
ABSTRACT
Cell proliferation and differentiation show a remarkable inverse relationship. Precursor cells continue division before acquiring a fully differentiated state, while terminal differentiation usually coincides with proliferation arrest and permanent exit from the division cycle. Mechanistic insight in the temporal coordination between cell cycle exit and differentiation has come from studies of cells in culture and genetic animal models. As initially described for skeletal muscle differentiation, temporal coordination involves mutual antagonism between cyclin-dependent kinases that promote cell cycle entry and transcription factors that induce tissue-specific gene expression. Recent insights highlight the contribution of chromatin-regulating complexes that act in conjunction with the transcription factors and determine their activity. In particular SWI/SNF chromatin remodelers contribute to dual regulation of cell cycle and tissue-specific gene expression during terminal differentiation. We review the concerted regulation of the cell cycle and cell type-specific transcription, and discuss common mutations in human cancer that emphasize the clinical importance of proliferation versus differentiation control.
Introduction
The formation of a complete organism from a single fertilized egg is an intriguing and highly complex process. It requires the generation of large numbers of cells, which at the appropriate times acquire specialized functions and morphologies, while assembling into well-defined structures, tissues, and organs. Most cells follow a gradual process of specialization with a final step, terminal differentiation, characterized by acquisition of a fully differentiated post-mitotic state. Examples include neurons, muscles, and bone cells formed from proliferating precursor cells that shut down the cell cycle machinery while activating cell type-specific transcriptional programs. The temporal coupling between cell cycle withdrawal and differentiation is crucial for normal growth and development, and continues to be critical for tissue homeostasis and cell replacement throughout life. In contrast, a failure to arrest proliferation or loss of differentiation can lead to a variety of diseases and are hallmarks of cancer cells. Deregulation of proliferation has been long known to contribute to carcinogenesis. Recent insights in the frequency of genetic alterations in human cancer also highlight a widespread disruption of differentiation-related chromatin regulators and cell type-specific transcription factors in human cancer.Citation1 Here, we examine the molecular mechanisms that connect cell cycle exit to cell differentiation, and consider the potential implications for human cancer.
Cell cycle entry and exit
Cyclin Dependent Kinases (CDKs) in association with cyclin regulatory subunits are the master regulators of the cell division cycle. Dependent on the activity of CDK-cyclin complexes in the G1 phase, cells may arrest cell division or commit to go through a division cycle ().Citation2 This decision depends on extracellular signals and cell-intrinsic information, which determine CDK reactivation after the previous cell cycle.Citation3 Stimulation of quiescent cells with mitogens and growth factors induces expression of D-type cyclins (D1, D2, D3 in mammals) and activation of CDK4 or CDK6 (together: CDK4/6) ().Citation4 CDK4/6-cyclin D is responsible for limited phosphorylation of the retinoblastoma tumor suppressor (Rb) protein. This phosphorylation is thought to weaken the interaction between pRb and the heterodimeric transcription factor E2F/DP (together referred to as E2F). As a consequence, the repression of activating E2Fs by pRb is reduced, which allows initial transcription of E2F-dependent genes that include cyclin E and other cell cycle genes. Subsequent activation of CDK2-cyclin E leads to further phosphorylation and inactivation of pRb, release of E2F, and full commitment to S-phase entry ().Citation5
Figure 1. Regulation of the G1/S transition. Phosphorylation by CDK-cyclin complexes counteracts the binding between retinoblastoma tumor suppressor (Rb) family proteins and E2F transcription factors, thereby allowing transcriptional activation of S-phase genes. E2F refers to heterodimeric transcription factors that contain an E2F and DP family protein. Some E2F subunits are primarily transcriptional activators (E2F1, E2F2, E2F3a), and are blocked by pRb binding. Other E2Fs are transcriptional repressors and act in conjunction with pRb (E2F4, E2F5), or independent of the pRb protein family (E2F6–8). The pRb protein family consists of pRb, p107, and p130. Inhibition of APC/C-FZR1 may involve association with the Emi1 inhibitor (not shown) or phosphorylation by CDKs.Citation145 See text for further information and references.
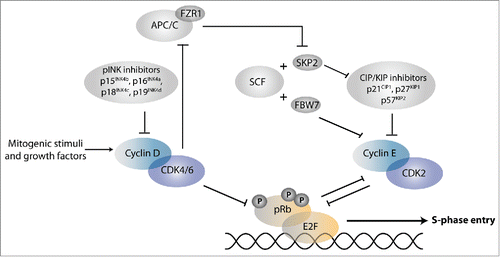
In addition to pRb-mediated transcriptional repression, several other levels of control counteract progression from G1 into S-phase. This includes members of 2 different families of CDK inhibitory proteins (CKIs) that associate with CDKs ().Citation6 CKIs of the INK4 protein family, such as p16INK4A, bind specifically to CDK4/6 kinases and prevent their interaction with D-type cyclins. In contrast, CKIs of the CIP/KIP family associate with CDK-cyclin complexes and block their activity. The CIP/KIP family consists of p21Cip1, p27Kip1, and p57Kip2 and is particularly important for temporal control of CDK2-cyclin E. Proteins of both CKI families contribute to cell cycle exit and show increased expression in differentiating cells. The importance of CKIs is further underscored by the functional inactivation of p16INK4A in a wide variety of human cancers.Citation1,6
Entry into the cell cycle is also controlled by ubiquitin-dependent protein degradation. This is primarily regulated at the level of substrate recognition by E3 ubiquitin ligases. E3 ligases with important functions in G1/S inhibition are the Anaphase Promoting Complex/Cyclosome (APC/C) in association with the FZR1/Cdh1 coactivator, and Skp1, Cullin, F-box factor (SCF) complexes. SCF in complex with the Fbw7 substrate-recognition factor targets cyclin E for degradation and inhibits cell cycle progression. In contrast, SCF in association with Skp2 directs the destruction of p21Cip1 and p27Kip1 and promotes cell cycle entry ().Citation7
Quiescence or permanent arrest
Induction of negative cell cycle regulators and inhibition of positive regulators can cause temporary or permanent cell cycle arrest. A major question is how the reversible non-dividing state, known as quiescence, differs from permanent cell cycle arrest. Interestingly, quiescent cells may retain the ability to resume proliferation by actively preventing differentiation.Citation8,9 It has been proposed that increased expression of the transcriptional repressor Hairy and Enhancer of Split1 (HES1) antagonizes senescence and terminal differentiation of quiescent cells.Citation9 HES1 is a target of Notch signaling and has also been implicated in p57Kip2 repression and maintenance of the proliferating state of muscle precursor cells.Citation9,10 While expression of cell cycle inhibitors contributes to both temporal and permanent cell cycle arrest, stable transcriptional repression of cell cycle-promoting genes may be specific for the irreversible cell cycle arrest that coincides with terminal differentiation.Citation11
Proliferation-differentiation decisions in G1
As cells respond to external signals during the G1 phase, developmental variations in the cell division cycle may influence proliferation vs. differentiation decisions. Early studies of embryonal carcinoma cells indicated that differentiation can be rapidly induced in G1, but not in S phase.Citation12 Since, developmental control over the length of G1 phase has been proposed as a differentiation-regulating mechanism. Undifferentiated cells in the early embryo of many animal systems, including flies, frogs, and zebrafish, undergo rapid cell divisions that entirely lack G1 and G2 phases. During mammalian embryogenesis, cell division cycles become very short after preimplantation embryos reach the blastocyst stage, with a subset of cells completing the division cycle in only 3 hours.Citation13,14 Similarly, embryonic stem cells established from the inner cell mass of preimplantation embryos have unusual cell cycles with a short G1 phase of approximately 2 hours.
Various studies have addressed whether the short G1 phase is relevant for maintenance of the undifferentiated state. The short embryonic cycles are characterized by the absence of active CDK inhibitors and constitutively high levels of CDKs and cyclins, especially CDK2-cyclin E.Citation15-17 These cell cycle profiles start to change during the induction of differentiation or developmental transitions such as the specification of the 3 germ layers in mice. During these processes, the CDK-cyclin associated kinase activity drops and becomes more cell cycle dependent. This coincides with establishment of a functional CDK4/6-cyclin D–pRb pathway and increased expression of cell cycle inhibitors.Citation14 As a consequence, cell cycles become longer and include an extended G1 phase. Experimentally increasing the levels of CDK2-cyclin E activity in mouse or human embryonic stem cells reduced the cell cycle length and obstructed differentiation. Decreasing CDK2-cyclin E levels had the opposite effect.Citation15,16 Studies with sorted populations of human and murine embryonic stem cells established that cells with the shortest G1 phase are in the naïve pluripotent state, and that G1 corresponds to a higher susceptibility to differentiate, compared to S and G2.Citation18-20
Thus, high G1 CDK-cyclin activity, a short G1 phase, or the combination of both promotes the undifferentiated state of embryonic stem cells. The converse is seen during later stages of development, in particular in the nervous system. As neural stem cell-like progenitors switch from proliferative divisions to neurogenic divisions in the developing mouse brain, the time in G1 phase increases from approximately 3 to 12 hours.Citation16 Cyclin E inhibition in the progenitors resulted in a longer G1 phase and a premature switch from proliferative to neurogenic divisions. Comparable effects were obtained after interfering with CDK4-cyclin D activity. Moreover, cyclin D1 knockout in the retina caused increased neurogenesis at the expense of progenitor expansion, whereas overexpression of cyclin D1 and CDK4 led to shortening of G1 phase and a delay in neurogenesis.Citation21,22 The increased length of G1 may allow time to respond to external signals and to accumulate differentiation-inducing transcription factors.Citation16
Probably contributing to differentiation susceptibility in G1, cyclin-dependent kinases cannot oppose differentiation-inducing mechanisms during part of G1 phase. As such, signals received in early versus late G1 can have different outcomes.Citation23 In response to TGFβ-related signaling, pluripotent human embryonic stem cells in early G1 were observed to form endoderm, while late G1 cells showed neurectodermal specification. The difference was traced to activation of CDK4/6-cyclin D, which phosphorylated and blocked nuclear import of Smad2/3, thereby preventing endoderm and allowing neurectodermal differentiation.Citation23 These data emphasize the intimate connection between G1 length, G1 CDK-cyclin activation and the response to developmental signals.
CDK inhibition of differentiation-inducing transcription factors
Muscle differentiation
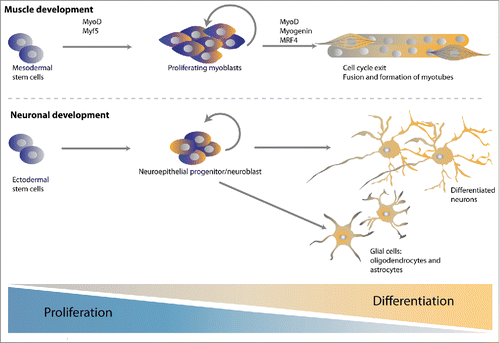
A direct antagonism between cell cycle-promoting CDKs and differentiation-inducing transcription factors has been long proposed. Insight has come from studies of skeletal muscle formation from differentiating C2C12 mouse myoblast cells in culture, and from primary myoblasts or progenitors such as satellite cells in model systems.Citation24-26 Skeletal muscle differentiation starts with a determination step, formation of proliferative myoblasts, which is followed by induction of muscle-specific gene expression, exit from the cell cycle, and fusion to form multinucleated myotubes (). A network of helix-loop-helix (bHLH) myogenic regulatory factors (MRFs) controls the determination and differentiation steps. The myoblast determination protein (MyoD) and myogenic factor 5 (Myf5) serve as the main myogenic determination factors, while myogenin, Mrf4 and MyoD promote the induction of terminal differentiation (). These MRFs act in concert with general transcription factors, including their E protein heterodimeric binding partners, and the myocyte enhancer factor MEF2.
Figure 2. Model systems to study the coordination of cell cycle exit and differentiation. Both muscle and neuronal development has served as powerful model systems to study the temporal coupling between cell cycle exit and differentiation. Differentiated muscle cells and neurons are formed from precursor cells that first become lineage restricted, then committed precursors that continue proliferative divisions, and finally terminally differentiated post-mitotic cells. This requires the activation of myogenic and pro-neuronal transcription factors and is often accompanied by the upregulation of negative cell cycle regulators.
Ectopic expression of MyoD has long been known to trigger muscle-specific gene expression in many cell types.Citation27 Nevertheless, MyoD is present in proliferating myoblasts and associated with regulatory regions of a substantial number of target genes that remain silent until differentiation is induced and cell proliferation seizes. Thus, additional regulators control MyoD-dependent transcriptional activation and execution of a muscle differentiation program. Overexpression of the cell cycle regulator Cyclin D1 was found to inhibit MyoD-induced myogenesis and transcriptional activation.Citation28-30 This effect appeared CDK4-cyclin D kinase-activity dependent and partly Rb-independent. As an attractive hypothesis, CDK4-cyclin D might phosphorylate and inhibit MyoD, but evidence for such phosphorylation was not found. Instead, mutual inhibition of CDK4 and MyoD through direct protein association has been proposed.Citation31 It remains unclear, however, if this interaction is physiologically relevant.
Some evidence indicates that CDK4-cyclin D phosphorylates the transcription factor MEF2, which acts together with MyoD in muscle differentiation.Citation32,33 This could explain the observed pRb-independent CDK4-cyclin D effect, while the pRb-dependent contribution is likely to result from E2F-dependent transcriptional activation of cyclin E. Several groups reported direct phosphorylation of MyoD by CDK2-cyclin E at serine 200, one of multiple in vivo phosphorylated residues of MyoD.Citation34-36 In support of in vivo phosphorylation, roscovitin, a chemical CDK2 and CDK1 inhibitor, and overexpression of p57Kip2 each prevented MyoD-Ser200 phosphorylation. MyoD-Ser200 phosphorylation was found to correspond to increased turnover of MyoD at the end of G1 phase.Citation34,36,37 By preventing MyoD accumulation and concomitant muscle differentiation, this mechanism may contribute to continued myoblast proliferation. Nevertheless, the exact contributions of CDK-dependent phosphorylation remain incompletely understood, and the switch from transcriptional repression to activation of muscle specific genes by MyoD, MEF2 and associated transcriptional regulators clearly includes many additional levels of control (see below).Citation38
Figure 3. Chromatin remodeling by the SWI/SNF complex. (A) The SWI/SNF chromatin-remodeling complex uses ATP hydrolysis to alter the chromatin state and DNA accessibility. Binding of the SWI/SNF complex to chromatin reduces the interaction between DNA and histones, allowing either sliding or ejection of histones. This creates a more open and accessible chromatin structure, or possibly the reverse. (B) Schematic overview of BAF and PBAF SWI/SNF complexes. SWI/SNF complexes consist of several core subunits (yellow) in association with additional accessory subunits (gray) and signature subunits (red) which are mutually exclusive for either the BAF complex (BAF250/ARID1) or the PBAF complex (BAF180/PBRM, BRD7 and BAF200/ARID2). All subunits are known by multiple names. While not used in the text, the systematic SMARC (SWI/SNF-related, Matrix-associated, Actin-dependent Regulator of Chromatin) nomenclature is included. For simplicity, only the BAF names are used in (C). (C) The core subunits (yellow) of the SWI/SNF complex assemble together with accessory subunits to ultimately form a functional complex. Dependent on the associated subunits, the complex may interact with specific transcription factors and exert cell type or differentiation-specific functions. Here, some of the variants described to be specifically incorporated during neuronal or muscle differentiation are shown in blue.
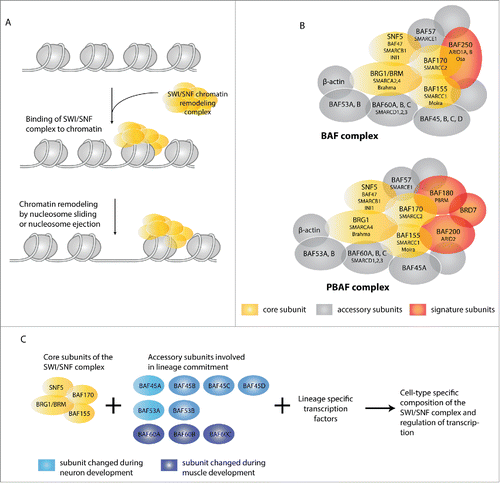
Figure 4. Robust control over the proliferation versus differentiation decision. A regulatory network of cell cycle regulators, transcription factors, lineage-specific SWI/SNF complexes and chromatin modification complexes mediates the all-or-nothing transition from proliferating precursors to differentiated post-mitotic cells.
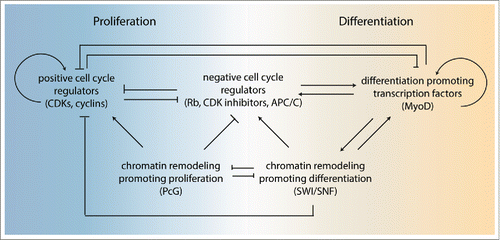
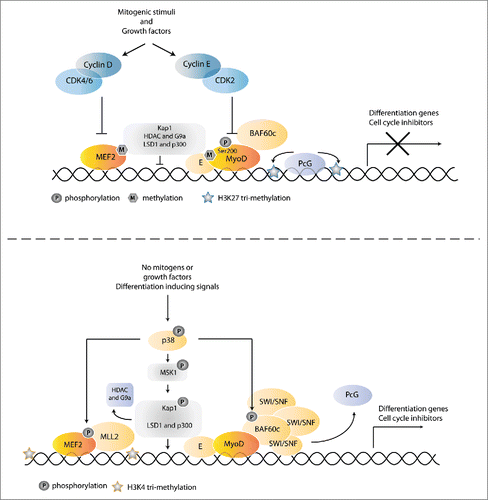
Figure 5. Coordinating muscle-specific gene expression with cell cycle exit. (A) In proliferating cells, muscle-specific gene expression is prevented by phosphorylation of MyoD by CDK2-cyclin E, as well as inhibition of the collaborating MEF2 transcription factor by CDK4/6-cyclin D. In addition the scaffolding protein KAP1 is bound to MyoD-E12 and MEF2 transcription factors, which results in the recruitment of co-repressor complexes that also methylate and inhibit MyoD and MEF2. The presence of PcG complexes further inhibits transcription of muscle specific genes. Reduction of growth factors allows differentiation, in part through downregulation of the inhibitory phosphorylations. Simultaneously, differentiation signals induce p38 MAPK activation. Activation of p38 results in phosphorylation of BAF60c bound to MyoD, which triggers the assembly of a functional SWI/SNF complex onto the preassembled MyoD/BAF60c pioneer complex. Recruitment of the SWI/SNF chromatin remodeler contributes to the replacement of PcG complexes and results in the formation of an open and active chromatin state. In addition, p38 MAPK phosphorylates MEK2, which induces recruitment of an MLL2-menin HMT transcriptional activator. As a third mechanism, activation of the p38 MAPK pathway leads to phosphorylation of KAP1, thereby triggering release of co-repressors and recruitment of co-activators at target gene promoters. See text for further information and references.
Neuronal differentiation
Like muscle formation, neuronal differentiation has been studied in a variety of systems, ranging from embryonic carcinoma, neuroblastoma and pluripotent stem cells induced to differentiate in culture, to sophisticated animal systems. Neuronal development usually starts from a neuroepithelial progenitor or stem cell, which gives rise to neuronal-restricted and glia-restricted progenitors (). Glia-restricted precursors can generate oligodendrocytes and astrocytes, while neuronal progenitors contribute to the formation of the various neurons of the central and peripheral nervous system.Citation40 The pro-neuronal bHLH transcription factors of the Neurogenin (Neurog), NeuroD, and Achaete scute-like 1 (Ascl1) families are critical for neurogenesis. Interfering with these transcription factors influences the coordination between proliferation and differentiation and thereby the final number of differentiated neurons in the brain.Citation41,42
Examination of the proneuronal differentiation factor neurogenin 2 (Ngn2) in Xenopus and mouse neuronal precursors revealed extensive phosphorylation in vivo.Citation43,44 Both mouse and Xenopus Ngn2 contain 9 potential CDK-phosphorylated residues, all serines followed by proline, and cyclin A and cyclin B kinases efficiently phosphorylated Ngn2 in vitro. Mutating these residues to non-phosphorylatable alanine increased the activity of Ngn2 as an inducer of neurogenesis. Moreover, progressive phosphorylation in mitotic extracts correlated with reduced DNA binding of Ngn2 in mobility shift assays.Citation43,44 Similar results were obtained for the bHLH neurogenic transcription factor Ascl1/Mash1.Citation43 As an interesting hypothesis, progressive multisite phosphorylation could affect the affinity of these transcription factors in a cell cycle-dependent fashion, allowing binding of only highly accessible sites in proliferating precursor cells, while association with more closed regulatory regions of differentiation promoting genes would be postponed until CDK levels drop.Citation45
Other examples from genetic model systems
Inhibition of differentiation inducing factors by CDK2-cyclin E phosphorylation may be used broadly, as indicated by examples in highly diverse systems. One of these systems is the Drosophila neuroblast.Citation46-47 Drosophila neuroblasts typically divide asymmetrically, combining self-renewal with the generation of a ganglion mother cell, which divides again to form 2 differentiated neurons. The transcription factor Prospero is deposited exclusively to the ganglion mother cell during the asymmetric neuroblast division. Prospero enters the nucleus of this cell and induces a transcriptional program required for neuronal differentiation. In the absence of cyclin E, nuclear localization of Prospero is observed in both neuroblast daughter cells, leading to premature neuronal differentiation.Citation47,48 In contrast, ectopic cyclin E expression induces asymmetric Prospero distribution in a precursor that normally divides symmetrically. Thus, cyclin E controls Prospero localization and antagonizes differentiation, though it remains to be established if this involves direct phosphorylation.
CDK2-cyclin E has also been implicated in antagonizing cell differentiation in C. elegans.Citation49,50 One example is reminiscent of Drosophila Prospero and involves an asymmetric cell division in the somatic gonad.Citation49 Upon loss of cyclin E, some of these divisions become symmetric, with the daughter cell that normally remains temporally quiescent also becoming a differentiated Distal Tip Cell, a fate normally acquired only by its sister cell. A quite distinct example of CDK2-cyclin E regulated differentiation relates to germ line stem cells that form differentiated gametes.Citation50 This transition involves a switch from mitotic cell division to entry into meiotic prophase. Meiotic entry and arrest of cell division are promoted by the GLD-1 (defective in Germ Line Development) protein, which associates with mRNA targets and inhibits their translation. Several lines of evidence indicate that GLD-1 is a direct substrate of CDK2-cyclin E in vivo and in vitro. As a consequence of CDK2-cylin E dependent GLD-1 phosphorylation, GLD-1 levels remain low in the stem cells of the germ line, and premature cell cycle exit and meiotic entry are prevented.Citation50 Interestingly, GLD-1 also represses cyclin E mRNA translation, allowing a double negative feedback loop and abrupt transition from mitosis to meiosis.Citation50-52 These examples illustrate how CDKs, in particular CDK2-cyclin E, can directly counter the activity of differentiation inducing factors.
Negative regulators of the cell cycle promote differentiation
While positive cell cycle regulators prevent differentiation, negative cell cycle regulators promote differentiation. In particular, members of the CIP/KIP family of CDK inhibitors are important for connecting cell cycle exit and differentiation. Obviously, these cell cycle inhibitors interfere with the activity of CDKs, and thus may overcome the CDK-mediated inhibition of differentiation-inducing transcription factors described above. In addition, CKIs may contribute functions independent from CDK regulation. For example, direct binding of p57Kip2 to MyoD has been reported to stabilize MyoD, thereby promoting differentiation.Citation35 In addition, p27Kip1 expression was shown to arrest mouse oligodendrocyte precursors, but p21Cip1 was still needed for full differentiation.Citation53 Furthermore, the N-terminal part of Xenopus p27 (Xic1) has been shown to contribute a cell-cycle independent function in the differentiation of multiple cell types.Citation45 These functions of CIP/KIP family members are not well understood, but may relate to stabilization of differentiation-inducing transcription factors.
In cooperation with CIP/KIP family members, transcriptional co-repressors of the pRb protein family promote cell differentiation. This role resides at least in part in the inhibition of cell cycle entry by complexes of pRb and E2F family proteins.Citation5 However, pRb complexes have been reported to also promote transcription of cell type-specific genes.Citation54 The best-described example is the differentiation of liver macrophages, which are critical for completion of erythropoiesis.Citation55 In this process, pRb can bind the ETS-domain transcription-activating factor PU.1 as well as its antagonist, the bHLH-domain transcriptional repressor Id2. PU.1 is needed for myeloid gene expression, which is repressed by pRb association. Nevertheless, homozygous deletion of the Rb gene caused reduced expression of specific myeloid genes, indicating a positive role for pRb in transcription. As a likely explanation, loss of Rb also disrupts pRb-mediated sequestering of Id2, and Id2 antagonizes PU.1-activated transcription in the absence of pRb.Citation55 What mechanisms control whether pRb associates with PU.1 or Id2 is currently not understood.
In addition to CKIs and pRb family members, targeted protein degradation plays an important role in coordinating cell cycle progression with differentiation. The APC/CCdh1 E3 ligase contributes to cell cycle arrest and muscle fiber formation of C2C12 cells. APC/CCdh1 targets both the cell cycle regulator Skp2 and the early myogenic differentiation factor Myf5 for degradation.Citation56 The degradation of Skp2 results in elevated levels of the CDK inhibitors p21Cip1 and p27Kip1, while degradation of Myf5 is required for myogenic fusion. Thus, by promoting cell cycle arrest and ensuring the presence of the correct combination of transcription factors, the APC/CCdh1 can act as a dual function regulator that links cell cycle exit to differentiation.Citation56
The examples above illustrate direct regulation of differentiation-related transcription factors by G1/S inhibitors. CIP/KIP inhibitors, pRb and APC/CCdh1 are all well-established inhibitors of G1 progression, which also promote differentiation through inhibition of G1 CDK-cyclins and lengthening of G1. Thus, regulators of the cell cycle contribute both directly and indirectly to regulation of cell differentiation, thereby coordinating cell cycle exit and terminal differentiation.
Differentiation-inducing transcription factors induce cell cycle arrest
To coordinate differentiation with cell cycle exit, the induction of cell type-specific genes needs to go along with altered expression of cell cycle regulators. Importantly, several transcription factors that promote differentiation of muscle cells, neurons, or blood cells also control expression of cell cycle genes. Such a dual function in transcriptional regulation was first observed for MyoD, which triggers expression of the cell cycle inhibitors p21Cip1 and p57Kip2.Citation57-59 Similar to muscle-specific genes, MyoD associates with p21Cip1 and p57Kip2 promoter sequences in committed precursor cells, but transcriptional activation only occurs coincident with differentiation.Citation60,10 Knockout of p21Cip1 and p57Kip2 impairs skeletal muscle development in vivo, with defective muscle fibers resembling those of myogenin knockout mice.Citation61 These data point to an essential role for MyoD-induced expression of CIP/KIP inhibitors in cell cycle arrest during muscle formation.
Similar mechanisms are likely used during differentiation of other cell types. For instance, 2 key transcription factors required for erythrocyte development and maturation, EKLF and GATA-1, directly control the expression levels of p21Cip1.Citation62,63 In addition, many studies have observed a correlation between cell type-specific transcription factors and cell cycle gene expression without examining direct regulation. For instance, the Drosophila neurogenic transcription factor Prospero, described above, promotes induction of neuron-specific genes together with inactivation of positive cell cycle genes. Prospero mutant embryos show continued proliferation despite initiation of early steps of neuronal differentiation. Loss of Prospero was found to coincide with severe upregulation of cyclin E, cyclin A, and String/Cdc25, while ectopic Prospero expression reduced expression of these positive cell cycle regulators.Citation64 While Prospero promotes cell cycle exit, it remains to be demonstrated whether it acts directly at the promoters of cell cycle genes. In contrast, a study in chicken spinal cords addressed whether Neurogenin 2 (Ngn2) directly regulates cell cycle genes.Citation65 Gene expression was characterized immediately following ectopic Ngn2 expression in early embryos, at the time of neuronal commitment and before full differentiation. This revealed Ngn2-dependent inhibition of patterning genes, induction of differentiation-related genes, and simultaneously reduced expression of positive cell cycle regulators. Cyclin E2 was identified as the only candidate direct target of transcriptional repression by Ngn2 at this early stage, while the strongly reduced expression of cyclin D and A-type cyclins appeared indirect.Citation65
Although further studies are needed, it is clear that multiple cell cycle regulators directly control differentiation, while several cell type-specific transcription regulators directly control cell cycle gene expression. By making the expression of differentiation-specific genes dependent on cell cycle regulators, and the activity of cell cycle regulators on differentiation factors, tight coordination between cell cycle exit and differentiation can be established.
Chromatin regulators coordinate cell cycle exit with differentiation
The transcriptional changes involved in differentiation are not achieved by transcription factors alone, but rather in coordination with a large variety of chromatin regulating factors. Gene transcription requires binding of transcription factors to regulatory DNA sequences in promoter and enhancer elements. Whether these transcription factors can bind and activate transcription depends on the chromatin structure. Open chromatin allows transcription factor and RNA polymerase access, whereas densely packed nucleosomes inhibit transcription. The chromatin composition and packaging of DNA is regulated by multiple mechanisms, such as DNA methylation, histone modification (including phosphorylation, methylation, acetylation and ubiquitylation), incorporation of histone variants and ATP-dependent chromatin remodeling.Citation66 Multi-subunit chromatin modification complexes covalently attach methyl groups and other modifications to DNA or histones, or remove such modifications. This either directly affects chromatin compaction or contributes to the binding of molecules that recognize or respond to these modifications. ATP-dependent chromatin remodelers, on the other hand, use the energy of ATP hydrolysis to exchange, move, or eject histones. Together, these regulators determine the accessibility of the DNA to transcription factors, co-regulators, and RNA polymerase complexes. Chromatin regulators that are particularly important in regulating cell cycle control, lineage commitment, and terminal differentiation include pRb-containing modification complexes and proteins of the Polycomb group and Trithorax group. We discuss these regulators below, with special attention for SWI/SNF ATPase-dependent chromatin remodeling complexes because of their central role in cell division and differentiation decisions.
pRb-dependent chromatin remodeling
Proteins of the pRb family promote the transcriptional silencing of cell cycle genes during differentiation. This is accomplished at least in part through association with E2F family proteins, thereby blocking activating E2Fs (E2F1, E2F2, E2F3a) and acting in conjunction with repressive E2Fs (E2F4, E2F5).Citation5 pRb has also been reported to associate with cell type-specific transcription factors, including MyoD, Myogenin, C/EBP, PU.1, NF-IL6, Pax-3, and AP-2.Citation54,67 While poorly understood, interactions of pRb family members with cell-type specific transcription factors may prevent transcriptional activation of differentiation-specific genes in proliferating cells, as described for Rb-E2F related complexes.Citation68 It is thought that pRb fulfills these transcriptional repressor functions in part via the recruitment of various chromatin regulators.
Chromatin regulatory proteins reported to associate with pRb include histone deacetylases (HDACs), ATPases of the SWI/SNF chromatin remodeling complex, histone methyltransferases (HMTs), the DNA methyltransferase DNMT1, and histone binding proteins such as HP1.Citation5,69-71 Whether these interactions, often detected by co-immunoprecipitation, reflect functional cooperation in vivo has beens difficult to proof. Perhaps best documented is the contribution of pRb complexes in histone deacetylation, which corresponds to pRb/E2F localization at gene regulatory sequences and contributes to transcriptional repression of cyclin E, cyclin A, and other E2F targets during cell cycle exit.Citation69,71 HDAC recruitment to pRb/E2F-associated promoters has been reported to depend on SWI/SNF function.Citation72 Genetic interactions in flies and worms support cooperation between the Rb pathway and SWI/SNF complex, likely by acting in parallel in transcriptional repression.Citation73–75
The histone methyltransferases (HMTs) found associated with pRb contribute to the formation of general gene silencing marks. This includes the HMT Suv39h1, which is responsible for trimethylation of lysine 9 of histone H3 (H3K9me3), a repressive histone methylation mark associated with heterochromatin. H3K9 trimethylation allows binding of the heterochromatin binding protein HP1, which is an important step in transcriptional repression. Notably, deacetylation, trimethylation and HP1 binding of H3K9 are 3 sequential steps in gene silencing, carried out by 3 different chromatin regulators found in association with pRb.Citation76 The HMTs Suv4–20 h1 and h2 also associate with pRb and are responsible for bi- and tri-methylation of H4K20. These methylations create a binding site for the lethal malignant brain tumor protein L(3)MBT.Citation77 L(3)MBT has been reported to form part of a silencing complex that contains pRb as well as a HP1 family member. Thus, through enzymatic activities and protein association, the chromatin regulators associated with pRb have the potential to induce chromatin compaction and repression of gene transcription. These combined activities likely contribute to the stable silencing of cell cycle genes during cell cycle exit.
DREAM/synMuvB
Genetic studies in C. elegans and biochemical experiments in Drosophila have identified another Rb-related repressor complex. C. elegans genes encoding components of this complex are needed to silence transcription of an epidermal growth factor (EGF) related gene in the epidermis. Only simultaneous mutation of genes of different classes (class A, B, or C genes) causes de-repression of lin-3 EGF and abnormal expression of EGF in the epidermis. This induces the formation of extra vulval tissue at abnormal positions. This multi-vulva phenotype is “synthetic” (synMuv), because it arises when mutations that individually do not cause a multivulva phenotype are combined. C. elegans Rb-related (LIN-35), E2F-like (EFL-1), DP-like (DPL-1) and some additional conserved transcriptional regulatory proteins all belong to the same synMuv B class, indicating that these proteins function within a complex or genetic pathway. Biochemical purification of Rb/E2F complexes from Drosophila embryos revealed a repressor complex containing fly Rb (RBF), E2F2, MYB and in addition 4 conserved synMuv B proteins.Citation78 This DREAM (Drosophila RBF, E2F and Myb) complex is not only similar to the C. elegans synMuv B complex, but also closely related to a mammalian complex that contains repressive E2F4 and E2F5 and the pRb-related protein p130.Citation79-81 Importantly, the components of these complexes remained associated through multiple biochemical purification steps, and were genetically identified because of their overlapping functions in C. elegans. Therefore, the DREAM and synMuvB RB/E2F-related protein complexes are likely physiologically relevant in vivo.
Surprisingly, the Drosophila and human DREAM complex did not appear to contain obvious histone modification activities. This could question whether chromatin regulators are truly required for the Rb-related repressor functions in vivo. An independently purified Myb-MuvB complex that largely corresponds to DREAM was found to contain associated Rpd3 HDAC and L(3)MBT proteins.Citation82 These proteins were not detected in Drosophila DREAM, human p130-E2F4/5, or C. elegans synMuv B complexes.Citation78-81 However, C. elegans hda-1 HDAC1, hpl-1 HP1 and lin-61 L(3)MBT all showed substantial phenotypic overlap with synMuv B genes, suggesting overlapping functions. Moreover, Drosophila DREAM bound specifically to deacetylated histone H4 tails, and L(3)MBT required Myb-MuvB/DREAM for its chromosomal recruitment.Citation78,83 The combined data support that DREAM/MuvB and chromatin modifying complexes function together in transcriptional repression and likely engage in low affinity physical interactions.
Only a subset of the C. elegans synMuv B genes contributes to cell cycle arrest.Citation84 In contrast, all prevent inappropriate lin-3 EGF expression and several synMuv B genes are needed for the silencing of germline-specific genes in somatic cells.Citation5,80,85 De-repression of germline genes in synMuv B mutants is most obvious in the intestine and skin, 2 tissues with continued S-phases and thus without cell cycle arrest during larval development. Similarly, Drosophila DREAM subunits are required for the permanent repression of sex- and differentiation-specific genes in proliferating S2 cells and embryos. Importantly, the fly dE2F2/RBF complexes remained chromatin bound in S phase when E2F1 is active and CDK activity high.Citation68,86 In contrast, mammalian p130-E2F4/5 DREAM mediates gene silencing specifically in quiescent or G0 arrested cells.Citation81 In summary, some Rb/E2F-related complexes repress cell cycle genes during cell cycle exit, while other Rb/E2F-related complexes repress cell type-specific gene expression in cycling cells.
Polycomb and trithorax group genes and developmental control of differentiation
Chromatin regulators of the Polycomb group (PcG) and the antagonistically acting Trithorax group (TrxG) have emerged as key players in cell cycle control, stem cell maintenance, cell fate determination and terminal differentiation.Citation87-89 PcG and TrxG genes were originally identified in Drosophila, through their roles in the maintenance of Homeobox (HOX) transcription factor expression. While established in the early embryo, HOX gene expression needs to be maintained during later developmental stages to determine the identity of body segments. Mutations in PcG genes result in expression of HOX genes outside the normal domains, which causes body segments to adopt the wrong identity. TrxG genes were defined through mutants that fail to maintain HOX gene expression, or as antagonists of PcG. Since, homologues of Drosophila PcG and TrxG genes have been found to be conserved in a wide variety of multicellular organisms and are now known to repress or activate, respectively, transcription of many genes.
PcG proteins form multi-subunit protein complexes that include the well-characterized Polycomb Repressor Complex 1 and 2. Both of these complexes contain a subunit with catalytic activity. The dRing protein of PRC1 catalyzes ubiquitylation of H2A at lysine residue K119. EZH (Enhancer of Zeste), one of the core subunits of the PRC2 complex, is an HMT responsible for the methylation of lysine 27 of histone H3 (H3K27).Citation90 Other PRC2 subunits are needed for this HMT activity, and one of the PRC1 subunits recognizes H3K27me3. Thus, PRC1 recruitment is likely coupled to PRC2 function and binding of H3K27me3 by PRC1 probably contributes to chromatin compaction.Citation89 Nevertheless, the relation between PRC1 and PRC2 activities and mechanisms of PcG-mediated transcriptional repression remain poorly understood.
Trithorax group proteins oppose PcG protein-mediated transcriptional repression and generally contribute to maintenance of an active chromatin state. TrxG proteins form a highly diverse class, which includes proteins with histone modifying activities, components of chromatin remodeling complexes and DNA binding proteins.Citation88 The best-known TrxG representative is the histone methyltransferase MLL1. MLL1 catalyzes histone H3K4 trimethylation (H3K4me3), which is associated with active transcription. Several MLL complexes also contain histone acetyltransferase (HAT) activity, while other TrxG proteins form a variety of ATP-dependent chromatin remodeling complexes, including SWI/SNF complexes (see below).
PcG and TrxG in differentiation
In light of proliferation versus differentiation control, PcG and TrxG genes are of particular interest because of their important contribution to lineage commitment. As embryonic stem cells become committed progenitors, remarkable changes in their chromatin structure occur that even can be observed by light and electron microscopy.Citation91 The chromatin structure of embryonic stem cells is generally open and allows transcriptional activation, while the chromatin of lineage-committed cells becomes more compact and less accessible to transcriptional activators. Nevertheless, maintenance of the pluripotent stem cell state requires that developmental genes remain silenced. This dual goal may be achieved by marking the regulatory regions of differentiation-related genes with bivalent domains, which contain both the activating H3K4me3 and inactivating H3K27me3 histone modifications.Citation92 While bivalent domains prevent gene transcription, they also maintain the potential for later activation. During differentiation, most of the bivalent domains lose one of the 2 modifications and become monovalent, leaving either activating or inactivating marks.Citation92,93
In addition to their role in lineage commitment, the antagonism between PcG and TrxG proteins is critical for terminal differentiation and cell cycle exit. The ink4A tumor suppressor locus provides the best-characterized example of direct cell cycle regulation by PcG and TrxG proteins. As first observed for the bmi1 PRC1 component, PcG protein overexpression results in repression of this locus, while PcG gene knockout in mice leads to increased expression of p16INK4A and p19ARF, and interferes with cell proliferation.Citation94-96 TrxG proteins counteract PcG repression of this ink4A locus, as is further described below.
Genome-wide analysis of human embryonic fibroblasts showed PcG protein binding and H3K27me3 modification of many genes involved in neuronal, bone, muscle, hematopoietic and epithelial differentiation.Citation97 During retinoic acid induced neuronal differentiation, PcG binding and the H3K27me3 marks of neuron-specific genes decreased progressively.Citation97,98 These and other results support that PcG-mediated gene silencing prevents premature expression of cell type-specific genes.
PcG-mediated repression can be reinstalled to allow progenitor cells to switch fate. This was demonstrated for the sequential production of neurons and astrocytes from neural precursor cells in the mouse neocortex. Conditional knockout of the mouse Ring1 PRC1 and EZH2 PRC2 genes interfered with downregulation of neurogenin1, and delayed the switch from divisions that produce neurons to those producing astrocytes.Citation99 Thus PcG-mediated gene repression is an important mechanism in the timing of developmental decisions. In contrast to PcG, the TrxG gene Mll1 promotes neurogenesis in the mouse post-natal brain.Citation100 Conditional knockout of Mll1 in neural precursor cells in the subventricular zone prevented their neuronal differentiation. The transcription factor Dlx2 was identified as the critical target of MLL1 in this process. The Dlx2 gene contains a bivalently marked promoter, which depends on MLL1 binding for activation.Citation100 This example illustrates how the combined activity of PcG and TrxG proteins determines the expression of genes that control important developmental transitions, including neuronal differentiation.
As shown for neurons, muscle differentiation also involves PcG protein-mediated chromatin remodeling. In proliferating mouse C2C12 myoblasts, H3K27me3 marks muscle-specific genes, while this silencing mark is absent from the promoters of cell cycle genes at this stage. This is in contrast to later stages of muscle differentiation, when differentiation genes become expressed and cell cycle genes acquire the H3K27me3 repressive mark.Citation101,102 Interestingly, Rb knockdown by RNAi caused loss of H3K27 trimethylation, re-expression of cell cycle genes, and cell cycle re-entry of terminally differentiated muscle cells.Citation102 Additional experimental evidence supports that silencing of cell cycle genes coincides with trimethylation of H3K27, and not H3K9 trimethylation, in this system.Citation102,103
PcG proteins may be recruited to MyoD-bound promoters in proliferating myoblasts to prevent premature differentiation. In agreement with this idea, ectopic expression of MyoD induced muscle differentiation of C. elegans precursor germ cells, but only when combined with knockdown of PcG-gene function.Citation104 ln mouse muscle formation, PcG gene expression is downregulated during terminal differentiation, while overexpression of the PcG protein EZH inhibits muscle differentiation.Citation101 Chromatin immunoprecipitation-sequencing (ChIP-seq) analysis of differentiating C2C12 cells revealed MyoD binding at the promoters of thousands of genes, of which only a subset showed altered expression during muscle differentiation.Citation60 Focusing on repressive chromatin marks in differentiating C2C12 cells, H3K27 trimethylation was strongly associated with gene silencing but different gene classes could be distinguished.Citation103 Genes induced during muscle differentiation required continuous PRC2 complex activity for transcriptional repression in myoblasts, and appeared to lack PRC1 at their regulatory sequences. In contrast, loci that remained continuously silenced during differentiation were associated with PRC1 and retained H3K27me3 even after PRC2 knockdown.Citation103 These and other findings indicate that PcG proteins act broadly as global transcriptional repressors, maintaining cellular homeostasis, suppressing cell type-specific gene expression, antagonizing cell cycle arrest and sustaining the undifferentiated state.
SWI/SNF dependent chromatin remodeling in differentiation and cell Cycle Regulation
The switching/sucrose non-fermenting (SWI/SNF) subclass of TrxG proteins currently receives much scientific attention. This was triggered by the recent discovery of an unexpectedly high frequency of mutations in SWI/SNF subunit genes in human cancer.Citation1,105,106 It has become clear that SWI/SNF complexes play critical roles in lineage commitment and terminal differentiation in a wide variety of tissues and cell-types.Citation107 SWI/SNF-complexes are found in all eukaryotes, predominantly promote transcriptional activation, and are needed for proper expression of approximately 6% of all genes in yeast.Citation108 They act as chromatin remodelers that use the energy of ATP hydrolysis to mobilize nucleosomes, thereby altering DNA accessibility and regulating transcription (). The complex consists of several core subunits, including the ATPase subunit Brahma (BRM) in Drosophila, and either BRM or BRM-related Gene 1 (BRG1) in mammals (). The other core components are the BRM/BRG1-associated factors (BAFs) BAF155, BAF170 and SNF5 (BAF47, INI1). These subunits appear present in all SWI/SNF complexes, and when combined as purified proteins reconstitute remodeling activity to the same level as the entire SWI/SNF complex.Citation109 In addition to this catalytically active core, SWI/SNF complexes contain a large number of associated subunits. Based on the associated subunits, 2 subclasses of SWI/SNF complexes have been distinguished, BAF and PBAF, which contain the BAF250/ARID1/Osa and BAF180/PBRM signature subunits, respectively ().
Importantly, several of the associated SWI/SNF subunits are encoded by multiple genes (). At least some of these variants show lineage-specific and differentiation-stage specific expression and complex association. For example, BAF45a and BAF53a need to be associated with the SWI/SNF complex in proliferating neuronal progenitors, but are exchanged for BAF45b/c and BAF53b when these cells exit the cell cycle and differentiate.Citation110 Moreover, 3 different genes encode BAF60 variants (), but BAF60c is the only subunit expressed in the mesodermal precursors of cardiomyocytes and myoblasts.Citation111 BAF60c interacts with the GATA4 cardiac transcription factor and with MyoD, and is required for the formation of differentiated heart and skeletal muscle. As such, BAF60c is currently the best-described example of a variant subunit that bridges the SWI/SNF complex to differentiation-inducing transcription factors.Citation24,111
The SWI/SNF complex might fulfill its role in differentiation by increasing DNA accessibility for transcription factor binding, or by binding transcription factors in order to locally change the chromatin to provide access for additional factors, co-activators and the RNA polymerase complex. Both mechanisms have been proposed for muscle differentiation. On the one hand, association of the SWI/SNF BRG1 subunit was reported to precede association of MyoD with the myogenin promoter.Citation112 In contrast, a more recent study found BRG1-SWI/SNF complex recruitment to follow induction of differentiation, after DNA binding of a preassembled complex of MyoD with the muscle-specific BAF60c subunit.Citation113 While not fully resolved, recruitment of SWI/SNF components may precede, coincide with or follow promoter binding of cell type-specific transcription factors, dependent on the target gene and cell-type.
SWI/SNF and cell-cycle arrest
SWI/SNF-mediated chromatin remodeling is critical not only for cell differentiation but also for coordinating cell cycle arrest with cell-type-specific gene expression. The SWI/SNF complex has been implicated in the regulation of cell cycle genes that include the INK4A-ARF locus, p21Cip1, cMyc, cyclin D, and cyclin E. At least some of these genes appear directly SWI/SNF regulated, as demonstrated in malignant rhabdoid tumor cells that contain inactivating mutations in the human SNF5/BAF47 gene. Reintroduction of SNF5 into these tumor cells resulted in upregulation of p16INK4a and p15INK4b expression and arrest of cell division.Citation114,115 SNF5 expression induced extensive chromatin remodeling and removal of PRC1 and PRC2 PcG complexes from the INK4a-INK4b locus. As described above, PcG-complexes silence the INK4A-ARF locus, hence PcG gene knockout and SNF5 activation both lead to increased expression of p16INK4A. In contrast, loss of SWI/SNF activity was found to result in PcG-mediated silencing of the INK4A locus, leading to continued proliferation and a stem cell-like signature.Citation114,115 These observations fit with the identification of SWI/SNF genes as members of the Trithorax group. Mechanistically, the SWI/SNF complex antagonizes PcG-mediated transcriptional repression by replacing PcG proteins at the promoters of their joint target genes.Citation115
Similar to the p16INK4A CDK4/6 inhibitor, expression of p21Cip1 is also directly controlled by the SWI/SNF complex. Down-regulation of several SWI/SNF components led to loss of p21Cip1 expression, while re-expression of SNF5 in malignant rhabdoid tumor cells induced p21Cip1 levels, independently of p53.Citation116,117 ChIP experiments showed strong p21Cip1 promoter association of SNF5, in the proximity of the transcription start site and together with RNA Polymerase II association. The BAF-specific subunit BAF250b expressed in HeLa cells also directly controlled p21Cip1 expression and induced cell cycle arrest.Citation118 These data support direct transcriptional activation of p21Cip1 by the SWI/SNF BAF complex, and indicate that p21Cip1 is an important mediator of SWI/SNF dependent cell cycle arrest.
The SWI/SNF complex predominantly acts as a transcriptional activator but has been reported to silence gene expression also. This could indirectly result from induction of a silencing factor, such as Hamlet in Drosophila neural stem cells.Citation119 Support for direct SWI/SNF-mediated transcriptional repression of cell cycle genes comes from studies in C. elegans. Chromatin immunoprecipitation experiments revealed SWI/SNF association with promoter regions of nearly all negative cell cycle regulators in C. elegans, as well as a subset of positive regulators.Citation74,120 Moreover, detection of mRNA expression during muscle differentiation demonstrated that induction of negative regulators and silencing of the positive regulators cyclin E and Cdk4 both require SWI/SNF function.Citation74 While the mechanism of repression remained unclear, mammalian SWI/SNF has been proposed to repress cyclin D and cyclin E through recruitment of HDAC1 and concomitant inhibition of transcription through histone deacetylation.Citation121 Nucleosome remodeling and HDAC recruitment to SWI/SNF-bound promoters may be used more broadly for SWI/SNF-mediated gene repression.Citation72,122
In summary, the chromatin-remodeling complexes described above contribute directly to the regulation of cell cycle genes and differentiation-associated genes. In particular the SWI/SNF complex acts as a dual regulator of general cell cycle genes and cell type-specific genes. As global transcriptional regulators of many cell cycle and differentiation genes, chromatin-remodeling complexes fulfill a critical role in coordinating cell cycle exit with terminal differentiation.
All-or-nothing transition from proliferating precursor to differentiated cell
Robust control over the proliferation versus differentiation decision likely results from redundant regulators and feedback control mechanisms (). For example, 3 Rb-related transcriptional repressors and 3 CIP/KIP family CDK inhibitors contribute to cell cycle exit in mammals. Redundancies are observed between the 2 families, and between members of each individual family of G1/S inhibitors. Notably, triple knockout mice missing the entire Rb or CIP/KIP family are viable until approximately day 10 or day 13 of gestation, respectively.Citation123,124 As many differentiated cell types and tissues have been formed at these stages, cell cycle exit and differentiation can occur in the absence of an entire class of cell cycle inhibitors. Our results in C. elegans emphasize this conclusion and showed that cell cycle exit of muscle precursors involves cooperation between at least 5 different levels of regulation, including Rb, SCFβTrCP, APC/CFZR1, CKI, and SWI/SNF complexes ().Citation74 These regulators display a high degree of redundancy, are individually not sufficient for reliable control, yet in combination create highly robust “all-or-nothing” decisions in cell cycle exit.
An important double negative feedback appears to operate in the proliferation-differentiation decision: G1 CDK-cyclins antagonize differentiation-inducing transcription factors, while these transcription factors in turn antagonize CDK-cyclin activation. The mutual antagonism between PcG and TrxG genes, in particular the TrxG-SWI/SNF class, is an integral part of this network. Several other levels of control likely help reinforce the 2 mutually exclusive states, such as regulation of protein degradation by E3 ubiquitin ligases, and regulation of mRNA stability by miRNAs. The mutual antagonism is expected to create bistability: cells either adopt a state of proliferation without differentiation, or shut down the cell cycle and acquire a differentiated state.
If this is true, how can cells make the transition from proliferating precursor cells to post-mitotic differentiated cells? Undergoing such a switch requires a substantial disruption of the initial equilibrium. This can be accomplished by overexpression of a single critical regulator, for instance as discussed for the ectopic expression of MyoD or CIP/KIP inhibitors. In vivo, developmental transitions are likely accomplished through coinciding alterations of multiple players. The trigger for these alterations comes from external signals, cell intrinsic information, or a combination of both. Many external factors can induce transitions, such as cytokines, mitogens, growth factors, Notch, Wnt/Wg, Hedgehog, and TGFβ-BMP ligands. Cell intrinsic information includes transcription regulators that become unequally distributed during asymmetric cell division. External or internal factors can disrupt an initially stable proliferative state by inducing expression of CDK inhibitors, suppression of cyclin transcription, activation of differentiation-inducing transcription factors, recruitment of chromatin modification complexes, and displacement of PcG proteins from promoters by SWI/SNF complexes, ultimately leading to cell cycle arrest and induction of cell type-specific gene expression.
Substantial disruption of a differentiation transcriptional program allows reversion to an undifferentiated proliferative state, as best illustrated by induction of pluripotent stem cells from post-mitotic neurons or mature lymphocytes.Citation125 At the same time, the mutual antagonism between cell cycle regulators and cell-type specific transcription precludes that proliferation and differentiation coincide. CDK2-cyclin E expression in differentiated C. elegans muscle cells induced expression of a defined set of genes with strongly enriched signatures of E2F targets and cell cycle functions.Citation11 CDK4-Cyclin D expression altered a broader set of genes, but still could not overcome silencing of critical cell cycle regulators. Thus, manipulation of cell cycle regulators readily alters the proliferation-differentiation decision in precursor cells, but controls that resist CDK-cyclin regulation appear to promote maintenance of the arrested state in terminally differentiated cells.
Nevertheless, some specific examples have been reported of coincident occurrence of cell division and a highly differentiated state. For instance, loss of Rb allowed proliferation of post-mitotic hair cells in the mouse inner ear, and part of these hair cells remained functionalCitation126 Similarly, conditional inactivation of multiple Rb-family members in the mouse eye caused terminally differentiated neurons to resume proliferation while maintaining a differentiated state.Citation127 In Drosophila, simultaneous activation of E2F and G1 CDK-cyclins caused overproliferation of terminally differentiated cells in the eyes and wingsCitation128,129 In addition, cardiac muscle cells are notoriously post-mitotic, but mouse HL-1 cells derived from cardiac muscle combine proliferation with maintenance of a differentiated phenotype. Even normal cardiomyocytes can proliferate to promote regeneration of the neonatal mouse heart, a capacity that is lost soon after birth.Citation130,131 Much insight may be gained from understanding how the feedback mechanisms that coordinate cell cycle exit and differentiation are overcome in these specific situations.
Switching from proliferation to differentiation
While some mechanisms apply to differentiation in general, much of the regulation is tissue specific, which greatly adds to the complexity of the proliferation-differentiation decision. Summarizing some of the insight in muscle fiber formation from proliferating myoblasts illustrates this point (). Myogenic-regulatory factors act in a switch-like circuitry, with external proliferation and differentiation-inducing factors determining expression of MRFs as well as the switch from transcriptional repression to activation. Growth factors and mitogenic signals prevent MyoD-induced activation of muscle-specific genes in proliferating myoblasts. As described above, this involves phosphorylation of MyoD by CDK2-cyclin E, as well as phosphorylation of the collaborating MEF2 transcription factor by CDK4/6-cyclin D.Citation32-37 Reduction of growth factors allows differentiation, in part through downregulation of the inhibitory phosphorylations. Simultaneously, differentiation signals induce p38 MAPK activation with critical regulatory functions in muscle formation (). In proliferating C2C12 myoblasts, MyoD and MEF2 are already present at the promoters of many differentiation induced genes, and MyoD is associated with the BAF60c SWI/SNF subunit.Citation113 Activated p38 MAPK phosphorylates BAF60c, which triggers the assembly of a functional SWI/SNF complex onto the preassembled MyoD/BAF60c pioneer complex.Citation113 In addition, p38 MAPK phosphorylates MEK2, which induces recruitment of an MLL2-menin TrxG activation complex.Citation132 An additional p38 MAPK pathway leads to phosphorylation of a scaffolding protein, KAP1, associated with MyoD-E12 and MEF2 transcription factors, release of co-repressors and recruitment of co-activators at target gene promoters.Citation38 Thus, growth factor reduction and differentiation-inducing signals both contribute to differentiation by activating transcription from MyoD-MEF2 occupied promoters.
In conclusion, a regulatory network of cell cycle regulators, transcription factors, lineage-specific SWI/SNF complexes and chromatin modification complexes induce the all-or-nothing transition from proliferating precursors to differentiated post-mitotic cells (). While complex, a deeper understanding of these regulatory networks and their cell type-specific variations has the potential to lead to improved possibilities for regenerative medicine and cancer treatment.
Proliferation vs. differentiation in cancer
The mechanisms that maintain the balance between cell proliferation and differentiation are often compromised in cancer cells, leading to unperturbed proliferation and a failure to differentiate.Citation133 Nevertheless, the link between proliferation and differentiation still seems to exist, at least in some cancers. Overexpression of cell cycle inhibitors can be sufficient for differentiation induction. For instance, overexpression of the CDK inhibitors p21Cip1, p27Kip1, p19ARF and p16INK4A inhibited proliferation and simultaneously induced differentiation in a variety of tumor cells.Citation134-136 Similarly, modulating the activity of transcription factors can promote both differentiation and cell cycle arrest and has been used in the treatment of leukemia.Citation137 These examples demonstrate that the tight link between proliferation and differentiation can still exist in malignancies, with differentiation following from inhibition of the cell cycle, and cell cycle arrest resulting from differentiation induction.
While transcriptional regulators are becoming attractive targets in anti-cancer therapy, tissue- and cell type-specific differences complicate a general therapeutic strategy. The contribution of the transcription factor PU.1 in acute myeloid leukemia (AML) and mouse erythroleukemia (MEL) illustrates this point. PU.1 is required for the generation of myeloid and common lymphoid precursor cells.Citation137 Early loss of PU.1 can prevent myeloid differentiation, allowing continuous proliferation of precursor cells without differentiation and contributing to cancer development. However, PU.1 has an opposite role during erythroid differentiation, where it prevents differentiation into mature erythrocytes by inhibiting the transcription factor GATA-1. In agreement, loss of PU.1 stimulated MEL cells to re-enter a differentiation program and to undergo terminal growth arrest.
The contribution and composition of PcG complexes is also cell type dependent. Deregulation of PcG genes is strongly associated with cancer, in agreement with their roles in stem cell maintenance, inhibition of differentiation, and transcriptional repression of the INK4 locus. PcG genes act as oncogenes and have been found upregulated in some cancers, while downregulated and showing tumor suppressor activity in others.Citation138 Context-dependent contributions of critical PcG target genes likely explain this discrepancy.
While various SWI/SNF subunits are established tumor suppressors, the mutation frequency of individual subunits shows remarkable variation dependent on the cancer type. Biallelic inactivation of the SNF5/SMARCB1 core subunit is found almost invariably in malignant rhabdoid tumors, and likely is the single causative mutation in these aggressive pediatric malignancies.Citation106 However, mutation of SNF5 or of the SMARCC1 (BAF155) and SMARCC2 (BAF170) core subunits is not common in other cancer types.Citation1,105,139 The BRG1 (SMARCA4) enzymatic subunit shows substantial mutation frequencies, in particular in melanomas. Overall, ARID1A (BAF250a) is the most commonly mutated SWI/SNF subunit gene, with particularly high inactivation rates in a subset of bladder and endometrial carcinomas, as well as ovarian and renal clear cell carcinoma. While ARID1A is specifically associated with the BAF complex, genes encoding the PBAF-specific subunits PBRM and ARID2 also show significant mutation frequencies. Collectively, SWI/SNF subunit genes have been found to be mutated in nearly 20% of all tumors reported in a large number of studies, placing them among the most frequently mutated genes in human cancer.Citation1,105,106,139,140
The subunit specific mutation frequencies may reside in selective pressure against cell type-specific functions in cell cycle arrest and differentiation, while complete elimination of SWI/SNF function may be detrimental or even cell lethal. Gene alterations that are mutually exclusive with inactivation of SWI/SNF subunits, affecting other chromatin remodeling components, cell type-specific transcription factors or cell cycle regulators, could help identify critical tumor suppressor functions of SWI/SNF complexes. However, SWI/SNF-mediated chromatin remodeling provides global transcriptional regulation, hence, its loss of function is unlikely to create all-or-nothing gene expression changes. Therefore, cooperation with oncogenic or tumor suppressor mutations in other transcriptional regulators and cell cycle genes can still be expected, as supported by recent results in C. elegans.Citation74
Does disrupted SWI/SNF function create cancer vulnerabilities and possibilities for treatment? Loss of SNF5 has been shown to result in downregulation of the INK4A locus and upregulation of cyclin D1 expression. Pharmacological inhibition or genetic ablation of cyclin D1 in rhabdoid tumor cells inhibited tumor growth, indicating that the elevated levels of cyclin D in these tumors contribute to their uncontrolled proliferation.Citation141-144 Based on the intimate connection between proliferation and differentiation, simultaneous targeting of cell cycle and transcriptional regulators may provide efficient and specific future anti-cancer therapies.
Disclosure of potential conflicts of interest
The authors declare no conflicting financial interests.
Acknowledgments
The authors thank A. Thomas and I. The for critically reading the manuscript.
Funding
This work was supported by the Netherlands Organization for Scientific Research (NWO), research program 819.02.016, and Worldwide Cancer Research grant 14–1294 to SvdH.
Related Research Data
References
- Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, et al. Mutational landscape and significance across 12 major cancer types. Nature 2013; 502:333-9; http://dx.doi.org/10.1038/nature12634
- Blagosklonny MV, Pardee AB. The restriction point of the cell cycle. Cell Cycle 2002; 1:103-10
- Spencer SL, Cappell SD, Tsai F-C, Overton KW, Wang CL, Meyer T. The proliferation-quiescence decision is controlled by a bifurcation in CDK2 activity at mitotic exit. Cell 2013; 155:369-83; http://dx.doi.org/10.1016/j.cell.2013.08.062
- Choi YJ, Anders L. Signaling through cyclin D-dependent kinases. Oncogene 2014; 33:1890-903; http://dx.doi.org/10.1038/onc.2013.137
- Van den Heuvel S, Dyson NJ. Conserved functions of the pRB and E2F families. Nat Rev Mol Cell Biol 2008; 9:713-24; http://dx.doi.org/10.1038/nrm2469
- Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev 1999; 13:1501-12; http://dx.doi.org/10.1101/gad.13.12.1501
- Bassermann F, Eichner R, Pagano M. The ubiquitin proteasome system - implications for cell cycle control and the targeted treatment of cancer. Biochim Biophys Acta 2014; 1843:150-62; http://dx.doi.org/10.1016/j.bbamcr.2013.02.028
- Coller HA, Sang L, Roberts JM. A new description of cellular quiescence. PLoS Biol 2006; 4:e83; http://dx.doi.org/10.1371/journal.pbio.0040083
- Sang L, Coller HA, Roberts JM. Control of the reversibility of cellular quiescence by the transcriptional repressor HES1. Science 2008; 321:1095-100; http://dx.doi.org/10.1126/science.1155998
- Zalc A, Hayashi S, Auradé F, Bröhl D, Chang T, Mademtzoglou D, Mourikis P, Yao Z, Cao Y, Birchmeier C, et al. Antagonistic regulation of p57kip2 by Hes/Hey downstream of Notch signaling and muscle regulatory factors regulates skeletal muscle growth arrest. Dev 2014; 141:2780-90
- Korzelius J, The I, Ruijtenberg S, Prinsen MBW, Portegijs V, Middelkoop TC, Groot Koerkamp MJ, Holstege FCP, Boxem M, van den Heuvel S. Caenorhabditis elegans cyclin D/CDK4 and cyclin E/CDK2 induce distinct cell cycle re-entry programs in differentiated muscle cells. PLoS Genet 2011; 7:e1002362; http://dx.doi.org/10.1371/journal.pgen.1002362
- Mummery CL, van den Brink CE, de Laat SW. Commitment to differentiation induced by retinoic acid in P19 embryonal carcinoma cells is cell cycle dependent. Dev Biol 1987; 121:10-9; http://dx.doi.org/10.1016/0012-1606(87)90133-3
- Mac Auley A, Werb Z, Mirkes PE. Characterization of the unusually rapid cell cycles during rat gastrulation. Dev 1993; 117:873-83
- Ciemerych MA, Sicinski P. Cell cycle in mouse development. Oncogene 2005; 24:2877-98; http://dx.doi.org/10.1038/sj.onc.1208608
- Filipczyk AA, Laslett AL, Mummery C, Pera MF. Differentiation is coupled to changes in the cell cycle regulatory apparatus of human embryonic stem cells. Stem Cell Res 2007; 1:45-60; http://dx.doi.org/10.1016/j.scr.2007.09.002
- Lange C, Calegari F. Cdks and cyclins link G1 length and differentiation of embryonic, neural and hematopoietic stem cells. Cell Cycle 2010; 9:1893-900; http://dx.doi.org/10.4161/cc.9.10.11598
- Stead E, White J, Faast R, Conn S, Goldstone S, Rathjen J, Dhingra U, Rathjen P, Walker D, Dalton S. Pluripotent cell division cycles are driven by ectopic Cdk2, cyclin A/E and E2F activities. Oncogene 2002; 21:8320-33; http://dx.doi.org/10.1038/sj.onc.1206015
- Calder A, Roth-Albin I, Bhatia S, Pilquil C, Lee JH, Bhatia M, Levadoux-Martin M, McNicol J, Russell J, Collins T, et al. Lengthened G1 phase indicates differentiation status in human embryonic stem cells. Stem Cells Dev 2013; 22:279-95; http://dx.doi.org/10.1089/scd.2012.0168
- Coronado D, Godet M, Bourillot P-Y, Tapponnier Y, Bernat A, Petit M, Afanassieff M, Markossian S, Malashicheva A, Iacone R, et al. A short G1 phase is an intrinsic determinant of naïve embryonic stem cell pluripotency. Stem Cell Res 2013; 10:118-31; http://dx.doi.org/10.1016/j.scr.2012.10.004
- Sela Y, Molotski N, Golan S, Itskovitz-Eldor J, Soen Y. Human embryonic stem cells exhibit increased propensity to differentiate during the G1 phase prior to phosphorylation of retinoblastoma protein. Stem Cells 2012; 30:1097-108; http://dx.doi.org/10.1002/stem.1078
- Lange C, Huttner WB, Calegari F. Cdk4/cyclinD1 overexpression in neural stem cells shortens G1, delays neurogenesis, and promotes the generation and expansion of basal progenitors. Cell Stem Cell 2009; 5:320-31; http://dx.doi.org/10.1016/j.stem.2009.05.026
- Calegari F, Huttner WB. An inhibition of cyclin-dependent kinases that lengthens, but does not arrest, neuroepithelial cell cycle induces premature neurogenesis. J Cell Sci 2003; 116:4947-55; http://dx.doi.org/10.1242/jcs.00825
- Pauklin S, Vallier L. The cell-cycle state of stem cells determines cell fate propensity. Cell 2013; 155:135-47; http://dx.doi.org/10.1016/j.cell.2013.08.031
- Puri PL, Mercola M. BAF60 A, B, and Cs of muscle determination and renewal. Genes Dev 2012; 26:2673-83; http://dx.doi.org/10.1101/gad.207415.112
- Braun T, Gautel M. Transcriptional mechanisms regulating skeletal muscle differentiation, growth and homeostasis. Nat Rev Mol Cell Biol 2011; 12:349-61; http://dx.doi.org/10.1038/nrm3118
- Buckingham M, Rigby PWJ. Gene regulatory networks and transcriptional mechanisms that control myogenesis. Dev Cell 2014; 28:225-38; http://dx.doi.org/10.1016/j.devcel.2013.12.020
- Weintraub H, Tapscott SJ, Davis RL, Thayer MJ, Adam MA, Lassar AB, Miller AD. Activation of muscle-specific genes in pigment, nerve, fat, liver, and fibroblast cell lines by forced expression of MyoD. Proc Natl Acad Sci U S A 1989; 86:5434-8; http://dx.doi.org/10.1073/pnas.86.14.5434
- Skapek SX, Rhee J, Spicer DB, Lassar AB. Inhibition of myogenic differentiation in proliferating myoblasts by cyclin D1-dependent kinase. Science 1995; 267:1022-4; http://dx.doi.org/10.1126/science.7863328
- Rao SS, Chu C, Kohtz DS. Ectopic expression of cyclin D1 prevents activation of gene transcription by myogenic basic helix-loop-helix regulators. Mol Cell Biol 1994; 14:5259-67; http://dx.doi.org/10.1128/MCB.14.8.5259
- Guo K, Walsh K. Inhibition of myogenesis by multiple cyclin-Cdk complexes. Coordinate regulation of myogenesis and cell cycle activity at the level of E2F. J Biol Chem 1997; 272:791-7; http://dx.doi.org/10.1074/jbc.272.2.791
- Zhang JM, Zhao X, Wei Q, Paterson BM. Direct inhibition of G(1) cdk kinase activity by MyoD promotes myoblast cell cycle withdrawal and terminal differentiation. EMBO J 1999; 18:6983-93; http://dx.doi.org/10.1093/emboj/18.24.6983
- Di Giorgio E, Gagliostro E, Clocchiatti A, Brancolini C. The control operated by the cell cycle machinery on MEF2 stability contributes to the downregulation of CDKN1A and entry into S phase. Mol Cell Biol 2015; 35:1633-47; http://dx.doi.org/10.1128/MCB.01461-14
- Lazaro J-B, Bailey PJ, Lassar AB. Cyclin D-cdk4 activity modulates the subnuclear localization and interaction of MEF2 with SRC-family coactivators during skeletal muscle differentiation. Genes Dev 2002; 16:1792-805; http://dx.doi.org/10.1101/gad.U-9988R
- Kitzmann M, Vandromme M, Schaeffer V, Carnac G, Labbé JC, Lamb N, Fernandez A. cdk1- and cdk2-mediated phosphorylation of MyoD Ser200 in growing C2 myoblasts: role in modulating MyoD half-life and myogenic activity. Mol Cell Biol 1999; 19:3167-76.http://dx.doi.org/10.1128/MCB.19.4.3167
- Reynaud EG, Pelpel K, Guillier M, Leibovitch MP, Leibovitch SA. p57(Kip2) stabilizes the MyoD protein by inhibiting cyclin E-Cdk2 kinase activity in growing myoblasts. Mol Cell Biol 1999; 19:7621-9; http://dx.doi.org/10.1128/MCB.19.11.7621
- Tintignac LA, Leibovitch MP, Kitzmann M, Fernandez A, Ducommun B, Meijer L, Leibovitch SA. Cyclin E-cdk2 phosphorylation promotes late G1-phase degradation of MyoD in muscle cells. Exp Cell Res 2000; 259:300-7; http://dx.doi.org/10.1006/excr.2000.4973
- Song A, Wang Q, Goebl MG, Harrington MA. Phosphorylation of nuclear MyoD is required for its rapid degradation. Mol Cell Biol 1998; 18:4994-9; http://dx.doi.org/10.1128/MCB.18.9.4994
- Singh K, Cassano M, Planet E, Sebastian S, Jang SM, Sohi G, Faralli H, Choi J, Youn H-D, Dilworth FJ, et al. A KAP1 phosphorylation switch controls MyoD function during skeletal muscle differentiation. Genes Dev 2015; 29:513-25; http://dx.doi.org/10.1101/gad.254532.114
- Dehay C, Kennedy H. Cell-cycle control and cortical development. Nat Rev Neurosci 2007; 8:438-50; http://dx.doi.org/10.1038/nrn2097
- Holland EC. Gliomagenesis: genetic alterations and mouse models. Nat Rev Genet 2001; 2:120-9; http://dx.doi.org/10.1038/35052535
- Paridaen JTML, Huttner WB. Neurogenesis during development of the vertebrate central nervous system. EMBO Rep 2014; 15:351-64; http://dx.doi.org/10.1002/embr.201438447
- Wilkinson G, Dennis D, Schuurmans C. Proneural genes in neocortical development. Neuroscience 2013; 253:256-73; http://dx.doi.org/10.1016/j.neuroscience.2013.08.029
- Ali F, Hindley C, McDowell G, Deibler R, Jones A, Kirschner M, Guillemot F, Philpott A. Cell cycle-regulated multi-site phosphorylation of Neurogenin 2 coordinates cell cycling with differentiation during neurogenesis. Dev 2011; 138:4267-77
- Hindley C, Ali F, McDowell G, Cheng K, Jones A, Guillemot F, Philpott A. Post-translational modification of Ngn2 differentially affects transcription of distinct targets to regulate the balance between progenitor maintenance and differentiation. Dev 2012; 139:1718-23
- Hardwick LJA, Philpott A. Nervous decision-making: to divide or differentiate. Trends Genet TIG 2014; 30:254-61; http://dx.doi.org/10.1016/j.tig.2014.04.001
- Homem CCF, Knoblich JA. Drosophila neuroblasts: a model for stem cell biology. Dev 2012; 139:4297-310
- Berger C, Pallavi SK, Prasad M, Shashidhara LS, Technau GM. A critical role for cyclin E in cell fate determination in the central nervous system of Drosophila melanogaster. Nat Cell Biol 2005; 7:56-62; http://dx.doi.org/10.1038/ncb1203
- Berger C, Kannan R, Myneni S, Renner S, Shashidhara LS, Technau GM. Cell cycle independent role of Cyclin E during neural cell fate specification in Drosophila is mediated by its regulation of Prospero function. Dev Biol 2010; 337:415-24; http://dx.doi.org/10.1016/j.ydbio.2009.11.012
- Fujita M, Takeshita H, Sawa H. Cyclin E and CDK2 repress the terminal differentiation of quiescent cells after asymmetric division in C. elegans. PloS One 2007; 2:e407; http://dx.doi.org/10.1371/journal.pone.0000407
- Jeong J, Verheyden JM, Kimble J. Cyclin E and Cdk2 control GLD-1, the mitosis/meiosis decision, and germline stem cells in Caenorhabditis elegans. PLoS Genet 2011; 7:e1001348; http://dx.doi.org/10.1371/journal.pgen.1001348
- Fox PM, Vought VE, Hanazawa M, Lee M-H, Maine EM, Schedl T. Cyclin E and CDK-2 regulate proliferative cell fate and cell cycle progression in the C. elegans germline. Dev 2011; 138:2223-34
- Biedermann B, Wright J, Senften M, Kalchhauser I, Sarathy G, Lee M-H, Ciosk R. Translational repression of cyclin E prevents precocious mitosis and embryonic gene activation during C. elegans meiosis. Dev Cell 2009; 17:355-64; http://dx.doi.org/10.1016/j.devcel.2009.08.003
- Zezula J, Casaccia-Bonnefil P, Ezhevsky SA, Osterhout DJ, Levine JM, Dowdy SF, Chao M V, Koff A. p21cip1 is required for the differentiation of oligodendrocytes independently of cell cycle withdrawal. EMBO Rep 2001; 2:27-34; http://dx.doi.org/10.1093/embo-reports/kve008
- Korenjak M, Brehm A. E2F-Rb complexes regulating transcription of genes important for differentiation and development. Curr Opin Genet Dev 2005; 15:520-7; http://dx.doi.org/10.1016/j.gde.2005.07.001
- Iavarone A, King ER, Dai X-M, Leone G, Stanley ER, Lasorella A. Retinoblastoma promotes definitive erythropoiesis by repressing Id2 in fetal liver macrophages. Nature 2004; 432:1040-5; http://dx.doi.org/10.1038/nature03068
- Li W, Wu G, Wan Y. The dual effects of Cdh1/APC in myogenesis. FASEB J 2007; 21:3606-17
- Busanello A, Battistelli C, Carbone M, Mostocotto C, Maione R. MyoD regulates p57kip2 expression by interacting with a distant cis-element and modifying a higher order chromatin structure. Nucleic Acids Res 2012; 40:8266-75; http://dx.doi.org/10.1093/nar/gks619
- Halevy O, Novitch BG, Spicer DB, Skapek SX, Rhee J, Hannon GJ, Beach D, Lassar AB. Correlation of terminal cell cycle arrest of skeletal muscle with induction of p21 by MyoD. Science 1995; 267:1018-21; http://dx.doi.org/10.1126/science.7863327
- Parker SB, Eichele G, Zhang P, Rawls A, Sands AT, Bradley A, Olson EN, Harper JW, Elledge SJ. p53-independent expression of p21Cip1 in muscle and other terminally differentiating cells. Science 1995; 267:1024-7; http://dx.doi.org/10.1126/science.7863329
- Cao Y, Yao Z, Sarkar D, Lawrence M, Sanchez GJ, Parker MH, MacQuarrie KL, Davison J, Morgan MT, Ruzzo WL, et al. Genome-wide MyoD binding in skeletal muscle cells: a potential for broad cellular reprogramming. Dev Cell 2010; 18:662-74; http://dx.doi.org/10.1016/j.devcel.2010.02.014
- Zhang P, Wong C, Liu D, Finegold M, Harper JW, Elledge SJ. p21(CIP1) and p57(KIP2) control muscle differentiation at the myogenin step. Genes Dev 1999; 13:213-24; http://dx.doi.org/10.1101/gad.13.2.213
- Papetti M, Wontakal SN, Stopka T, Skoultchi AI. GATA-1 directly regulates p21 gene expression during erythroid differentiation. Cell Cycle 2010; 9:1972-80; http://dx.doi.org/10.4161/cc.9.10.11602
- Siatecka M, Lohmann F, Bao S, Bieker JJ. EKLF directly activates the p21WAF1/CIP1 gene by proximal promoter and novel intronic regulatory regions during erythroid differentiation. Mol Cell Biol 2010; 30:2811-22; http://dx.doi.org/10.1128/MCB.01016-09
- Li L, Vaessin H. Pan-neural Prospero terminates cell proliferation during Drosophila neurogenesis. Genes Dev 2000; 14:147-51
- Lacomme M, Liaubet L, Pituello F, Bel-Vialar S. NEUROG2 drives cell cycle exit of neuronal precursors by specifically repressing a subset of cyclins acting at the G1 and S phases of the cell cycle. Mol Cell Biol 2012; 32:2596-607; http://dx.doi.org/10.1128/MCB.06745-11
- Chen T, Dent SYR. Chromatin modifiers and remodellers: regulators of cellular differentiation. Nat Rev Genet 2014; 15:93-106; http://dx.doi.org/10.1038/nrg3607
- Morris EJ, Dyson NJ. Retinoblastoma protein partners. Adv Cancer Res 2001; 82:1-54; http://dx.doi.org/10.1016/S0065-230X(01)82001-7
- Dimova DK, Stevaux O, Frolov M V, Dyson NJ. Cell cycle-dependent and cell cycle-independent control of transcription by the Drosophila E2F/RB pathway. Genes Dev 2003; 17:2308-20; http://dx.doi.org/10.1101/gad.1116703
- Gordon GM, Du W. Conserved RB functions in development and tumor suppression. Protein Cell 2011; 2:864-78; http://dx.doi.org/10.1007/s13238-011-1117-z
- Sadasivam S, DeCaprio JA. The DREAM complex: master coordinator of cell cycle-dependent gene expression. Nat Rev Cancer 2013; 13:585-95; http://dx.doi.org/10.1038/nrc3556
- Talluri S, Dick FA. Regulation of transcription and chromatin structure by pRB: here, there and everywhere. Cell Cycle 2012; 11:3189-98; http://dx.doi.org/10.4161/cc.21263
- Gunawardena RW, Fox SR, Siddiqui H, Knudsen ES. SWI/SNF activity is required for the repression of deoxyribonucleotide triphosphate metabolic enzymes via the recruitment of mSin3B. J Biol Chem 2007; 282:20116-23; http://dx.doi.org/10.1074/jbc.M701406200
- Cui M, Fay DS, Han M. lin-35/Rb Cooperates With the SWI/SNF Complex to Control Caenorhabditis elegans Larval Development. Genetics 2004; 167:1177-85; http://dx.doi.org/10.1534/genetics.103.024554
- Ruijtenberg S, van den Heuvel S. G1/S Inhibitors and the SWI/SNF Complex Control Cell-Cycle Exit during Muscle Differentiation. Cell 2015; 162:300-13; http://dx.doi.org/10.1016/j.cell.2015.06.013
- Staehling-Hampton K, Ciampa PJ, Brook A, Dyson N. A genetic screen for modifiers of E2F in Drosophila melanogaster. Genetics 1999; 153:275-87
- Nielsen SJ, Schneider R, Bauer UM, Bannister AJ, Morrison A, O'Carroll D, Firestein R, Cleary M, Jenuwein T, Herrera RE, et al. Rb targets histone H3 methylation and HP1 to promoters. Nature 2001; 412:561-5; http://dx.doi.org/10.1038/35087620
- Trojer P, Li G, Sims RJ, Vaquero A, Kalakonda N, Boccuni P, Lee D, Erdjument-Bromage H, Tempst P, Nimer SD, et al. L3MBTL1, a histone-methylation-dependent chromatin lock. Cell 2007; 129:915-28; http://dx.doi.org/10.1016/j.cell.2007.03.048
- Korenjak M, Taylor-Harding B, Binné UK, Satterlee JS, Stevaux O, Aasland R, White-Cooper H, Dyson N, Brehm A. Native E2F/RBF complexes contain Myb-interacting proteins and repress transcription of developmentally controlled E2F target genes. Cell 2004; 119:181-93; http://dx.doi.org/10.1016/j.cell.2004.09.034
- Harrison MM, Ceol CJ, Lu X, Horvitz HR. Some C. elegans class B synthetic multivulva proteins encode a conserved LIN-35 Rb-containing complex distinct from a NuRD-like complex. Proc Natl Acad Sci U S A 2006; 103:16782-7; http://dx.doi.org/10.1073/pnas.0608461103
- Harrison MM, Lu X, Horvitz HR. LIN-61, one of two Caenorhabditis elegans malignant-brain-tumor-repeat-containing proteins, acts with the DRM and NuRD-like protein complexes in vulval development but not in certain other biological processes. Genetics 2007; 176:255-71; http://dx.doi.org/10.1534/genetics.106.069633
- Litovchick L, Sadasivam S, Florens L, Zhu X, Swanson SK, Velmurugan S, Chen R, Washburn MP, Liu XS, DeCaprio JA. Evolutionarily conserved multisubunit RBL2/p130 and E2F4 protein complex represses human cell cycle-dependent genes in quiescence. Mol Cell 2007; 26:539-51; http://dx.doi.org/10.1016/j.molcel.2007.04.015
- Lewis PW, Beall EL, Fleischer TC, Georlette D, Link AJ, Botchan MR. Identification of a Drosophila Myb-E2F2/RBF transcriptional repressor complex. Genes Dev 2004; 18:2929-40; http://dx.doi.org/10.1101/gad.1255204
- Blanchard DP, Georlette D, Antoszewski L, Botchan MR. Chromatin reader L(3)mbt requires the Myb-MuvB/DREAM transcriptional regulatory complex for chromosomal recruitment. Proc Natl Acad Sci U S A 2014; 111:E4234-43; http://dx.doi.org/10.1073/pnas.1416321111
- Boxem M, van den Heuvel S. C. elegans class B synthetic multivulva genes act in G(1) regulation. Curr Biol CB 2002; 12:906-11; http://dx.doi.org/10.1016/S0960-9822(02)00844-8
- Petrella LN, Wang W, Spike CA, Rechtsteiner A, Reinke V, Strome S. synMuv B proteins antagonize germline fate in the intestine and ensure C. elegans survival. Dev 2011; 138:1069-79
- Dimova DK, Dyson NJ. The E2F transcriptional network: old acquaintances with new faces. Oncogene 2005; 24:2810-26; http://dx.doi.org/10.1038/sj.onc.1208612
- Schuettengruber B, Chourrout D, Vervoort M, Leblanc B, Cavalli G. Genome regulation by polycomb and trithorax proteins. Cell 2007; 128:735-45; http://dx.doi.org/10.1016/j.cell.2007.02.009
- Schuettengruber B, Martinez A-M, Iovino N, Cavalli G. Trithorax group proteins: switching genes on and keeping them active. Nat Rev Mol Cell Biol 2011; 12:799-814; http://dx.doi.org/10.1038/nrm3230
- Laugesen A, Helin K. Chromatin Repressive Complexes in Stem Cells, Development, and Cancer. Cell Stem Cell 2014; 14:735-51; http://dx.doi.org/10.1016/j.stem.2014.05.006
- Müller J, Hart CM, Francis NJ, Vargas ML, Sengupta A, Wild B, Miller EL, O'Connor MB, Kingston RE, Simon JA. Histone methyltransferase activity of a Drosophila Polycomb group repressor complex. Cell 2002; 111:197-208; http://dx.doi.org/10.1016/S0092-8674(02)00976-5
- Efroni S, Duttagupta R, Cheng J, Dehghani H, Hoeppner DJ, Dash C, Bazett-Jones DP, Le Grice S, McKay RDG, Buetow KH, et al. Global transcription in pluripotent embryonic stem cells. Cell Stem Cell 2008; 2:437-47; PMID:18462694; http://dx.doi.org/10.1016/j.stem.2008.03.021
- Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006; 125:315-26; PMID:16630819; http://dx.doi.org/10.1016/j.cell.2006.02.041
- Voigt P, Tee W-W, Reinberg D. A double take on bivalent promoters. Genes Dev 2013; 27:1318-38; PMID:23788621; http://dx.doi.org/10.1101/gad.219626.113
- Jacobs JJ, Kieboom K, Marino S, DePinho RA, van Lohuizen M. The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature 1999; 397:164-8; PMID:9923679; http://dx.doi.org/10.1038/16476
- Martinez A-M, Cavalli G. The role of Polycomb Group Proteins in Cell Cycle Regulation During Development. Cell Cycle 2014; 5:1189-97; http://dx.doi.org/10.4161/cc.5.11.2781
- Voncken JW, Roelen BAJ, Roefs M, de Vries S, Verhoeven E, Marino S, Deschamps J, van Lohuizen M. Rnf2 (Ring1b) deficiency causes gastrulation arrest and cell cycle inhibition. Proc Natl Acad Sci U S A 2003; 100:2468-73; PMID:12589020; http://dx.doi.org/10.1073/pnas.0434312100
- Bracken AP, Dietrich N, Pasini D, Hansen KH, Helin K. Genome-wide mapping of Polycomb target genes unravels their roles in cell fate transitions. Genes Dev 2006; 20:1123-36; PMID:16618801; http://dx.doi.org/10.1101/gad.381706
- Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim T-K, Koche RP, et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 2007; 448:553-60; PMID:17603471; http://dx.doi.org/10.1038/nature06008
- Hirabayashi Y, Suzki N, Tsuboi M, Endo TA, Toyoda T, Shinga J, Koseki H, Vidal M, Gotoh Y. Polycomb limits the neurogenic competence of neural precursor cells to promote astrogenic fate transition. Neuron 2009; 63:600-13; PMID:19755104; http://dx.doi.org/10.1016/j.neuron.2009.08.021
- Lim DA, Huang Y-C, Swigut T, Mirick AL, Garcia-Verdugo JM, Wysocka J, Ernst P, Alvarez-Buylla A. Chromatin remodelling factor Mll1 is essential for neurogenesis from postnatal neural stem cells. Nature 2009; 458:529-33; PMID:19212323; http://dx.doi.org/10.1038/nature07726
- Caretti G, Di Padova M, Micales B, Lyons GE, Sartorelli V. The Polycomb Ezh2 methyltransferase regulates muscle gene expression and skeletal muscle differentiation. Genes Dev 2004; 18:2627-38; PMID:15520282; http://dx.doi.org/10.1101/gad.1241904
- Blais A, van Oevelen CJC, Margueron R, Acosta-Alvear D, Dynlacht BD. Retinoblastoma tumor suppressor protein-dependent methylation of histone H3 lysine 27 is associated with irreversible cell cycle exit. J Cell Biol 2007; 179:1399-412; PMID:18166651; http://dx.doi.org/10.1083/jcb.200705051
- Asp P, Blum R, Vethantham V, Parisi F, Micsinai M, Cheng J, Bowman C, Kluger Y, Dynlacht BD. Genome-wide remodeling of the epigenetic landscape during myogenic differentiation. Proc Natl Acad Sci U S A 2011; 108:E149-58; PMID:21551099; http://dx.doi.org/10.1073/pnas.1102223108
- Patel T, Tursun B, Rahe DP, Hobert O. Removal of Polycomb repressive complex 2 makes C. elegans germ cells susceptible to direct conversion into specific somatic cell types. Cell Rep 2012; 2:1178-86; PMID:23103163; http://dx.doi.org/10.1016/j.celrep.2012.09.020
- Kadoch C, Hargreaves DC, Hodges C, Elias L, Ho L, Ranish J, Crabtree GR. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat Genet 2013; 45:592-601; PMID:23644491; http://dx.doi.org/10.1038/ng.2628
- Wang X, Haswell JR, Roberts CWM. Molecular pathways: SWI/SNF (BAF) complexes are frequently mutated in cancer–mechanisms and potential therapeutic insights. Clin Cancer Res 2014; 20:21-7; http://dx.doi.org/10.1158/1078-0432.CCR-13-0280
- Wilson BG, Roberts CWM. SWI/SNF nucleosome remodellers and cancer. Nat Rev Cancer 2011; 11:481-92; PMID:21654818; http://dx.doi.org/10.1038/nrc3068
- Holstege FC, Jennings EG, Wyrick JJ, Lee TI, Hengartner CJ, Green MR, Golub TR, Lander ES, Young RA. Dissecting the regulatory circuitry of a eukaryotic genome. Cell 1998; 95:717-28; PMID:9845373; http://dx.doi.org/10.1016/S0092-8674(00)81641-4
- Phelan ML, Sif S, Narlikar GJ, Kingston RE. Reconstitution of a core chromatin remodeling complex from SWI/SNF subunits. Mol Cell 1999; 3:247-53; PMID:10078207; http://dx.doi.org/10.1016/S1097-2765(00)80315-9
- Lessard J, Wu JI, Ranish JA, Wan M, Winslow MM, Staahl BT, Wu H, Aebersold R, Graef IA, Crabtree GR. An essential switch in subunit composition of a chromatin remodeling complex during neural development. Neuron 2007; 55:201-15; PMID:17640523; http://dx.doi.org/10.1016/j.neuron.2007.06.019
- Lickert H, Takeuchi JK, Von Both I, Walls JR, McAuliffe F, Adamson SL, Henkelman RM, Wrana JL, Rossant J, Bruneau BG. Baf60c is essential for function of BAF chromatin remodelling complexes in heart development. Nature 2004; 432:107-12; PMID:15525990; http://dx.doi.org/10.1038/nature03071
- De la Serna IL, Ohkawa Y, Berkes CA, Bergstrom DA, Dacwag CS, Tapscott SJ, Imbalzano AN. MyoD targets chromatin remodeling complexes to the myogenin locus prior to forming a stable DNA-bound complex. Mol Cell Biol 2005; 25:3997-4009; PMID:15870273; http://dx.doi.org/10.1128/MCB.25.10.3997-4009.2005
- Forcales SV, Albini S, Giordani L, Malecova B, Cignolo L, Chernov A, Coutinho P, Saccone V, Consalvi S, Williams R, et al. Signal-dependent incorporation of MyoD-BAF60c into Brg1-based SWI/SNF chromatin-remodelling complex. EMBO J 2012; 31:301-16; PMID:22068056; http://dx.doi.org/10.1038/emboj.2011.391
- Kia SK, Gorski MM, Giannakopoulos S, Verrijzer CP. SWI/SNF mediates polycomb eviction and epigenetic reprogramming of the INK4b-ARF-INK4a locus. Mol Cell Biol 2008; 28:3457-64; PMID:18332116; http://dx.doi.org/10.1128/MCB.02019-07
- Wilson BG, Wang X, Shen X, McKenna ES, Lemieux ME, Cho Y-J, Koellhoffer EC, Pomeroy SL, Orkin SH, Roberts CWM. Epigenetic antagonism between polycomb and SWI/SNF complexes during oncogenic transformation. Cancer Cell 2010; 18:316-28; PMID:20951942; http://dx.doi.org/10.1016/j.ccr.2010.09.006
- Kuwahara Y, Charboneau A, Knudsen ES, Weissman BE. Reexpression of hSNF5 in malignant rhabdoid tumor cell lines causes cell cycle arrest through a p21(CIP1/WAF1)-dependent mechanism. Cancer Res 2010; 70:1854-65; PMID:20179200; http://dx.doi.org/10.1158/0008-5472.CAN-09-1922
- Kuwahara Y, Wei D, Durand J, Weissman BE. SNF5 reexpression in malignant rhabdoid tumors regulates transcription of target genes by recruitment of SWI/SNF complexes and RNAPII to the transcription start site of their promoters. Mol Cancer Res MCR 2013; 11:251-60; PMID:23364536; http://dx.doi.org/10.1158/1541-7786.MCR-12-0390
- Inoue H, Giannakopoulos S, Parkhurst CN, Matsumura T, Kono EA, Furukawa T, Tanese N. Target genes of the largest human SWI/SNF complex subunit control cell growth. Biochem J 2011; 434:83-92; PMID:21118156; http://dx.doi.org/10.1042/BJ20101358
- Eroglu E, Burkard TR, Jiang Y, Saini N, Homem CCF, Reichert H, Knoblich JA. SWI/SNF complex prevents lineage reversion and induces temporal patterning in neural stem cells. Cell 2014; 156:1259-73; PMID:24630726; http://dx.doi.org/10.1016/j.cell.2014.01.053
- Riedel CG, Dowen RH, Lourenco GF, Kirienko NV, Heimbucher T, West JA, Bowman SK, Kingston RE, Dillin A, Asara JM, et al. DAF-16 employs the chromatin remodeller SWI/SNF to promote stress resistance and longevity. Nat Cell Biol 2013; 15:491-501; PMID:23604319; http://dx.doi.org/10.1038/ncb2720
- Zhang HS, Gavin M, Dahiya A, Postigo AA, Ma D, Luo RX, Harbour JW, Dean DC. Exit from G1 and S phase of the cell cycle is regulated by repressor complexes containing HDAC-Rb-hSWI/SNF and Rb-hSWI/SNF. Cell 2000; 101:79-89; PMID:10778858; http://dx.doi.org/10.1016/S0092-8674(00)80625-X
- Flowers S, Nagl NG, Beck GR, Moran E. Antagonistic roles for BRM and BRG1 SWI/SNF complexes in differentiation. J Biol Chem 2009; 284:10067-75; PMID:19144648; http://dx.doi.org/10.1074/jbc.M808782200
- Wirt SE, Adler AS, Gebala V, Weimann JM, Schaffer BE, Saddic L a, Viatour P, Vogel H, Chang HY, Meissner A, et al. G1 arrest and differentiation can occur independently of Rb family function. J Cell Biol 2010; 191:809-25; PMID:21059851; http://dx.doi.org/10.1083/jcb.201003048
- Tateishi Y, Matsumoto A, Kanie T, Hara E, Nakayama K, Nakayama KI. Development of mice without Cip/Kip CDK inhibitors. Biochem Biophys Res Commun 2012; 427:285-92; PMID:23000166; http://dx.doi.org/10.1016/j.bbrc.2012.09.041
- Jaenisch R, Young R. Stem cells, the molecular circuitry of pluripotency and nuclear reprogramming. Cell 2008; 132:567-82; PMID:18295576; http://dx.doi.org/10.1016/j.cell.2008.01.015
- Sage C, Huang M, Karimi K, Gutierrez G, Vollrath MA, Zhang D-S, García-Añoveros J, Hinds PW, Corwin JT, Corey DP, et al. Proliferation of functional hair cells in vivo in the absence of the retinoblastoma protein. Science 2005; 307:1114-8; PMID:15653467; http://dx.doi.org/10.1126/science.1106642
- Ajioka I, Martins R a P, Bayazitov IT, Donovan S, Johnson D a, Frase S, Cicero S a, Boyd K, Zakharenko SS, Dyer M a. Differentiated horizontal interneurons clonally expand to form metastatic retinoblastoma in mice. Cell 2007; 131:378-90; PMID:17956737; http://dx.doi.org/10.1016/j.cell.2007.09.036
- Buttitta LA, Katzaroff AJ, Perez CL, de la Cruz A, Edgar BA. A double-assurance mechanism controls cell cycle exit upoN-terminal differentiation in Drosophila. Dev Cell 2007; 12:631-43; PMID:17419999; http://dx.doi.org/10.1016/j.devcel.2007.02.020
- Buttitta L a, Katzaroff AJ, Edgar B a. A robust cell cycle control mechanism limits E2F-induced proliferation of terminally differentiated cells in vivo. J Cell Biol 2010; 189:981-96; PMID:20548101; http://dx.doi.org/10.1083/jcb.200910006
- Porrello ER, Mahmoud AI, Simpson E, Hill JA, Richardson JA, Olson EN, Sadek HA. Transient Regenerative Potential of the Neonatal Mouse Heart. Science 2011; 331:1078-80; PMID:21350179; http://dx.doi.org/10.1126/science.1200708
- Mahmoud AI, Kocabas F, Muralidhar SA, Kimura W, Koura AS, Thet S, Porrello ER, Sadek HA. Meis1 regulates postnatal cardiomyocyte cell cycle arrest. Nature 2013; 497:249-53; PMID:23594737; http://dx.doi.org/10.1038/nature12054
- Rampalli S, Li L, Mak E, Ge K, Brand M, Tapscott SJ, Dilworth FJ. p38 MAPK signaling regulates recruitment of Ash2L-containing methyltransferase complexes to specific genes during differentiation. Nat Struct Mol Biol 2007; 14:1150-6; PMID:18026121; http://dx.doi.org/10.1038/nsmb1316
- Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell 2011; 144:646-74; PMID:21376230; http://dx.doi.org/10.1016/j.cell.2011.02.013
- Adachi M, Roussel MF, Havenith K, Sherr CJ. Features of macrophage differentiation induced by p19INK4d, a specific inhibitor of cyclin D-dependent kinases. Blood 1997; 90:126-37; PMID:9207446
- Kranenburg O, Scharnhorst V, Van der Eb AJ, Zantema A. Inhibition of cyclin-dependent kinase activity triggers neuronal differentiation of mouse neuroblastoma cells. J Cell Biol 1995; 131:227-34; PMID:7559779; http://dx.doi.org/10.1083/jcb.131.1.227
- Matushansky I, Radparvar F, Skoultchi AI. Reprogramming leukemic cells to terminal differentiation by inhibiting specific cyclin-dependent kinases in G1. Proc Natl Acad Sci U S A 2000; 97:14317-22; PMID:11114185; http://dx.doi.org/10.1073/pnas.250488697
- Rosenbauer F, Tenen DG. Transcription factors in myeloid development: balancing differentiation with transformation. Nat Rev Immunol 2007; 7:105-17; PMID:17259967; http://dx.doi.org/10.1038/nri2024
- Koppens M, van Lohuizen M. Context-dependent actions of Polycomb repressors in cancer. Oncogene 2015. http://www.nature.com.proxy.library.uu.nl/onc/journal/vaop/ncurrent/full/onc2015195a.html; http://dx.doi.org/10.1038/onc.2015.195
- Shain AH, Pollack JR. The Spectrum of SWI/SNF Mutations, Ubiquitous in Human Cancers. PLoS ONE 2013; 8:e55119; PMID:23355908; http://dx.doi.org/10.1371/journal.pone.0055119
- Kim KH, Roberts CWM. Mechanisms by which SMARCB1 loss drives rhabdoid tumor growth. Cancer Genet 2014; 207:365-72; PMID:24853101; http://dx.doi.org/10.1016/j.cancergen.2014.04.004
- Alarcon-Vargas D, Zhang Z, Agarwal B, Challagulla K, Mani S, Kalpana GV. Targeting cyclin D1, a downstream effector of INI1/hSNF5, in rhabdoid tumors. Oncogene 2006; 25:722-34; PMID:16302003; http://dx.doi.org/10.1038/sj.onc.1209112
- Lünenbürger H, Lanvers-Kaminsky C, Lechtape B, Frühwald MC. Systematic analysis of the antiproliferative effects of novel and standard anticancer agents in rhabdoid tumor cell lines. Anticancer Drugs 2010; 21:514-22; PMID:20147838; http://dx.doi.org/10.1097/CAD.0b013e3283375d5c
- Smith ME, Cimica V, Chinni S, Jana S, Koba W, Yang Z, Fine E, Zagzag D, Montagna C, Kalpana GV. Therapeutically targeting cyclin D1 in primary tumors arising from loss of Ini1. Proc Natl Acad Sci U S A 2011; 108:319-24; PMID:21173237; http://dx.doi.org/10.1073/pnas.0913297108
- Tsikitis M, Zhang Z, Edelman W, Zagzag D, Kalpana GV. Genetic ablation of Cyclin D1 abrogates genesis of rhabdoid tumors resulting from Ini1 loss. Proc Natl Acad Sci U S A 2005; 102:12129-34; PMID:16099835; http://dx.doi.org/10.1073/pnas.0505300102
- The I, Ruijtenberg S, Bouchet BP, Cristobal A, Prinsen MBW, van Mourik T, Koreth J, Xu H, Heck AJR, Akhmanova A, et al. Rb and FZR1/Cdh1 determine CDK4/6-cyclin D requirement in C. elegans and human cancer cells. Nat Commun 2015; 65906; PMID:25562820; http://www.nature.com/ncomms/2015/150106/ncomms6906/full/ncomms6906.html.