Wassmann K, Benezra R. Mad2 transiently associates with an APC/p55Cdc complex during mitosis. Proc Natl Acad Sci U S A 1998; 95:11193-8; PMID:9736712; http://dx.doi.org/10.1073/pnas.95.19.11193
- Reimann JD, Gardner BE, Margottin-Goguet F, Jackson PK. Emi1 regulates the anaphase-promoting complex by a different mechanism than Mad2 proteins. Genes Dev 2001; 15:3278-85; PMID:11751633; http://dx.doi.org/10.1101/gad.945701
- Sudakin V, Chan GK, Yen TJ. Checkpoint inhibition of the APC/C in HeLa cells is mediated by a complex of BUBR1, BUB3, CDC20, and MAD2. J Cell Biol 2001; 154:925-36; PMID:11535616; http://dx.doi.org/10.1083/jcb.200102093
- Iwai H, Kim M, Yoshikawa Y, Ashida H, Ogawa M, Fujita Y, Muller D, Kirikae T, Jackson PK, Kotani S, et al. A bacterial effector targets Mad2L2, an APC inhibitor, to modulate host cell cycling. Cell 2007; 130:611-23; PMID:17719540; http://dx.doi.org/10.1016/j.cell.2007.06.043
- Izawa D, Pines J. Mad2 and the APC/C compete for the same site on Cdc20 to ensure proper chromosome segregation. J Cell Biol 2012; 199:27-37; PMID:23007648; http://dx.doi.org/10.1083/jcb.201205170
- Zhou Y, Ching YP, Chun AC, Jin DY. Nuclear localization of the cell cycle regulator CDH1 and its regulation by phosphorylation. J Biol Chem 2003; 278:12530-6; PMID:12560341; http://dx.doi.org/10.1074/jbc.M212853200
- Michel L, Diaz-Rodriguez E, Narayan G, Hernando E, Murty VV, Benezra R. Complete loss of the tumor suppressor MAD2 causes premature cyclin B degradation and mitotic failure in human somatic cells. Proc Natl Acad Sci U S A 2004; 101:4459-64; PMID:15070740; http://dx.doi.org/10.1073/pnas.0306069101
- Homer HA, McDougall A, Levasseur M, Yallop K, Murdoch AP, Herbert M. Mad2 prevents aneuploidy and premature proteolysis of cyclin B and securin during meiosis I in mouse oocytes. Genes Dev 2005; 19:202-7; PMID:15655110; http://dx.doi.org/10.1101/gad.328105
- Sotillo R, Hernando E, Díaz-Rodríguez E, Teruya-Feldstein J, Cordón-Cardo C, Lowe SW, Benezra R. Mad2 overexpression promotes aneuploidy and tumorigenesis in mice. Cancer Cell 2007; 11:9-23; PMID:17189715; http://dx.doi.org/10.1016/j.ccr.2006.10.019
- Morgan-Lappe SE, Tucker LA, Huang X, Zhang Q, Sarthy AV, Zakula D, Vernetti L, Schurdak M, Wang J, Fesik SW. Identification of Ras-related nuclear protein, targeting protein for xenopus kinesin-like protein 2, and stearoyl-CoA desaturase 1 as promising cancer targets from an RNAi-based screen. Cancer Res 2007; 67:4390-8; PMID:17483353; http://dx.doi.org/10.1158/0008-5472.CAN-06-4132