
ABSTRACT
We found that inhibitors of mitochondrial respiratory chain complexes III (myxothiazol) and I (piericidin A) in some epithelial carcinoma cell lines induce transcription of the p53-responsive SESN2 gene that plays an important role in stress response and homeostatic regulation. However, the effect did not depend on p53 because i) there was no induction of p53 after the treatment with piericidin A; ii) after the treatment with myxothiazol the peak of SESN2 gene upregulation occurred as early as 5h, before the onset of p53 activation (13h); iii) a supplementation with uridine that abolishes the p53 activation in response to myxothiazol did not abrogate the induction of SESN2 transcripts; iv) in the p53 negative HCT116 p53 -/- cells SESN2 transcription could be also induced by myxothiazol. In response to the respiratory chain inhibitors we observed an induction of ATF4, the key transcription factor of the integrated stress response (ISR). We found that the induction of SESN2 transcripts could be prevented by the ISR inhibitory small molecule ISRIB. Also, by inhibiting or overexpressing ATF4 with specific shRNA or ATF4-expressing constructs, respectively, we have confirmed the role of ATF4 in the SESN2 gene upregulation induced by mitochondrial dysfunction. At a distance of 228 bp upstream from the SESN2 transcription start site we found a candidate sequence for the ATF4 binding site and confirmed its requirement for the induction of SESN2 in luciferase reporter experiments. We suggest that the upregulation of SESN2 by mitochondrial dysfunction provides a homeostatic feedback that attenuates biosynthetic processes during temporal losses of energy supply from mitochondria thereby assisting better adaptation and viability of cells in hostile environments.
Introduction
Sestrins represent a small family of proteins involved in metabolic regulation and stress response. In mammals sestrins are encoded by 3 genes (SESN1, SESN2 and SESN3) presumably sharing similar activities. Sestrins have antioxidant functions and enforce homeostasis, stress resistance and protection against deleterious oxidation-induced DNA modifications and mutations.Citation1,2 The functions of sestrins are mediated by several different mechanisms. The excess of hydrogen peroxide is eliminated by peroxiredoxins, which are regenerated by a sestrin-induced sulfinic reductase Srx.Citation1,3 Sesn1 and Sesn2 proteins induce an activity of the transcription factor Nrf2 that controls a set of key antioxidant enzymes, including the Srx, through an activation of the p62-dependent autophagic degradation of Kelch-like ECH-associated protein 1 (Keap1), the inhibitor of Nfr2.Citation3,4 Besides, the antioxidant effect is achieved through the ability of sestrins to inhibit mTORC1 leading to overall decrease in metabolic activity Citation5-7 and stimulation of the autophagy.Citation8,9 Finally sestrins further attenuate metabolism by assisting the LKB1-dependent activation of the AMP-dependent protein kinase.Citation10 Collectively, the activities of sestrins enforce homeostasis under conditions of changing workloads and facilitate cells’ viability and adaptation to different stresses.Citation11,12
The expression of sestrins is controlled by a set of transcription factors. In response to genotoxic stresses SESN1 and SESN2 are induced by р53,Citation13,14 and SESN3 – by FOXOCitation15,16 transcription factors. The induction of SESN1 and SESN2 by hypoxia is mediated both by p53 Citation14 and by the transcription factor HIF1.Citation17,18 In addition, oxidative stresses induce an Nrf2-mediated stimulation of SESN2 through the antioxidant response element (ARE) present within the gene.Citation19 Nutrients availability also affects the expression of sestrins. A deficiency of nutrients, such as low glucose, stimulates SESN2 expression along with some other p53-regulated genes involved in metabolic regulation and cell cycle arrest through the formation of a complex between p53 and PPARγ coactivator–1α (PGC-1α).Citation20 On the contrary, the excess of saturated fatty acids that result in the endoplasmic reticulum (ER) stress in hepatocytes is accompanied by the SESN2 induction mediated by the transcription factor c/EBP-β.Citation21 Remarkably, treatment of cells with the anticancer drugs nelfinavir and bortezomib that induce the ER stress and the autophagy were also shown to activate the expression of SESN2, along with the known ER stress markers, such as transcription factors ATF3, ATF4 and CHOP. It was found that ATF4 was solely responsible for the induction of SESN2.Citation22
ATF4 (Activating transcription factor 4) plays a pivotal role in responses of cells to stresses.Citation23,24 Different stressful conditions induce 4 protein kinases: the PKR-like endoplasmic reticulum kinase (PERK), the general control nonderepressible 2 (GCN2) serine/threonine kinase, protein kinase R (PKR) and heme regulator inhibitor (HRI).Citation25 These 4 protein kinases induce phosphorylation of the alpha-subunit of translation factor eIF2. This blocks the exchange of eIF2-GDP to eIF2-GTP, thus reducing global translation initiation and subsequent protein synthesis. However, the eIF2α phosphorylation paradoxically enhances the translation of mRNAs containing upstream open reading frames (uORFs), in particular the transcription factor ATF4.Citation25,26 ATF4 target genes are involved in biosynthesis, folding and assembly of proteins, metabolism, transport of nutrients, protection against oxidative stress and regulation of apoptosis. As ATF4 integrates signals from different kinases upstream of eIF2α,Citation24 the eIF2α / ATF4 cascade is commonly referred to as the Integrated Stress Response (ISR). Besides the translational regulation in response to stresses, the ATF4 is upregulated at the level of transcription.Citation27,28 Presumably the combination of transcriptional and translational regulations allows the eIF2-kinase signaling pathway to selectively repress or activate key regulatory genes of the cell.Citation28
Mitochondrial dysfunction caused by an inhibition of the respiratory chain induces expression of stress-responsive genes. We have shown recently that the early response to the inhibition of mitochondrial respiratory complex III that enforces cell survival depends on the transcription factor ATF4.Citation29 However, the sustained inhibition of complex III induces stabilization and activation of the p53 tumor suppressor, due to the shut-off of dihydroorotate dehydrogenase (DHODH), a key enzyme of the de novo pyrimidine biosynthesis pathway.Citation30 We found that the activation of p53 abolishes and reverses the earlier ATF4-mediated pro-survival stress response and induces apoptotic cell death.Citation29 Upon the p53 activation the expression of many known ATF4-target genes is substantially downregulated, with the exception of SESN2 whose expression is upregulated not only early (4-5 h) after the addition of complex III inhibitor myxothiazol, but also at later stages (13-17 h) when p53 activity is increased.Citation29 SESN2 is known to be a transcriptional target of p53.Citation14 Here we show that in response to inhibition of mitochondrial respiratory chain the expression of SESN2 is induced not only by p53, but also by ATF4. We have identified a potential ATF4 binding site and confirmed experimentally its requirement for the induction of SESN2 in response to ATF4. We conclude that the induction of SESN2 expression in response to the mitochondrial respiratory chain dysfunction is mediated by ATF4, the key transcription factor of the integrated stress response. Presumably the mechanism provides a homeostatic feedback that attenuates metabolism in response to temporal loss of energy supply from mitochondria and assists in better adaptation and viability of cells in hostile environments.
Results
The induction of SESN2 expression in response to an inhibition of mitochondrial respiratory chain does not depend on the tumor suppressor p53
In the previous studyCitation29 we monitored quantitative changes in transcripts following a treatment of HCT116 colon carcinoma cells with the mitochondrial respiration chain complex III inhibitor myxothiazol (1μM). In particular, we found that levels of mRNA for SESN2 gene are substantially increased (). As SESN2 is a transcriptional target of the p53 tumor suppressorCitation14 we hypothesized that the observed transcriptional activation of SESN2 might depend on p53 that is activated by the inhibition of mitochondrial electron transport chain (ETC) complex III due to a deficiency of the ETC-dependent dihydroorotate dehydrogenase, a mitochondrial membrane enzyme of the de novo pyrimidine biosynthesis pathway.Citation30,31 To check the hypothesis, the cells were treated with myxothiazol in the presence of uridine that restores pools of uridylic and cytidylic nucleotides, and completely prevents the p53 stabilization and activation.Citation30 It was found that uridine did not affect the induction of SESN2 mRNA in response to myxothiazol treatment for 13 h, while completely preventing the p53 activation, which in particular was supported by the lack of induction of transcripts from the p53-reponsive TP53INP1 (). We concluded that the induction of SESN2 expression in this system was not dependent on p53. This was supported by the fact that by 5 h of myxothiazol treatment, before the onset of activation of p53, the expression level of SESN2 mRNA was higher than that after the 13 h treatment ().
Table 1. Fold change in mRNA levels of HCT116 cells treated with 1μM myxothiazol for 5 h (M5), 13h (M13), 17h (M17) or with 1μM myxothiazol and 1 mM uridine for 13 h (MU13). The data was obtained by RNA-seq;29 FDR <0.05.
By RT-qPCR we have confirmed the RNA-seq results. We have shown that a short term inhibition of complex III in HCT116 cells induces SESN2 mRNA substantially to the same extent as the p53 activation by nutlin-3, an inhibitor of the p53/Mdm2 interactionCitation32 (). A combined treatment with myxothiazol and nutlin-3 did not show further increase in SESN2 mRNA levels. In HCT116 p53-/- knockout cells the treatment with myxothiazol (but not with nutlin-3) induced SESN2 mRNA expression (). The respiratory chain complex I inhibitor piericidin A also induced SESN2 mRNA (), although no p53 activation was observed (). In HeLa and RKO human carcinoma cell lines as well as in human embryonic kidney cell line HEK293T the inhibition of mitochondrial respiratory chain also induced SESN2 mRNA ( and ), arguing against a cell line specific effect.
Figure 1. Induction of SESN2 mRNA in response to the inhibition of the mitochondrial respiratory chain is independent of p53. (A, B) Fold changes of SESN2 mRNA (A) and of the p53-target gene TP53INP1 mRNA (B) in wild-type or p53-/- HCT116 cells after myxothiazol (4 h) and/or nutlin-3 (16 h) treatment. (C, D) Fold changes of SESN2 (C) and TP53INP1 (D) mRNAs in HCT116 cells treated with piericidin A (4 h). The data was obtained by RT-qPCR, the normalization was to 18S rRNA. The means and standard deviations on the basis of at least 3 independent experiments are presented. Student's t-test was used to analyze statistical significance (*P < 0.05, **P<0.01, ***P< 0.001).
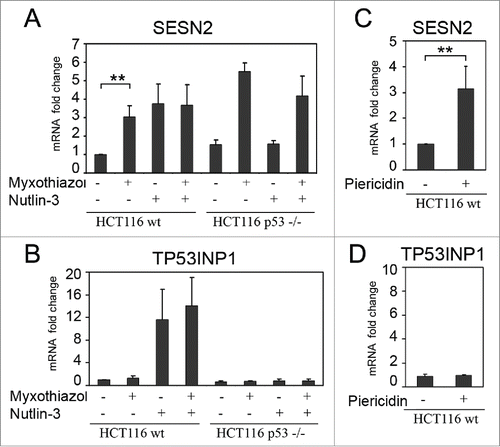
Figure 2. Induction of SESN2 mRNA in response to the inhibition of mitochondrial respiratory chain depends on ISR. SESN2 mRNA level in HeLa (A), HEK293T (B) and HCT116 (C) cells after treatment with 1μM myxothiazol, with and without 200 nM ISRIB (4h). The data was obtained by RT-qPCR, normalized to18S rRNA. The means and standard deviations on the basis of at least 3 independent experiments are presented. Student's t-test was used to analyze statistical significance (*P < 0.05, **P<0.01, ***P< 0.001).
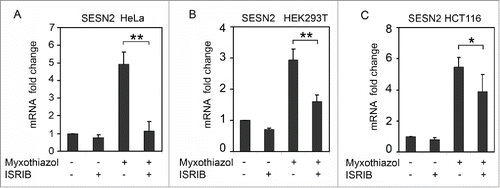
Figure 3. The effect of ATF4 knockdown on SESN2 mRNA expression. ATF4 (A, C) and SESN2 (B, D) mRNA fold changes in HeLa (A, B) and RKO (C, D) cells expressing ATF4 shRNA (ATF4) or a scrambled control shRNA (sc). The cells were treated with 1 μM myxothiazol for 4 h where indicated. The data was obtained by RT-qPCR and normalized to 18S rRNA. The means and standard deviations on the basis of 3 independent experiments are presented. Student's t-test was used to analyze statistical significance (*P < 0.05, **P < 0.01, ***P < 0.001).
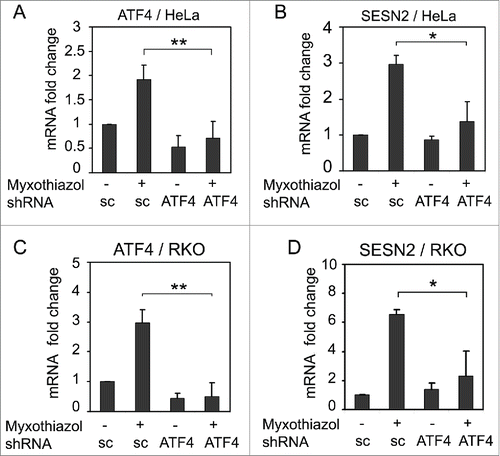
As we have previously detected the induction of ATF4 () and some of the ATF4-regulated genes after a short-term inhibition of complex III,Citation29 we hypothesized that ATF4, a key factor of ISR, could be responsible for the induction of SESN2 upon mitochondrial dysfunction. Therefore, we tested levels of SESN2 expression after a treatment with the ISR inhibitor ISRIB, as well as in the cells with either inhibited or overexpressed ATF4.
Effects of an ISR inhibitor and low and high ATF4 levels on SESN2 gene expression
ISRIB, a small molecule inhibitor of the integrated stress response,Citation33,34 was shown to counteract negative effects of eIF2α phosphorylation on translation by acting downstream of all eIF2-kinases. Phosphorylation of eIF2α attenuates protein synthesis by inhibiting eIF2B, a guanine nucleotide exchange factor (GEF), which accelerates the exchange of GDP for GTP in the eIF2 complex. ISRIB has been found to stabilize eIF2B, increase its GEF activity and restore protein synthesis even in the presence of the factors that cause cellular stress.Citation35,36 Besides ISRIB specifically inhibits translation of stress-inducible mRNAs, containing uORFs, including the ATF4 mRNA. Citation33
We found that ISRIB prevented in HeLa cells and reduced significantly in HEK293T and HCT116 cells the induction of SESN2 mRNA in response to myxothiazol treatment (). It confirms a role of ISR in the induction of SESN2 expression in response to mitochondrial dysfunction and suggests an involvement of the transcription factor ATF4.
This conclusion has been directly tested by specific inhibition of ATF4 expression by RNA interference.Citation37 Lentiviral constructs expressing shRNA to ATF4 mRNA were introduced into HeLa or RKO cells with subsequent puromycin selection. Using RT-qPCR, we confirmed that ATF4 mRNA levels were substantially decreased in these cells (). The expression of the ATF4-specific shRNA prevented the induction of ATF4 and SESN2 mRNAs in response to the short-term treatment with myxothiazol, but did not influence the basal levels of SESN2 mRNA (). We conclude that the induction of SESN2 gene expression in response to respiratory chain complex III inhibition depends on the ATF4 transcription factor.
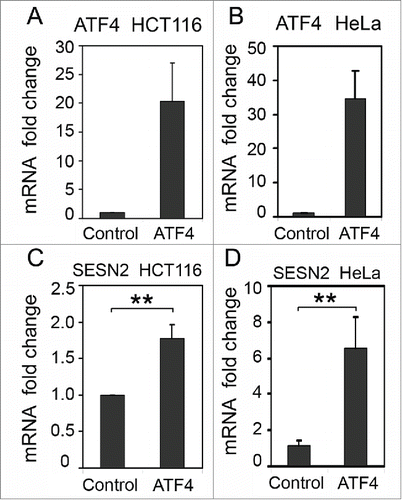
In a reciprocal experiment we overexpressed ATF4 mRNA in HCT116 and HeLa cells by introducing an ATF4 cDNA expression construct (). The ectopic expression of ATF4 mRNA in HCT116 cells resulted in a significant (1.8-fold, p-value 0.0023) increase in SESN2 mRNA levels () and even higher induction of SESN2 (6.5-fold) when ATF4 was overexpressed in HeLa cells (). The results confirm the ability of ATF4 to stimulate SESN2 expression.
Figure 4. The effect of ATF4 overexpression on SESN2 mRNA levels. ATF4 (A, B) and SESN2 (C, D) mRNA fold changes in HCT116 (A, C) and HeLa (B, D) cells with ectopic expression of ATF4 mRNA (ATF4) or transfected with a control empty vector (Control). The data was obtained by RT-qPCR and normalized to 18S rRNA. The means and standard deviations on the basis of 3 independent experiments are presented. Student's t-test was used to analyze statistical significance (*P < 0.05, **P<0.01, ***P< 0.001).
Molecular mechanism of regulation of SESN2 expression by ATF4
To identify DNA elements responsible for the ATF4-mediated regulation, we placed firefly luciferase reporter gene downstream of the promoter and 5’-UTR region of SESN2 gene. ATF4 regulates transcription of its target genes through the formation of homodimers or heterooligomers with the transcription factors Jun, AP-1 and C/EBPCitation38,39 that bind to CARE (C/EBP-ATF) responsive elements having the consensus sequence XTTXCATCA (where X = G, A or T).Citation39 In the region from -625 to -618 bp relative to the SESN2 translation start codon (from -228 to -221 bp relative to the transcription start site) we found a candidate sequence for the ATF4 binding site TTTTCATCA. To test whether the sequence is important for the ATF4-mediated transcription regulation we amplified by PCR the genome segment comprising 724 bp upstream of the translation start codon of Sesn2 ORF and cloned it into the pGL3-Basic reporter plasmid carrying firefly luciferase gene (). The reporter construct pGL3-SESN2 was co-transfected into HCT116 or HeLa cells along with the normalization control plasmid expressing beta-galactosidase driven by a constitutive promoter.
Figure 5. Overexpression of ATF4 (but not p53) stimulates the luciferase reporter controlled by the SESN2 gene promoter region. (A) The reporter constructions. (B) Stimulation of luciferase reporter by ATF4 expression in HCT116 cells compared to the control HCT116 cells transfected with the empty vector. (C) The activity of reporters pGL3-SESN2 or pGL3-SESN2_mut with a mutated putative ATF4 binding site in HeLa cells with ectopic expression of p53 or ATF4 compared to control cells transfected with an empty vector. The effects of ATF4 and p53 expression are presented as relative values in comparison to normalized reporter activities in control cells. The means and standard deviations on the basis of at least 3 independent experiments are presented. Student's t-test was used to analyze statistical significance (*P < 0.05, **P < 0.01, ***P < 0.001).
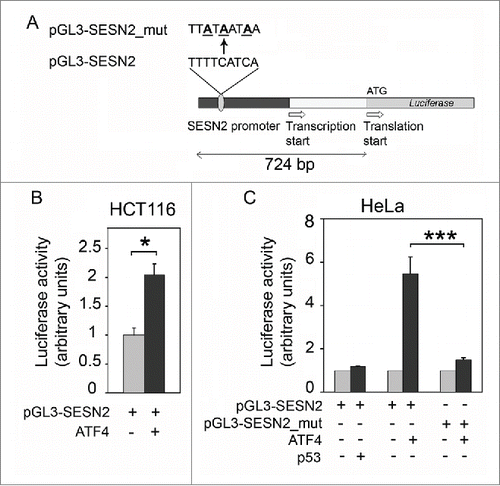
In response to the ectopic expression of ATF4 there was a 2-fold increase in the luciferase activity in HCT116 cells and almost a 6-fold increase in HeLa cells (), suggesting that the transcription factor ATF4 is capable of stimulating the SESN2 gene promoter. To test this hypothesis, the putative ATF4 binding site was mutated at the 3 most conserved positions by turning TTTTCATCA into TTATAATAA in the reporter plasmid pGL3-SESN2_mut (). As expected, the changes prevented the increase in reporter gene activity in response to the ATF4 overexpression (). We conclude that the regulation of SESN2 expression by the ATF4 transcription factor is mediated by the TTTTCATCA site located 221-228 bp upstream of the transcription start site. Also, despite the previously identified putative p53-binding site located between the transcription and translation start sites,Citation40 the ectopic expression of p53 did not result in the increase of the reporter activity ().
Discussion
In the present study we have demonstrated that in response to the mitochondrial dysfunction caused by the inhibition of mitochondrial respiratory chain there is a substantial induction of SESN2 gene expression. The induction presumably occurs as a part of the ISR and depends on the ATF4 transcription factor. The inhibition of ISR by ISRIB or the suppression of ATF4 by RNA-interference prevents the induction of SESN2 mRNA in response to the mitochondrial respiratory chain dysfunction. On the contrary, the ectopic expression of ATF4 is capable of increasing the level of SESN2 mRNA.
It has been recently reported that the expression of SESN2 gene is induced by the ER stress.Citation22,41 The peptidilarginine deiminase (PAM) family inhibitor YW3-56 that is capable of inducing the ER stress, was shown to stimulate the SESN2 gene expression in an ATF4-dependent manner in triple-negative breast carcinoma cells Citation41 that is in line with our results. The increased levels of Sesn2 protein were also observed in cells treated with the inducer of the ER stress nelfinavir and proteasome inhibitor bortezomib, and the effect was similar upon the ectopic expression of ATF4 in HeLa cells.Citation22 However, the molecular mechanism of this regulation has not been elucidated.Citation22,41 The results of our present experiments with reporter plasmids suggest that the mechanism of SESN2 regulation includes binding of the ATF4 transcription factor to TTTTCATCA site within 221-228 bp upstream of the transcription start site. ChIP-exo analysis Citation41 has recently detected ATF4 binding after YW3-56 treatment near the position -138 bp of SESN2 gene promoter relative to the transcription start site. The data has been confirmed by the conventional ChIP assay. Citation41 We suppose that the difference might be due to the lack of accuracy of the ChIP method (taking into account that for the ChIP experiments chromatin was fragmented to a range from 0.5 to 1 kb in ref. Citation41). It is also worth noting that the DNA sequence TTTGCAGCA located at the position −138 bp of SESN2 gene promoter does not match the canonical ATF4 consensus XTTXCATCA. Citation39,41
The ATF4-binding site TTTTCATCA within the SESN2 gene promoter identified and validated in this study matches the consensus CARE sequence. We found no absolutely identical sequences among the previously identified ATF4-responsive elements in the promoters of known ATF4-regulated genes.Citation39 There is only one nucleotide difference between the element in SESN2 gene and the nutrient-sensing response element 1 (NSRE1) in the ATF4-inducible asparagine synthase gene ASNS (GTTTCATCA), and the amino acid response element (AARE) in the pseudokinase gene TRIB3 (TTTGCATCA).Citation39 Both elements are responsible for transcriptional induction of the genes in response to amino acid starvation and ER stress.
Our data does not confirm the presence of functional p53-response elements between SESN2 transcription and translation start sites.Citation40 Perhaps the element is located in the region further downstream at position +733 bp or +10 kb, as has been previously suggested.Citation42,43
What could be the functional role of SESN2 gene induction in response to the mitochondrial dysfunction? The induction of SESN2 gene expression is known to occur in response to oxidative, electrophilic and genotoxic stresses.Citation3,5,14 Functions of the Sesn2 protein play important roles in the regulation of reactive oxygen species balance and in the control of energy metabolism. An inhibition of the mitochondrial respiratory chain may lead to the formation of reactive oxygen species Citation44 and a transient energy deficiency. Thus, the induction of SESN2 gene in response to a mitochondrial dysfunction may have adaptive and pro-survival roles through the ISR, by enforcing the detoxification of reactive oxygen species and aligning metabolic processes with the changing energy supplies. An inhibition of mitochondrial respiratory chain can lead to a shortage of energy required for protein biosynthesis. Being an inhibitor of mTORC1Citation5 Sesn2 protein attenuates overall biosynthetic activity and contributes to mitigation of the metabolic stress load. Also, during the ER stress a restoration of protein synthesis prior to a complete stress recovery may lead to apoptotic cell death.Citation45 Therefore, Sesn2 may promote stress resistance and viability of the cell through the inhibition of mTORC1 during energy shortages. Indeed, SESN2 was recently implicated as a sensor of energy stresses mediated by the reduction of ATP levels in cancer cells treated with the glycolysis inhibitor 2-deoxyglucose. The induction of SESN2 protein leading to the suppression of mTORC1 was shown to protect cells from energetic stress-induced apoptosis.Citation46
The results presented in this study suggest that the ATF4-dependent induction of SESN2 gene expression in response to mitochondrial dysfunction may contribute to integrated stress response and enforce better adaptation of cells to changing environmental conditions and energy supplies.
Materials and methods
Plasmid constructs
pHM-ATF4
ATF4-coding sequence was amplified by PCR using a total cDNA from the human colon carcinoma cell line RKO as a template and primers Atf4_dir (5′-AGGATCCGCAACATGACCGAAATG-3′) and Atf4_rev (5′-TTGAAGCTTGGTGCGCGCCAGGACCC-3′) containing sites for restriction nucleases BamHI and PstI, respectively. The amplified fragment was cloned into the pUC19 plasmid and sequenced. The sequence-verified insert of ATF4 cDNA was cloned into BamHI and PstI sites of pcDNA4/HisMax/B expression vector (Invitrogen) for a transient expression in mammalian cells.
pGL3-SESN2
To obtain SESN2 promoter fragment the PCR was performed on a total DNA from the human colon carcinoma cell line HCT116 with primers SESN2_dir (5′-GAGAGCTCTTAACCCTAGCCAGTCC-3′) and SESN2_rev (5′-TTGAAGCTTGGTGCGCG CCAGGACCC-3′). The PCR product was inserted into the pUC19 vector between SacI and HindIII restriction sites, then sequenced and recloned as a SacI-HindIII fragment into the vector pGL3-Basic (Promega).
pGL3-SESN2mut
Mutations within the putative ATF4-binding site were introduced by the megaprimer PCR. The first PCR was performed with primers SESN2_dir and SESN2_mut_rev (5′-GGGTTGCGTTATTATAAGGGACTTCACTG-3′), the plasmid pUC19 with inserted SESN2 gene promoter was used as a template. After agarose gel purification the PCR product was used as a forward primer in the second PCR with the other primer SESN2_rev. The PCR product was inserted into pUC19 between the sites SacI and HindIII, then sequenced and recloned as a SacI-HindIII fragment into the vector pGL3-Basic.
Cancer cell cultures
Cells were grown in DMEM, containing 10% fetal calf serum (FBS) at 37°C, 5% CO2 to 50-70% confluence. For various treatments we used 1 μM myxothiazol (Sigma-Aldrich Inc.), 1 mM uridine, 2 μM piericidin A (Sigma-Aldrich Inc.), 10 μM nutlin-3 (AdooQ BioScience); treatment conditions are indicated in figure legends. The RNA-seq conditions were previously described in ref. 29, raw sequencing data were deposited in NCBI (Bioproject accession number SRP043021).
Preparation of the lentiviral vector for the expression of ATF4-specific short hairpin RNA (with the sequence sh2 – gatccgGCCAAGCACTTCAAACCTCATCACGTGATGAGGTTTGAAGT GCTTGGCtttttg) and its introduction into RKO and HeLa cells was described in ref. 29.
Trancriptional reporters
HCT116 and HeLa cells were grown in 12-well plates to 50-70% confluency and transfected with the TurboFect transfection reagent (Thermo Scientific) according to the manufacturer′s protocols. Reporter plasmids pGL3-SESN2 or pGL3-SESN2_mut (0.1 μg), pcDNA4/HisMax/lacZ (Invitrogen), encoding β-galactosidase under control of the constitutive CMV promoter (0.5 μg), and the expression vectors pHM-ATF4 (0.5 μg), encoding the transcription factor ATF4, or pCMV-p53 (0.5 μg), encoding the tumor suppressor p53, were transfected into a single well of the 12-well culture plate. The total plasmid DNA was adjusted to 2 μg by addition of the “empty” vector pcDNA4/HisMax/B (Invitrogen). Cells were lysed in the Reporter lysis buffer (Luciferase Assay System, Promega) 44 h after transfection, the luciferase and β−galactosidase activities were measured as described previously.Citation47 Luciferase activity was normalized to β−galactosidase activity of the same lysate. For each measurement, at least 3 biological replicates were used. Mean values with standard deviations are presented.
Real-time PCR
RNA isolation, reverse transcription and real time PCR were performed as previously described.Citation29 Real-time PCR detection system CFX96 (Bio-Rad) and the following primers were used: ATF4_dir CTTCACCTTCTTACAACCTCTTC, ATF4_rev GTAGTCTGGCTTCCTATCTCC; 18S_dir CGGACAGGATTGACAGATTG, 18S_rev CAGAGTCTCGTTCGTTATCG; SESN2 dir: TTCGGATATGAGGACTTC; SESN2 rev: ATGGTATTGTAGGTGAGG; TP53INP1_dir TCAGCAGAAGAAGAAGAAGAAGAG, TP53INP1_rev AGCAGGAATCA CTTGTATCAGC. Quantification of the target genes was normalized using the reference 18 S rRNA to compensate for inter-PCR variations.
Abbreviations
| ATF4 | = | Activating Transcription Factor 4 |
| CARE | = | C/EBP-ATF responsive element |
| eIF2α | = | eukaryotic initiation factor 2, α subunit |
| ER | = | endoplasmic reticulum |
| FDR | = | false discovery rate |
| ISR | = | integrated stress response |
| ISRIB | = | inhibitor of the integrated stress response |
| qPCR | = | quantitative real-time PCR |
| RT | = | reverse transcription |
| shRNA | = | small hairpin RNA |
| TP53INP1 | = | tumor protein p53 inducible nuclear protein 1 |
| uORF | = | upstream open reading frame |
| UPR | = | un-folded protein response |
| 5’-UTR | = | 5’-untranslated region |
Disclosure of potential conflicts of interest
The authors declare no conflict of interest.
Acknowledgments
We thank Andrey Vartapetian for helpful discussions and Dmitry Andreev for kindly providing ISRIB.
Funding
Funding support was from the Russian Foundation for Basic Research grant 15-04-04945-a (A.G.E), and the Russian Science Foundation grant № 14-50-00060 (P.M.C.)
References
- Budanov AV, Sablina AA, Feinstein E, Koonin EV, Chumakov PM. Regeneration of peroxiredoxins by p53-regulated sestrins, homologs of bacterial AhpD. Science 2004; 304:596-600; PMID:15105503
- Sablina AA, Budanov AV, Ilyinskaya GV, Agapova LS, Kravchenko JE, Chumakov PM. The antioxidant function of the p53 tumor suppressor. Nat Med 2005; 11:1306-13; PMID:16286925
- Bae SH, Sung SH, Oh SY, Lim JM, Lee SK, Park YN, Lee HE, Kang D, Rhee SG. Sestrins activate Nrf2 by promoting p62-dependent autophagic degradation of Keap1 and prevent oxidative liver damage. Cell Metab 2013; 17:73-84; PMID:23274085
- Rhee SG, Bae SH. Antioxidant function of Sestrins mediated by promotion of autophagic degradation of Keap1 and Nrf2 activation and by inhibition of mTORC1. Free Radic Biol Med 2015; 88(Pt B):205-11: ePub; PMID:26117317; http://dx.doi.org/10.1016/j.freeradbio-med.2015.06.007
- Budanov AV, Karin M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell 2008; 134:451-60; PMID:18692468
- Lee JH, Budanov AV, Park EJ, Birse R, Kim TE, Perkins GA, Ocorr K, Ellisman MH, Bodmer R, Bier E, et al. Sestrin as a feedback inhibitor of TOR that prevents age-related pathologies. Science 2010; 327:1223-8; PMID:20203043
- Lee JH, Budanov AV, Talukdar S, Park EJ, Park HL, Park HW, Bandyopadhyay G, Li N, Aghajan M, Jang I, et al. Maintenance of metabolic homeostasis by sestrin2 and sestrin3. Cell Metab 2012; 16:311-21; PMID:22958918
- Alers S, Loffler AS, Wesselborg S, Stork B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol Cell Biol 2012; 32:2-11; PMID:22025673
- Maiuri MC, Malik SA, Morselli E, Kepp O, Criollo A, Mouchel PL, Carnuccio R, Kroemer G. Stimulation of autophagy by the p53 target gene Sestrin2. Cell Cycle 2009; 8:1571-6; PMID:19377293
- Morrison A, Chen L, Wang J, Zhang M, Yang H, Ma Y, Budanov A, Lee JH, Karin M, Li J. Sestrin2 promotes LKB1-mediated AMPK activation in the ischemic heart. FASEB J 2015; 29:408-17; PMID:25366347
- Zheltukhin AO, Chumakov PM. Constitutive and induced functions of the p53 gene. Biochemistry (Mosc) 2010; 75:1692-721; PMID:21418001
- Olovnikov IA, Kravchenko JE, Chumakov PM. Homeostatic functions of the p53 tumor suppressor: regulation of energy metabolism and antioxidant defense. Seminars in Cancer Biol 2009; 19:32-41; PMID:19101635
- Velasco-Miguel S, Buckbinder L, Jean P, Gelbert L, Talbott R, Laidlaw J, Seizinger B, Kley N. PA26, a novel target of the p53 tumor suppressor and member of the GADD family of DNA damage and growth arrest inducible genes. Oncogene 1999; 18:127-37; PMID:9926927
- Budanov AV, Shoshani T, Faerman A, Zelin E, Kamer I, Kalinski H, Gorodin S, Fishman A, Chajut A, Einat P, et al. Identification of a novel stress-responsive gene Hi95 involved in regulation of cell viability. Oncogene 2002; 21:6017-31; PMID:12203114
- Nogueira V, Park Y, Chen CC, Xu PZ, Chen ML, Tonic I, Unterman T, Hay N. Akt determines replicative senescence and oxidative or oncogenic premature senescence and sensitizes cells to oxidative apoptosis. Cancer Cell 2008; 14:458-70
- Hagenbuchner J, Kuznetsov A, Hermann M, Hausott B, Obexer P, Ausserlechner MJ. FOXO3-induced reactive oxygen species are regulated by BCL2L11 (Bim) and SESN3. J Cell Sci 2012; 125:1191-203
- Ishihara M, Urushido M, Hamada K, Matsumoto T, Shimamura Y, Ogata K, Inoue K, Taniguchi Y, Horino T, Fujieda M, et al. Sestrin-2 and BNIP3 regulate autophagy and mitophagy in renal tubular cells in acute kidney injury. Am J Physiol Renal Physiol 2013; 305:F495-509; PMID:23698117
- Olson N, Hristova M, Heintz NH, Lounsbury KM, van der Vliet A. Activation of hypoxia-inducible factor-1 protects airway epithelium against oxidant-induced barrier dysfunction. Am J Physiol Lung Cell Mol Physiol 2011; 301:L993-1002; PMID:21926263
- Shin BY, Jin SH, Cho IJ, Ki SH. Nrf2-ARE pathway regulates induction of Sestrin-2 expression. Free Radic Biol Med 2012; 53:834-41; PMID:22749810
- Sen N, Satija YK, Das S. PGC-1alpha, a key modulator of p53, promotes cell survival upon metabolic stress. Mol Cell 2011; 44:621-634; PMID:22099309
- Park HW, Park H, Ro SH, Jang I, Semple IA, Kim DN, Kim M, Nam M, Zhang D, Yin L, et al. Hepatoprotective role of Sestrin2 against chronic ER stress. Nature Communications 2014; 5:4233; PMID:24947615
- Bruning A, Rahmeh M, Friese K. Nelfinavir and bortezomib inhibit mTOR activity via ATF4-mediated sestrin-2 regulation. Mol Oncol 2013; 7:1012-8; PMID:23916134
- Teske BF, Wek SA, Bunpo P, Cundiff JK, McClintick JN, Anthony TG, Wek RC. The eIF2 kinase PERK and the integrated stress response facilitate activation of ATF6 during endoplasmic reticulum stress. Mol Biol Cell 2011; 22:4390-405; PMID:21917591
- Wengrod JC, Gardner LB. Cellular adaptation to nutrient deprivation: crosstalk between the mTORC1 and eIF2alpha signaling pathways and implications for autophagy. Cell Cycle 2015; 14:2571-2577; PMID:26039820
- Baird TD, Wek RC. Eukaryotic initiation factor 2 phosphorylation and translational control in metabolism. Adv Nutr 2012; 3:307-321; PMID:22585904
- Lu PD, Harding HP, Ron D. Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response. J Cell Biol 2004; 167:27-33; PMID:15479734
- Dey S, Baird TD, Zhou D, Palam LR, Spandau DF, Wek RC. Both transcriptional regulation and translational control of ATF4 are central to the integrated stress response. J Biol Chem 2010; 285:33165-74; PMID:20732869
- Dey S, Savant S, Teske BF, Hatzoglou M, Calkhoven CF, Wek RC. Transcriptional repression of ATF4 gene by CCAAT/enhancer-binding protein beta (C/EBPbeta) differentially regulates integrated stress response. J Biol Chem 2012; 287:21936-49; PMID:22556424
- Evstafieva AG, Garaeva AA, Khutornenko AA, Klepikova AV, Logacheva MD, Penin AA, Novakovsky GE, Kovaleva IE, Chumakov PM. A sustained deficiency of mitochondrial respiratory complex III induces an apoptotic cell death through the p53-mediated inhibition of pro-survival activities of the activating transcription factor 4. Cell Death & Disease 2014; 5:e1511; PMID:25375376
- Khutornenko AA, Roudko VV, Chernyak BV, Vartapetian AB, Chumakov PM, Evstafieva AG. Pyrimidine biosynthesis links mitochondrial respiration to the p53 pathway. Proc Natl Acad Sci U S A 2010; 107:12828-33; PMID:20566882
- Khutornenko AA, Dalina AA, Chernyak BV, Chumakov PM, Evstafieva AG. The Role of Dihydroorotate Dehydrogenase in Apoptosis Induction in Response to Inhibition of the Mitochondrial Respiratory Chain Complex III. Acta Naturae 2014; 6:69-75; PMID:24772329
- Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004; 303:844-8; PMID:14704432; http://dx.doi.org/10.1126/science.1092472
- Sidrauski C, Acosta-Alvear D, Khoutorsky A, Vedantham P, Hearn BR, Li H, Gamache K, Gallagher CM, Ang KK, Wilson C, et al. Pharmacological brake-release of mRNA translation enhances cognitive memory. Elife 2013; 2:e00498; PMID:23741617; http://dx.doi.org/10.7554/eLife.00498
- Sidrauski C, McGeachy AM, Ingolia NT, Walter P. The small molecule ISRIB reverses the effects of eIF2alpha phosphorylation on translation and stress granule assembly. Elife 2015; 4:e05033
- Sekine Y, Zyryanova A, Crespillo-Casado A, Fischer PM, Harding HP, Ron D. Stress responses. Mutations in a translation initiation factor identify the target of a memory-enhancing compound. Science 2015; 348:1027-30; PMID:25858979; http://dx.doi.org/10.1126/science.aaa6986
- Sidrauski C, Tsai JC, Kampmann M, Hearn BR, Vedantham P, Jaishankar P, Sokabe M, Mendez AS, Newton BW, Tang EL, et al. Pharmacological dimerization and activation of the exchange factor eIF2B antagonizes the integrated stress response. Elife 2015; 4:e07314; PMID:25875391
- Chumakov SP, Kravchenko JE, Prassolov VS, Frolova EI, Chumakov PM. Efficient downregulation of multiple mRNA targets with a single shRNA-expressing lentiviral vector. Plasmid 2010; 63:143-9; PMID:20064551; http://dx.doi.org/10.1016/j.plasmid.2009.12.003
- Horiguchi M, Koyanagi S, Okamoto A, Suzuki SO, Matsunaga N, Ohdo S. Stress-regulated transcription factor ATF4 promotes neoplastic transformation by suppressing expression of the INK4a/ARF cell senescence factors. Cancer Res 2012; 72:395-401; PMID:22102693; http://dx.doi.org/10.1158/0008-5472.CAN-11-1891
- Kilberg MS, Shan J, Su N. ATF4-dependent transcription mediates signaling of amino acid limitation. Trends Endocrinol Metab 2009; 20:436-443; PMID:19800252; http://dx.doi.org/10.1016/j.tem.2009.05.008
- Lee SO, Andey T, Jin UH, Kim K, Sachdeva M, Safe S. The nuclear receptor TR3 regulates mTORC1 signaling in lung cancer cells expressing wild-type p53. Oncogene 2012; 31:3265-76; PMID:22081070; http://dx.doi.org/10.1038/onc.2011.504
- Wang S, Chen XA, Hu J, Jiang JK, Li Y, Chan-Salis KY, Gu Y, Chen G, Thomas C, Pugh BF, et al. ATF4 gene network mediates cellular response to the anticancer PAD inhibitor YW3-56 in triple-negative breast Cancer cells. Molecular Cancer Therapeutics 2015; 14:877-88; PMID:25612620; http://dx.doi.org/10.1158/1535-7163.MCT-14-1093-T
- Wang Y, Li P, Wang S, Hu J, Chen XA, Wu J, Fisher M, Oshaben K, Zhao N, Gu Y, et al. Anticancer peptidylarginine deiminase (PAD) inhibitors regulate the autophagy flux and the mammalian target of rapamycin complex 1 activity. J Biol Chem 2012; 287:25941-15953; PMID:22605338; http://dx.doi.org/10.1074/jbc.M112.375725
- Wei CL, Wu Q, Vega VB, Chiu KP, Ng P, Zhang T, Shahab A, Yong HC, Fu Y, Weng Z, et al. A global map of p53 transcription-factor binding sites in the human genome. Cell 2006; 124:207-19; PMID:16413492; http://dx.doi.org/10.1016/j.cell.2005.10.043
- Young TA, Cunningham CC, Bailey SM. Reactive oxygen species production by the mitochondrial respiratory chain in isolated rat hepatocytes and liver mitochondria: studies using myxothiazol. Arch Biochem Biophys 2002; 405:65-72; PMID:12176058; http://dx.doi.org/10.1016/S0003-9861(02)00338-7
- Han J, Back SH, Hur J, Lin YH, Gildersleeve R, Shan J, Yuan CL, Krokowski D, Wang S, Hatzoglou M, et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol 2013; 15:481-90; PMID:23624402; http://dx.doi.org/10.1038/ncb2738
- Ben-Sahra I, Dirat B, Laurent K, Puissant A, Auberger P, Budanov A, Tanti JF, Bost F. Sestrin2 integrates Akt and mTOR signaling to protect cells against energetic stress-induced death. Cell Death Differ 2013; 20:611-619; PMID:23238567; http://dx.doi.org/10.1038/cdd.2012.157
- Zakharova NI, Sokolov VV, Rud′ko VV, Melnikov SV, Vartapetian AB, Evstafieva AG. Influence of prothymosin alpha and its mutants on activity of the p53 tumor suppressor. Mol Biol (Mosk) 2008; 42:673-84; PMID:18856068; http://dx.doi.org/10.1134/S002689330804016X