
ABSTRACT
Oxygenated cancer cells have a high metabolic plasticity as they can use glucose, glutamine and lactate as main substrates to support their bioenergetic and biosynthetic activities. Metabolic optimization requires integration. While glycolysis and glutaminolysis can cooperate to support cellular proliferation, oxidative lactate metabolism opposes glycolysis in oxidative cancer cells engaged in a symbiotic relation with their hypoxic/glycolytic neighbors. However, little is known concerning the relationship between oxidative lactate metabolism and glutamine metabolism. Using SiHa and HeLa human cancer cells, this study reports that intracellular lactate signaling promotes glutamine uptake and metabolism in oxidative cancer cells. It depends on the uptake of extracellular lactate by monocarboxylate transporter 1 (MCT1). Lactate first stabilizes hypoxia-inducible factor-2α (HIF-2α), and HIF-2α then transactivates c-Myc in a pathway that mimics a response to hypoxia. Consequently, lactate-induced c-Myc activation triggers the expression of glutamine transporter ASCT2 and of glutaminase 1 (GLS1), resulting in improved glutamine uptake and catabolism. Elucidation of this metabolic dependence could be of therapeutic interest. First, inhibitors of lactate uptake targeting MCT1 are currently entering clinical trials. They have the potential to indirectly repress glutaminolysis. Second, in oxidative cancer cells, resistance to glutaminolysis inhibition could arise from compensation by oxidative lactate metabolism and increased lactate signaling.
Introduction
Cancer cells display a high metabolic adaptability allowing them to optimally cope with fluctuating microenvironmental conditions while simultaneously fulfilling their bioenergetic and biosynthetic activities.Citation1,2 Glucose, glutamine and lactate are among their major metabolic substrates.
For high yield energy metabolism in oxygenated oxidative cancer cells, one molecule of glucose is converted into 2 molecules of pyruvate during glycolysis, and pyruvate fuels the TCA cycle and oxidative phosphorylation (OXPHOS) in cell mitochondria, thus providing a maximum of 38 molcules of ATP (2 from glycolysis + up to 36 from OXPHOS) per molecule of glucose. Matching glycolytic pyruvate production and its use in the TCA cycle is controlled by the Pasteur Effect, i.e., a repressive feedback allosteric control exerted by energy-rich metabolites (such as ATP and citrate) on rate-limiting glycolytic enzymes.Citation3 In addition, coupling glycolysis with OXPHOS is regulated at the first enzymatic step of the TCA cycle where pyruvate dehydrogenase (PDH) is under the repressive control of PDH kinase 1 (PDK1).Citation4,5 Decoupling arises when hypoxia-inducible factor-1 (HIF-1) transcriptionally induces PDK1 expression in hypoxic and proliferating cells.Citation5 It goes along with a glycolytic compensation under the control of HIF-1 that upregulates the expression of most glycolytic transporters and enzymes, among which embryonic isoforms are re-expressed.Citation6,7 Glycolytic cancer cells indeed often express hexokinase 2, an embryonic hexokinase isoform that, compared to hexokinase 1, possesses 2 catalytic sites and no allosteric site, rendering it insensitive to allosteric feed-back inhibition by glucose-6-phosphate.Citation8,9 They also often re-express pyruvate kinase M2 (PKM2) that, compared to PKM1, exists either as an active tetramer promoting ATP production or as an inactive dimer promoting biosynthesis.Citation10,11 The balance between tetrameric and dimeric forms of PKM2 is under allosteric control.Citation12
Cancer cell proliferation necessitates metabolic plasticity. Macromolecule biosynthesis first depends on the ability of the cells to shunt carbohydrates from glycolysis to the pentose phosphate pathway (for NADPH and ribonucleide synthesis) and to aminoacid production pathways.Citation13 It also depends on glutamine uptake and metabolism, with glutamine serving as a precursor for glutathione synthesis, as a source of nitrogen (for DNA synthesis) and glutamate (for aminoacid synthesis and exchanges), and as a fuel for the oxidative TCA cycle.Citation14 When used in the bioreductive mode glutamine also yields citrate and NADPH that serve for lipid biosynthesis.Citation15 In cancer cells, these processes are under the control of transcription factor c-Myc. c-Myc indeed promotes the mRNA splicing of pyruvate kinase (PK) to PKM2,Citation16 producing a versatile PK isoform subject to allosteric regulation.Citation12 With respect to glutaminolysis, high affinity glutamine transporter SLC1A5/ASCT2 is a target gene product of c-Myc, and c-Myc further indirectly upregulates glutaminase 1 (GLS1) expression.Citation17,18
Lactate in tumors originates from glycolysis and glutaminolysis,Citation19 but it is not a dead-end product. From a bioenergetic standpoint, lactate has indeed been shown to be a preferred oxidative substrate to glucose in oxygenated/oxidative cancer cells, thereby supporting cooperativeness between glycolytic and oxidative cellsCitation20,21 and commensalism of oxidative cancer cells for glycolytic fibroblasts.Citation22 Lactate also acts as a paracrine signaling agent creating a pseudohypoxic response in oxidative cancer cells where it activates HIF-1 following its oxidation to pyruvate and pyruvate-mediated inhibition of prolylhydroxylases (PHDs).Citation23-25
The possibility for cancer cells to use different resources necessitates integration and coordination. Hence, glycolysis and glutaminolysis cooperate to support cancer cell proliferation.Citation26 In oxidative cancer cells, the use of lactate inhibits glucose metabolism through allosteric feedback repression of glycolysis,Citation27 supporting an intercellular metabolic symbiosis based on substrate exchanges.Citation20,21 In this context, whether and how lactate metabolism and glutaminolysis influence each other in oxidative cancer cells remains an open question. Therefore, this study aimed to test the relationship between lactate and glutamine metabolism in oxidative SiHa and HeLa cancer cells that, contrarily to glycolytic ones, are sensitive to lactate signaling.Citation25 Indeed, lactate transport across the plasma membrane is facilitated by passive lactate-proton symporters of the monocarboxylate family (MCTs): owing to an inward gradient of lactate and protons, oxidative cancer cells are capable of importing lactate for signaling, whereas glycolytic cancer cells that produce and export lactic acid do not take up lactate and do not display intracellular signaling in response to exogenous lactate.Citation25
Results
Selection of SiHa and HeLa human cancer cells as models of oxidative cancer cell metabolism
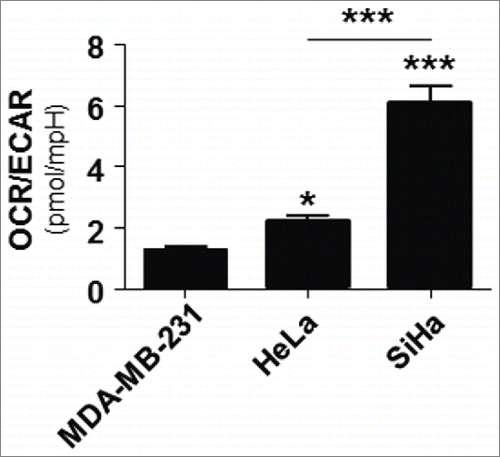
Lactate transport across cells membranes is primarily facilitated by passive transporters of the MCT family,Citation28 implying that oxidative cells consuming lactate are prone to lactate uptake, whereas glycolytic cells that produce lactate do not significantly import lactate and do not display intracellular signaling by exogenous lactate.Citation25 Because this study aimed to test the influence of oxidative lactate metabolism on glutamine metabolism, we first selected oxidative cancer cell lines as main models. Bioenergetic measurements using a Seahorse analyzer showed that SiHa and HeLa human cervix cancer cells are more oxidative than MDA-MB-231 human breast cancer cells (): SiHa had the highest OCR/ECAR ratio (indicative of a highly oxidative metabolism),Citation29 and HeLa were less oxidative than SiHa but more than MDA-MB-231 that were used as a glycolytic control in this experiment (Warburg phenotype).Citation30 These data are in good agreement with the previous observation that SiHa and HeLa cells take up lactate and activate HIF-1 in response to exogenous lactate.Citation20,25,31
Figure 1. SiHa and HeLa are more oxidative than MDA-MB-231 human cancer cells. Ratio between oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) in SiHa, HeLa and MDA-MB-231 cells (n = 5–8, *p < 0.05, ***p < 0.001).
Lactate stimulates glutaminolysis in oxidative cancer cells
To identify a possible crosstalk between lactate and glutamine metabolism, we first analyzed biopsies of SiHa tumors grown in Matrigel plugs in mice. Each mouse received 2 plugs: one containing sodium lactate (30 mM) and the other one an equal volume of saline. When evaluating the glutamine-processing pathway, we found that lactate delivered from Matrigel during 12 d stimulated the protein expression of glutamine transporter ASCT2 (, left panel) and of glutaminase 1 (GLS1) (, left panel), compared to paired tumors grown in the absence of lactate. Membrane-bound ASCT2 is the main transporter mediating cellular glutamine uptake, whereas mitochondria-resident GLS1 is the first enzyme of the glutaminolytic pathway converting L-glutamine into L-glutamate.Citation14,32 Our data thus suggested that lactate metabolism could potentially stimulate glutaminolysis in cancer cells. Accordingly, lactate uptake was mandatory for the response as tumors deficient for the inward lactate transporter MCT1 (shMCT1, see reference Citation25 for target extinction) did not display lactate-induced ASCT2 () and GLS1 () protein expression.
Figure 2. Lactate promotes glutamine metabolism in cancer. (A) Representative immunoblots and bar graphs represent the expression of glutamine transporter ASCT2 in tumors collected 12 d after having been established in Matrigel plugs in mice using SiHa cancer cells transfected with a control shRNA (shCTR, left panel) or a shRNA targeting monocarboxylate transporter 1 (shMCT1, right panel). Matrigel plugs contained 30 mM of lactate or an equal volume of saline (n = 5; ns, not significant, *p < 0.05). (B) Same is in A but analyzing glutaminase 1 (GLS1) protein expression (n = 5; ns, not significant, *p < 0.05). (C) Wild-type SiHa cells were treated during 6-h with sodium lactate (10 mM) and incubated during 18 min in the presence of increasing doses of L-[3,4–3H(N)]-glutamine. The graph shows intracellular 3H incorporation (n = 8; ***p < 0.005). (D) Representative immunoblot showing GDH1 and β-actin protein expression in SiHa cancer cells transfected with a siRNA targeting GLUD1/GDH1 (siGDH1). (E) Same as C but using mock-transfected SiHa cells (left, n = 8; ***p < 0.005) or SiHa cells transfected with siGDH1 (right, n = 8; ***p < 0.005).
![Figure 2. Lactate promotes glutamine metabolism in cancer. (A) Representative immunoblots and bar graphs represent the expression of glutamine transporter ASCT2 in tumors collected 12 d after having been established in Matrigel plugs in mice using SiHa cancer cells transfected with a control shRNA (shCTR, left panel) or a shRNA targeting monocarboxylate transporter 1 (shMCT1, right panel). Matrigel plugs contained 30 mM of lactate or an equal volume of saline (n = 5; ns, not significant, *p < 0.05). (B) Same is in A but analyzing glutaminase 1 (GLS1) protein expression (n = 5; ns, not significant, *p < 0.05). (C) Wild-type SiHa cells were treated during 6-h with sodium lactate (10 mM) and incubated during 18 min in the presence of increasing doses of L-[3,4–3H(N)]-glutamine. The graph shows intracellular 3H incorporation (n = 8; ***p < 0.005). (D) Representative immunoblot showing GDH1 and β-actin protein expression in SiHa cancer cells transfected with a siRNA targeting GLUD1/GDH1 (siGDH1). (E) Same as C but using mock-transfected SiHa cells (left, n = 8; ***p < 0.005) or SiHa cells transfected with siGDH1 (right, n = 8; ***p < 0.005).](/cms/asset/1c4239ab-c5a0-45d7-b0fc-f2bae33e6d61/kccy_a_1120930_f0002_b.gif)
Tumors are highly heterogeneous metabolically and in their cellular composition. To document lactate-induced glutaminolysis in oxidative cancer cells, we therefore used an in vitro assay with SiHa cells.Citation20 After a 6-h pretreatment with 10 mM of sodium lactate, cells were exposed to increasing doses of [3H]-L-glutamine. Unexpectedly, 3H labeling was reduced in lactate-treated compared to control cells (). This could indicate either reduced glutamine uptake or increased glutamine metabolism associated with the export of downstream labeled metabolites. To discriminate between the 2 possibilities, we targeted mitochondrial glutamine metabolism using smartpool siRNAs directed against glutamate dehydrogenase 1 (GLUD1/GDH1, see for target extinction), the enzyme converting L-glutamate into α-ketoglutarate downstream of GLS1 in mitochondria. Compared to mock-transfected cells that behaved as wild-type ones, blocking glutamine metabolism unveiled increased [3H]-L-glutamine uptake in lactate-treated compared to lactate-untreated cells (). Taken together, our data thus indicated that lactate uptake triggers glutamine uptake and metabolism in oxidative cancer cells.
Lactate activates c-Myc and stabilizes HIF-2α in oxidative cancer cells
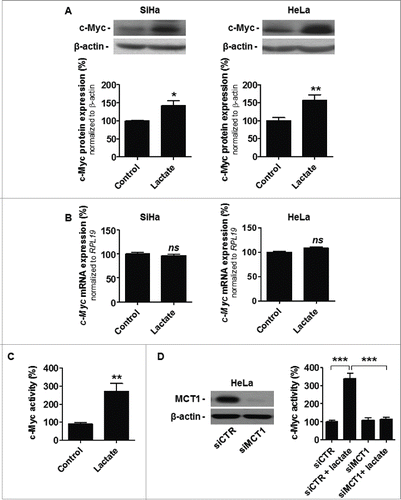
ASCT2 and GLS1 are well known to be under the transcriptional control of c-Myc,Citation17,18 but to our knowledge lactate has never been reported to regulate c-Myc activity. To establish a molecular link between lactate and glutamine pathways, we first documented that a 6-h treatment with 10 mM of sodium lactate induced c-Myc protein expression in SiHa cells, which was confirmed in HeLa (). In both cell lines, the response was posttranscriptional (). It was significant biologically: lactate, used as a sodium salt in pH-buffered medium, induced a ∼3-fold increase is c-Myc activity that was detected using a dual luciferase reporter assay (). Moreover, SLC16A1/MCT1 silencing repressed this response to lactate (; a representative immunoblot shows target extinction), thus demonstrating that MCT1-dependent lactate uptake stimulates c-Myc activity in these cancer cells.
Figure 3. MCT1-dependent lactate uptake stabilizes and activates c-Myc in oxidative cancer cells. (A-D) Cancer cells were treated ± 10 mM sodium lactate for 6-h. A, representative immunoblots and bar graphs represent c-Myc protein expression in oxidative SiHa (left) and HeLa (right) cancer cells (n = 4–8; *p < 0.05, **p < 0.01 ). (B) c-Myc mRNA expression detected using RT-qPCR in SiHa (left) and HeLa (right) cells (n = 6–9; ns, not significant). (C) c-Myc activity in wild-type HeLa cells was quantified using a dual reporter luciferase assay (n = 4; **p < 0.01). (D) Same as in C but using HeLa cells transfected with a control siRNA (siCTR) or with a siRNA targeting SLC16A1/MCT1 (siMCT1; n = 4; ***p < 0.005). The representative immunoblot shows MCT1 and β-actin protein expression in HeLa cells transfected with siCTR or siMCT1.
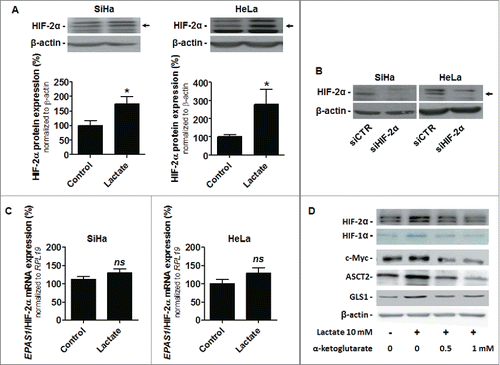
We next aimed to understand how lactate activates c-Myc. Lactate is well-known to act as a hypoxia-mimetic capable of activating HIF-1 in oxidative cancer cells, including SiHa and HeLa.Citation23-25 In the process, lactate-derived pyruvate competes with α-ketoglutarate to block HIF PHDs, thereby stabilizing HIF-1 subunit α and triggering HIF-1 activity. Interestingly, HIF-2α, similar to HIF-1α, is under PHD control.Citation33 Moreover, HIF-2α enhances c-Myc activity through binding to and stabilizing c-Myc:Max complexes in cell nuclei.Citation34 We therefore aimed to test the effect of lactate on HIF-2α in our models. That lactate stabilizes HIF-2α protein expression independently of hypoxia was verified in both SiHa and HeLa cells (; siRNA knockdown identified which of the bands corresponds to HIF-2α in ). Lactate did not modify EPAS1/HIF-2α mRNA level (), thus indicating that lactate activates HIF-1 and HIF-2 by a similar posttranslational mechanism in normoxic cancer cells. Accordingly, increasing doses of α-ketoglutarate repressed the stabilization of HIF-1α and HIF-2α by lactate in normoxic HeLa cells (). Moreover, the level of expression of c-Myc, ASCT2 and GLS1 followed the response of HIFs to lactate and α-ketoglutarate (). These data collectively pointed at the existence of a lactate signaling pathway that promotes glutamine uptake and use by sequentially inhibiting HIF PHDs, stabilizing HIF-2α, stabilizing and activating c-Myc and increasing the expression of ASCT2 and GLS1.
Figure 4. Lactate stabilizes HIF-2α in oxidative cancer cells. A-C, SiHa and HeLa cancer cells were treated ± 10 mM sodium lactate for 6-h. (A) Representative immunoblots and bar graphs represent HIF-2α protein expression (n = 4–8; *p < 0.05). (B) The representative immunoblot shows HIF-2α and β-actin protein expression in SiHa and HeLa cells transfected with a control siRNA (siCTR) or with a siRNA targeting EPAS1/HIF-2α (siHIF-2α). (C) EPAS1/HIF-2α mRNA expression was detected using RT-qPCR (n = 6; ns, not significant). (D) HeLa cells were treated ± 10 mM sodium lactate for 6-h in the presence or not of increasing doses of α-ketoglutarate. Immunoblots are representative of n = 3 and show HIF-2α, HIF-1α, c-Myc, ASCT2, GLS1 and β-actin expression.
Lactate triggers HIF-2α-dependent c-Myc activation in oxidative cancer cells
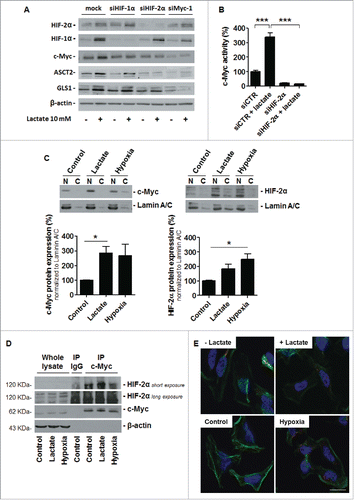
In normoxic SiHa and HeLa cells, lactate simultaneously stabilized HIF-1αCitation25 and HIF-2α (), 2 proteins that exert antagonistic effects on c-Myc.Citation35,36 Lactate activates c-Myc, indicating that HIF-2α activity predominates over HIF-1α for c-Myc induction. Accordingly, silencing EPAS1/HIF-2α (siHIF-2α) was sufficient to repress the lactate-induced expression of c-Myc, ASCT2 and GLS1 in HeLa cells (, compare lines 1 and 2 to lines 5 and 6); whereas HIF-1α stabilization by lactate was preserved. Comparatively, lactate-induced expression of c-Myc, ASCT2 and GLS1 was preserved upon HIF-1α silencing (siHIF-1α), and siHIF-1α did not interfere with HIF-2α protein stabilization by lactate (). Silencing c-Myc repressed the upregulation of ASCT2 and GLS1 expression by lactate in the cells, with preserved HIF-1α and HIF-2α induction (). This experiment thus indicated that HIF-2α controls c-Myc activation by lactate. Accordingly, silencing HIF-2α was sufficient to repress both lactate-induced and basal c-Myc activity in HeLa cells ().
Figure 5. HIF-2α controls c-Myc activation by lactate in oxidative cancer cells. (A) HIF-2α, HIF-1α, c-Myc, ASCT2, GLS1 and β-actin expression was detected in HeLa cells that were either mock-transfected, transfected with siHIF-1α, siHIF-2α or siMyc-1 and treated ± 10 mM sodium lactate for 6-h. Immunoblots are representative of n = 3. (B) c-Myc activity was quantified using a dual reporter luciferase assay in HeLa cells transfected with siCTR or siHIF-2α and cultured for 6-h ± 10 mM sodium lactate (n = 4; ***P < 0.005). (C) Representative immunoblots show c-Myc (left) and HIF-2α (right) protein expression in the nuclear (N) and cytoplasmic (C) fractions of HeLa cells treated for 24-h ± 10 mM sodium lactate or ± hypoxia (1% O2). Graphs show protein expression in the nuclear fraction normalized by nuclear marker lamin A/C expression (n = 3; *p < 0.05). (D) HeLa cells were treated during 24-h ± 10 mM sodium lactate or ± hypoxia (1% O2). Representative immunoblots of n = 3 show HIF-2α and c-Myc expression in whole cell lysate, in control IgG immunoprecipitate (IP) and in the c-Myc immunoprecipitate. E, HIF-2α:c-Myc heterocomplexes were detected using a proximity ligation assay in HeLa cells after a 6-h treatment ± 10 mM sodium lactate or ± hypoxia (1% O2). Heterocomplexes appear as red dots in the representative pictures where cell nuclei are stained in blue (DAPI) and F-actin in green (phalloidin). Omission of the primary antibody against c-Myc was used as a negative control. Bar = 50 μm.
Upon lactate treatment, HIF-2α and c-Myc simultaneously translocated to cell nuclei (), thus recapitulating the cell response to hypoxia (1% O2). Despite no commercially available antibody allowed us to verify lactate-induced HIF-2α:Max interaction in our human cell lines, we found that HIF-2α and c-Myc co-immunoprecipitated more abundantly in lactate-treated compared to untreated HeLa cells (). Hypoxia recapitulated the effects of lactate (). Increased interaction between HIF-2α and c-Myc was also visualized in a proximity ligation assay (PLA) where lactate triggered the formation of HIF-2α:c-Myc heterocomplexes ().
Lactate-induced HIF-2α -c-Myc signaling stimulates the glutamine pathway
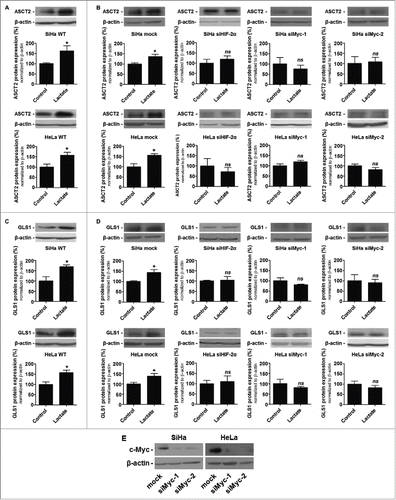
Lactate (10 mM) increased ASCT2 protein expression in SiHa and in HeLa cells () to the same extent as it did at a higher concentration (30 mM) in SiHa tumors in vivo (see ). Induction was fast (6 h, ) and the effect was persistent upon chronic lactate delivery (12 days, ). Either c-Myc silencing (with 2 different siRNAs, siMyc-1 and siMyc-2) or HIF-2α silencing repressed ASCT2 protein induction by lactate in SiHa and in HeLa cells (). Similarly, lactate-induced GLS1 expression in the cells () was totally and independently repressed by siMyc-1, siMyc-2 and siHIF-2α (). Target extinction is shown in for both cell lines. These data link lactate-induced HIF-2α and c-Myc expression to glutamine metabolism, thus confirming the existence of a functional lactate-HIF-2α-c-Myc signaling pathway in oxidative cancer cells.
Figure 6. Lactate promotes HIF-2α- and c-Myc-dependent ASCT2 and GLS1 protein expression in oxidative cancer cells. (A-D) Cancer cells were treated ± 10 mM sodium lactate for 6-h. (A), representative immunoblots and bar graphs show ASCT2 protein expression in wild-type SiHa (upper panel) and HeLa (lower panel) cells (n = 4–7; *p < 0.05). (B) Same as in A but using mock-transfected SiHa and HeLa cells (left panels), cells transfected with siHIF-2α (medium left), and cells transfected with 2 different siRNAs targeting c-Myc (siMyc-1 and siMyc-2, medium right and right) (n = 3–6; ns, not significant, *p < 0.05). (C) Representative immunoblots and bar graphs show GLS1 protein expression in wild-type SiHa (upper panel) and HeLa (lower panel) cells (n = 4–7; *p < 0.05). (D) Same as in C but using mock-transfected HeLa and SiHa cells (left), cells transfected with siHIF-2α (medium left), and cells transfected with siMyc-1 (medium right) or siMyc-2 (right) (n = 3–6; ns, not significant, *p < 0.05). (E) The representative immunoblot shows c-Myc and β-actin protein expression in SiHa and HeLa cells transfected with siMyc-1 or siMyc-2.
Lactate increases the oxidative metabolism of glutamine in a c-Myc-dependent manner
We finally aimed to understand the metabolic advantage for oxidative cancer cells of combining lactate and glutamine metabolism. We found that lactate (used as a pretreatment to discriminate its metabolic effects from that of glutamine) enhanced the oxygen consumption rate of both SiHa and HeLa cells fueled with glutamine (2 mM) (). Increased oxidative glutaminolysis in response to lactate was dependent on c-Myc as it was lost in cells transfected with siMyc-1 or siMyc-2 (). These results demonstrate that lactate stimulates the oxidative use of glutamine through c-Myc activation, as depicted in .
Figure 7. Lactate stimulates oxidative glutaminolysis. A-B, the oxygen consumption rate (OCR) of SiHa and HeLa cells was measured in a glucose-free fresh solution containing glutamine (2 mM), 6-h after cell treatment ± 10 mM sodium lactate. (A) OCR of wild-type SiHa and HeLa cells (n = 3 independent experiments; ***p < 0.005). (B) As in A but using mock-transfected SiHa and HeLa cells or the cells transfected with siMyc-1 or siMyc-2 (n = 3 independent experiments; ns, not significant; *p < 0.05, **p < 0.01). (C) Model depicting lactate-induced glutamine uptake and metabolism in oxidative cancer cells. According to the model, lactate enters into oxidative cancer cells using monocarboxylate transporter 1 (MCT1)-facilitated transport and is oxidized to pyruvate in the cytosol (the lactate dehydrogenase 1 [LDH1] reaction). Lactate-derived pyruvate mediates lactate signaling by competitively inhibiting the use of α-ketoglutarate (α-KG) by prolylhydroxylases (PHD), which results in HIF-2α protein stabilization. HIF-2α then stabilizes c-Myc protein expression (most probably by stabilizing c-Myc:Max complexes) in the cell nucleus where c-Myc promotes the transcription of target genes, among which SLC1A5/ASCT2 is the major membrane-bound glutamine transporter and glutaminase 1 (GLS1) catalyzes the conversion of L-glutamine to L-glutamate at the first step of glutaminolysis in the mitochondrion. Glutamine can be used either in the cytosol or in the mitochondrion. In the mitochondrial matrix, glutamate dehydrogenase 1 (GDH1) converts L-glutamate in α-KG to fuel oxidative glutaminolysis. Because silencing MCT1 blocks lactate-induced c-Myc activation, MCT1 inhibitors have the potential to indirectly inhibit glutamine uptake and metabolism in oxidative cancer cells.
![Figure 7. Lactate stimulates oxidative glutaminolysis. A-B, the oxygen consumption rate (OCR) of SiHa and HeLa cells was measured in a glucose-free fresh solution containing glutamine (2 mM), 6-h after cell treatment ± 10 mM sodium lactate. (A) OCR of wild-type SiHa and HeLa cells (n = 3 independent experiments; ***p < 0.005). (B) As in A but using mock-transfected SiHa and HeLa cells or the cells transfected with siMyc-1 or siMyc-2 (n = 3 independent experiments; ns, not significant; *p < 0.05, **p < 0.01). (C) Model depicting lactate-induced glutamine uptake and metabolism in oxidative cancer cells. According to the model, lactate enters into oxidative cancer cells using monocarboxylate transporter 1 (MCT1)-facilitated transport and is oxidized to pyruvate in the cytosol (the lactate dehydrogenase 1 [LDH1] reaction). Lactate-derived pyruvate mediates lactate signaling by competitively inhibiting the use of α-ketoglutarate (α-KG) by prolylhydroxylases (PHD), which results in HIF-2α protein stabilization. HIF-2α then stabilizes c-Myc protein expression (most probably by stabilizing c-Myc:Max complexes) in the cell nucleus where c-Myc promotes the transcription of target genes, among which SLC1A5/ASCT2 is the major membrane-bound glutamine transporter and glutaminase 1 (GLS1) catalyzes the conversion of L-glutamine to L-glutamate at the first step of glutaminolysis in the mitochondrion. Glutamine can be used either in the cytosol or in the mitochondrion. In the mitochondrial matrix, glutamate dehydrogenase 1 (GDH1) converts L-glutamate in α-KG to fuel oxidative glutaminolysis. Because silencing MCT1 blocks lactate-induced c-Myc activation, MCT1 inhibitors have the potential to indirectly inhibit glutamine uptake and metabolism in oxidative cancer cells.](/cms/asset/f307bf3c-8f26-409d-b515-f8cd64e28f33/kccy_a_1120930_f0007_c.gif)
Discussion
Cancer cells strive to optimally fulfill their metabolic needs in a microenvironment characterized by dynamic changes in nutrient availability. Oxygenated cancer cells close to perfused blood vessels are at a privileged location with high glucose and glutamine inputs (as the 2 nutrients are delivered from blood vessels) and additional lactate delivery from glycolytic/hypoxic cancer and host cells. From a bioenergetic standpoint, tumor growth is promoted when oxygenated cancer cells adopt an oxidative metabolism based on the preferential use of lactate compared to glucose, because thereby glucose spared at the vicinity of blood vessels is made available to fuel the glycolytic metabolism of distant hypoxic cancer cells.Citation20,21,25 Here, we report that cancer cells opting for the oxidative use of lactate further take advantage of intracellular lactate signaling to optimize glutamine metabolism, in particular oxidative glutaminolysis. Lactate indeed stimulates glutamine uptake and catabolism through PHD inhibition, HIF-2α stabilization, HIF-2α-mediated c-Myc transactivation, and c-Myc-mediated increase in the expression of the inward glutamine transporter ASCT2 and of GLS1, the latter catalyzing the conversion of L-glutamine in L-glutamate at the first step of glutaminolysis in mitochondria ().
A high level of lactate is of poor prognosis for many types of human tumors,Citation37,38 which not only reflects the glycolytic activity of cancer cells but also lactate-mediated activities. Lactate can indeed generally be used as a metabolic fuel by oxidative cancer cells.Citation20,21,39-44 It can also act as a signaling agent capable of activating transcription factors HIF-1,Citation23-25,45 NF-κB,Citation21,46 and c-Myc via posttranslational HIF-2α stabilization (this study), all independently of hypoxia.
Besides lactate binding to GPR81 (a G protein-coupled lactate receptor at the cell membrane),Citation47 at least 2 features are required for lactate signaling. First, lactate signaling depends on lactate uptake, a process primarily facilitated by MCT1 in oxidative cancer cells.Citation20,25,46 MCT1 is a passive lactate-proton symporter (Kmlactate ≈2.5–5 mM) driven by the gradient of lactate and protons across the cell membrane,Citation28 thus explaining why oxidative cancer cells are responsive to exogenous lactate signaling, whereas glycolytic cancer cells producing lactate are not.Citation25 The key role exerted by MCT1 in controlling lactate uptake is perhaps best illustrated by the fact that MCT1 inhibition, using pharmacological or genetic approaches, not only prevents lactate being used as an OXPHOS fuelCitation20 but also lactate-induced HIF-1,Citation25,45 NF-κBCitation46 and HIF-2α-c-Myc signaling (). A second key feature of lactate signaling is the ability of lactate to promote PHD inhibition independently of hypoxia. HIF-1 and HIF-2 activities are posttranslationally controlled by PHDs that catalyze the oxygen-dependent hydroxylation of key proline residues on HIF-1/2α subunits, targeting them for ubiquitylation and proteasome-mediated degradation under normoxia.Citation33,48 If PHDs are well known to be inactivated upon oxygen limitation (thus allowing rapid HIF-1α and HIF-2α protein stabilization under hypoxia), lactate has been documented to inhibit PHD activity as well.Citation23,24,45 It does so independently of hypoxia by supporting an inhibitory competition between lactate-derived pyruvate and α-ketoglutarate. Both HIF-1α and HIF-2α are under PHD control,Citation25,33 and we showed here that, similar to HIF-1α,Citation25 lactate stabilizes HIF-2α in oxidative cancer cells (). Involvement of PHD inhibition for lactate-induced HIF-1α and HIF-2α stabilization does not exclude additional effects of lactate on the cellular redox status, as lactate oxidation to pyruvate (the lactate dehydrogenase 1 reaction) increases the NADH/NAD+ ratio and pyruvate has antioxidant properties.Citation49,50 Considering that cancer cells chronically adapted to extracellular acidity activate HIF-2α and become addicted to glutamine,Citation51 the contribution of protons cotransported with lactate by MCT1 certainly deserves further consideration.
In hypoxic cancer cells and in von Hippel Lindau (VHL)-deficient cancer cells with high HIF-1α expression, HIF-1 opposes c-Myc activity. HIF-1 indeed increases the transcription of MXI1 that encodes a repressor of c-Myc and promotes c-Myc degradation.Citation35,36 This pathway has been proposed to prevent excessive c-Myc activation and c-Myc-induced apoptosis in hypoxic cancer cells.Citation35 Conversely, HIF-2α is well-known to exert an opposite effect: it increases c-Myc activity by binding to and stabilizing active c-Myc:Max complexes.Citation34 In response to lactate, we observed that HIF-2α exerts a predominant influence on c-Myc in normoxic oxidative cancer cells, which was illustrated by the facts that (i) lactate stabilized c-Myc expression posttranscriptionally (), (ii) silencing HIF-2α was sufficient to repress lactate-induced c-Myc stabilization and activation (), and (iii) lactate triggered the formation of HIF-2α:c-Myc heterocomplexes that localized almost exclusively in cell nuclei (). That HIF-2α predominates over HIF-1α to support c-Myc activation by lactate in normoxic cancer cells while HIF-1α-dependent c-Myc repression arises under hypoxia can be explained by different levels of HIF-1α stabilization in both conditions. Indeed, in SiHa cells, hypoxia induces a 10-fold higher expression of HIF-1α compared to lactate (10 mM),Citation25 which represents a 1 Log difference in the amplitude of the response. Thus, more generally, one can speculate that there is a threshold of HIF-1 activation below which HIF-2α-induced c-Myc activation is minimally repressed and above which it can be fully repressed. Additional studies are obviously needed to verify this hypothesis.
Radiolabelling experiments showed that lactate triggers glutamine uptake by oxidative SiHa cells, which necessitated glutaminolysis inhibition at the GDH1 step of oxidative glutaminolysis in mitochondria to be seen (). Glutamine metabolism controls different aspects of cell biology. With GDH1 inhibition and radioassays reflecting initial rate of glutamine uptake, the effects of lactate were quite subtle yet significant, both in terms of uptake rate () and glutamine-dependent oxygen consumption (). This strongly indicates that, even if a portion of the extra load of glutamine is used in oxidative glutaminolysis in mitochondria,Citation52 glutamine could provide additional advantages to oxidative cancer cells. First, glutamine is used as a precursor for glutathione synthesis and fuels NADPH production (malate to pyruvate conversion), improving the antioxidant defensesCitation26 that are highly solicited in oxidative cancer cells with enhanced metabolic activities. Second, bioreductive glutamine metabolism yields citrate that promotes lipid synthesis, thus sustaining cancer cell proliferation.Citation19 Finally, glutamine via glutamate is used as a nitrogen donor by aminotransferases, and it can further serve for aminoacid exchange across the plasma membrane.Citation26 Increased glutamine uptake in cancer cells using lactate as an oxidative fuel could thus optimize protein and nucleotide synthesis. The fact that we observed only a minor shift in glutamine uptake curves upon GDH1 inhibition suggests that glutamine is primarily used for anaplerosis rather than cataplerosis in oxidative cancer cells fueled with lactate. A significant fraction of glutamine and its metabolites could also ultimately be exported from the cells, which deserves to be investigated in future studies.
In tumors in vivo, we documented increased ASCT2 and GLS1 protein expression in response to lactate (), which was absent when cancer cells were deficient for SLC16A1/MCT1. Because wild-type SiHa cells were competent for metabolizing glutamine and as among the different metabolic phenotypes that naturally develop in vivo only oxidative cancer cells are capable of taking up lactate,Citation20,25 we did not observe increased tumor radiolabeling after L-[14C(U)]-glutamine delivery (data not shown). Nevertheless, the fact that lactate strongly activated the glutaminolytic machinery in a MCT1-dependent manner at the whole tumor level suggests that, in addition to antimetabolicCitation20 and anti-angiogenic effects,Citation25,45,46 glutamine metabolism inhibition could participate in the previously identified antitumor effects of MCT1 inhibition. Conversely, increased lactate signaling could be responsible for tumor resistance to antiglutaminolytic treatments targeting ASCT2 or GLS1 (see reference Citation9 for a recent review). In support of this, the ASCT2 inhibitor γ-L-glutamyl-p-nitroanilide (GPNA) has been reported to enhance MCT1 expression and activity in cancer.Citation53 Compensation could further involve c-Myc-induced SLC16A1/MCT1 transcription.Citation54 Thus, crosstalks between lactate and glutamine metabolism deserve special consideration for future therapeutic developments.
Conclusively, we report that lactate used at physiologically relevant doses (10–30 mM)Citation38 sequentially inhibits PHDs, stabilizes HIF-2α, activates c-Myc and stimulates glutamine metabolism in oxidative cancer cells. While our study focused on the coordination between oxidative lactate metabolism and glutaminolysis, it paves the way for future studies aimed to explore additional effects of lactate that could be conveyed by HIF-2α and c-Myc in cancer and in other pathologies. Because it can activate HIF-1, HIF-2, NF-κB and c-Myc in tumors, we propose lactate to be considered as an oncometabolite.
Experimental procedures
Cell lines and reagents
HeLa human cervix cancer adenocarcinoma cells, SiHa human cervix squamous cell carcinoma cells and MDA-MB-231 human breast cancer cells were from ATCC. HeLa were routinely cultured in RPMI containing FBS (10%), non-essential aminoacids (1%), sodium pyruvate (1%) and penicillin-streptomycin (1%); and SiHa and MDA-MB-231 in DMEM containing glucose (4.5 g/L), FBS (10%) and penicillin-streptomycin (1%). Media were buffered at pH 7.4 (3.7 g/L NaHCO3, 5% CO2). To avoid changes in extracellular pH, lactate was used as a sodium salt. Hypoxia (1% O2) was achieved in a Ruskinn hypoxic workstation. Unless specified otherwise, all reagents were from Sigma-Aldrich.
In vivo experiments
All in vivo experiments were performed with approval of UCL Comité d'Ethique pour l'Expérimentation Animale (approval ID TUMETABO) according to national animal care regulations. Tumor growth in Matrigel plugs ± lactate was achieved using a previously disclosed protocolCitation25 with SiHa cells expressing a control shRNA (shCTR, Addgene plasmid 1864) or a shRNA targeting SLC16A1/MCT1 (Open Biosystems shRNA clone TRCN0000038340). Briefly, anesthetized (ketamine/xylazine) 8 weeks-old male BALB/c nude mice (Elevage Janvier) received bilateral subcutaneous injections of 10Citation6 cancer cells in 300-μl of growth factor-reduced Matrigel containing 30 mM of sodium lactate (right flank) or an equal volume of saline (left flank). Twelve days after implantation, mice were sacrificed. Matrigel plugs were collected and snap-frozen in liquid nitrogen-cooled isopentane for further protein expression analyses. We also used samples collected in a previous study.Citation25
Co-immunoprecipitation, immunoblotting and immunolabeling
Co-immunoprecipitation - Cells were lysed in immunoprecipitation (IP) buffer (40 mM Hepes pH 7.4, 150 mM NaCl, 2 mM sodium pyrophosphate, 10 mM glycerol-3-phosphate, 0.5% Triton X-100, protease inhibitor cocktail [Sigma] and PhosSTOP phosphatase inhibitor [Roche]). Co-immunoprecipitation was performed using Dynabeads Protein G (Invitrogen). Manufacturer's recommendations were followed with the following modifications. Two micrograms of anti-c-Myc mouse monoclonal antibody (Santa Cruz) were admixed with 100 μl protein lysate at 4ºC under steering for ≥ 6 h prior to the addition of beads. IP buffer was used in place of Ab Binding & Washing Buffer. Beads were washed with IP buffer. Beads were added to the anti-c-Myc antibody-cell lysate mixture and incubated overnight at 4°C under steering. After binding, proteins were eluted from the beads using 200 μL of IP buffer, samples were diluted in 30 μL Laemmli (3X) and run on an 8% SDS-PAGE gel. Proteins in gels were transferred onto PVDF membranes for subsequent immunoblot analyses.
Immunoblotting - Cell fractionation was performed using the Nuclear Extract Kit from Active Motif according to manufacturer's guidelines. Tissue samples were homogenized in RIPA buffer. Cells were harvested in RIPA buffer. Western blotting was performed on an equal amount of total proteins as previously described,Citation21 except that lysates were not heat-denaturated. Primary antibodies were: rabbit polyclonals targeting HIF-2α (Novus Biologicals), MCT1 (Millipore), lamin A/C (Santa Cruz), GLS1 (Proteintech), ASCT2 (Millipore); mouse monoclonals against c-Myc (BD Biosciences), HIF-1α (BD Biosciences) and β-actin (Sigma); and a goat polyclonal against GDH1 (Novus biologicals).
Immunolabeling - The in situ detection of c-Myc:HIF-2α heterocomplexes was achieved using a proximity ligation assay (PLA, Duolink, Olink Bioscience) that detects interactions at a distance of less than 40 nm.Citation55 We followed manufacturer's recommendations on cells fixed with 4% paraformaldehyde and permeabilized with Triton X-100 (0.1%). Negative controls were run in the absence of the anti-c-Myc primary antibody. All images were acquired with the same settings on a Zeiss AxioImager microscope equipped with an Apotome module.
RT-qPCR
A previously disclosed protocolCitation51 was used for RT-qPCR with SYBR green on ViiA 7 Real-Time and Biorad IQ5 PCR systems. Primers were: human EPAS1/HIF-2α sense, 5′-GTC TCT CCA CCC CAT GTC TC-3′, antisense, 5′-GGT TCT TCA TCC GTT TCC AC-3′; human c-Myc sense, 5′-ATG AAA AGG CCC CCA AGG TA-3′, antisense, 5′-TTT CCG CAA CAA GTC CTC TTC-3′; housekeeping human ribosomal protein L19 (hRPL19) sense, 5′-CAA GCG GAT TCT CT GGA ACA-3′, antisense, 5′-TGG TCA GCC AGG AGC TTC TT-3′.
c-Myc activity assays
c-Myc activity was measured using the dual luciferase kit from Promega with pBV-Luc wt MBS1–4 (Addgene) as reporter of c-Myc activity and pRL Renilla Luciferase (Promega) as internal control. Cells were co-transfected using lipofectamine RNAiMax (Life Technologies).
RNA interference
Cells were transfected with siRNAs using Lipofectamine RNAiMax according to manufacturer's protocol. siRNAs targeted: human SLC16A1/MCT1 5′-AAG AGG CUG ACU UUU CCA AAU-3′ (siMCT1), human HIF-1α 5′-AAC UGG ACA CAG UGU GUU UGA-3′ (siHIF-1α), human EPAS1/HIF-2α 5′-CCC GGA UAG ACU UAU UGC CAA-3′ (siHIF-2α), and human c-Myc 5′-GAG AAC AGU UGA AAC ACA A-3′ (siMyc-1) and 5′-GAG AAC AGU UGA AAC ACA A-3′ (siMyc-2). ON-TARGETplus smartpool siRNAs (5′-CCC AAG AAC UAU ACU GAU A-3′, 5′-GCG AAG CGC UGU UGC UGU C-3′, 5′-GAA GAU CUA UGG UUG ACU A-3′, 5′-CCC AUG AAG UGC UAG AUA A-3′; Dharmacon) were used to silence human GLUD1/GDH1. Mock transfection and Allstar siRNA (Qiagen) were used as negative controls.
Metabolic assays
Mitochondrial oxygen consumption rates (OCR) and extracellular acidification rates (ECAR) were measured using a Seahorse XF96 Extracellular Flux Analyzer (Seahorse Bioscience). HeLa, SiHa and MDA-MB-231 cells (20,000 cells/well in 200 μL of medium) were seeded in 96-well Seahorse assay plates and allowed to adhere overnight. Cells were then treated ± 10 mM sodium lactate for 6 h. Prior to measurements, supernatants were replaced by non-buffered DMEM containing 2 mM glutamine without glucose and serum, and the cells were equilibrated for 1 h at 37°C without CO2 supplementation. Three baseline OCR measurements were acquired over 4 min and are reported as pmol/min after normalization for the amount of total proteins per well (Bradford assay). Eight wells per plate served as technical replicates, and each experiment was independently repeated at least 3 times.
Glutamine uptake assays
Glutamine uptake was determined using a radioactive assay that measures initial rates of uptake, as previously reported.Citation51 SiHa cells (500,000/well) were seeded in poly-L-lysine-coated 24-well plates. After overnight incubation, culture medium was replaced by 1 ml DMEM ± 10 mM sodium lactate, and cells were incubated for 6-h. Cells were washed 3 times with a modified Krebs solution containing 2 mM L-Glutamine without glucose, and incubated for 18 min at 37ºC in this solution. After equilibration, they were exposed to increasing concentrations of 2 mCi/ml L-[3,4–3H(N)]-glutamine (PerkinElmer) (0, 50, 100, 125, 200, 250 nM) at 37ºC for additional 18 min. Incubation medium was removed, then cells were washed 3 times with a glucose-free ice-cold Krebs solution containing 2 mM D-glutamine, and lysed with 0.1 M NaOH. Sample aliquots (100 μl) were transferred into 96-well Optiplates (PerkinElmer) and incubated in 150 μl of a Microscint 40 liquid scintillation solution (PerkinElmer) for 1 h at room temperature. Radioactivity was measured on a PerkinElmer Topcount reader, and is expressed as cpm normalized to total protein content (Bradford assay).
Statistical analysis
Results are expressed as means ± SEM (error bars are sometimes smaller than symbols). Student's t test, 1-way ANOVA (Tukey's post-hoc test) and 2-way ANOVA were used where appropriate. P < 0.05 was considered to be statistically significant.
Abbreviations
| α-KG | = | α-ketoglutarate |
| ASCT2 | = | amino acid transporter 2 |
| ECAR | = | extracellular acidification rate |
| EPAS1/HIF-2α | = | endothelial PAS domain-containing protein 1 |
| GLS1 | = | glutaminase 1 |
| GLUD1/GDH1 | = | glutamate dehydrogenase 1 |
| GPNA | = | γ-L-glutamyl-p-nitroanilide |
| HIF | = | hypoxia-inducible factor |
| hRPL19 | = | human ribosomal protein L19 |
| SLC16A1/MCT1 | = | monocarboxylate transporter 1 |
| NF-κB | = | nuclear factor-κB |
| OCR | = | oxygen consumption rate |
| OXPHOS | = | oxidative phosphorylation |
| PDH | = | pyruvate dehydrogenase |
| PDK1 | = | pyruvate dehydrogenase kinase 1 |
| PHD | = | prolylhydroxylase |
| PK | = | pyruvate kinase |
| PLA | = | proximity ligation assay |
| VHL | = | von Hippel Lindau (protein) |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Elise Beneteau and Thibaut Vazeille for excellent technical assistance.
Funding
This work was supported by grants from the European Research Council (FP7/2007–2013 European Research Council Independent Researcher Starting Grant No. 243188 TUMETABO), Interuniversity Attraction Pole (IAP) grant #UP7–03 from the Belgian Science Policy Office (Belspo), the Fonds National de la Recherche Scientifique (F.R.S.-FNRS), an Action de Recherche Concertée from the Communauté Française de Belgique (ARC 14/19–058), the Fonds Joseph Maisin, and the Fondation Belge contre le Cancer. A.C. is a Télévie Postdoctoral fellow. V.F.V.H., C.J.D. and M.S. are Télévie PhD fellows. P.S. is a Research Associate and P.E.P. a Postdoctoral Researcher of the F.R.S.-FNRS.
References
- Porporato PE, Dhup S, Dadhich RK, Copetti T, Sonveaux P. Anticancer targets in the glycolytic metabolism of tumors: a comprehensive review. Front Pharmacol 2011; 2:49; PMID:21904528; http://dx.doi.org/10.3389/fphar.2011.00049
- Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell 2012; 21:297-308; PMID:22439925; http://dx.doi.org/10.1016/j.ccr.2012.02.014
- Wu R, Racker E. Regulatory mechanisms in carbohydrate metabolism. IV. Pasteur effect and Crabtree effect in ascites tumor cells. J Biol Chem 1959; 234:1036–41; PMID:13654314
- Kolobova E, Tuganova A, Boulatnikov I, Popov KM. Regulation of pyruvate dehydrogenase activity through phosphorylation at multiple sites. Biochem J 2001; 358:69–77; PMID:11485553; http://dx.doi.org/10.1042/bj3580069
- Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab 2006; 3:187–97; PMID:16517406; http://dx.doi.org/10.1016/j.cmet.2006.01.012
- Dewhirst MW, Cao Y, Moeller B. Cycling hypoxia and free radicals regulate angiogenesis and radiotherapy response. Nat Rev Cancer 2008; 8:425–37; PMID:18500244; http://dx.doi.org/10.1038/nrc2397
- Sowter HM, Raval RR, Moore JW, Ratcliffe PJ, Harris AL. Predominant role of hypoxia-inducible transcription factor (Hif)-1alpha versus Hif-2alpha in regulation of the transcriptional response to hypoxia. Cancer Res 2003; 63:6130–4; PMID:14559790
- Bustamante E, Pedersen PL. High aerobic glycolysis of rat hepatoma cells in culture: role of mitochondrial hexokinase. Proc Natl Acad Sci U S A 1977; 74:3735–9; PMID:198801; http://dx.doi.org/10.1073/pnas.74.9.3735
- Mathupala SP, Rempel A, Pedersen PL. Glucose catabolism in cancer cells: identification and characterization of a marked activation response of the type II hexokinase gene to hypoxic conditions. J Biol Chem 2001; 276:43407–12; PMID:11557773; http://dx.doi.org/10.1074/jbc.M108181200
- Mazurek S, Boschek CB, Hugo F, Eigenbrodt E. Pyruvate kinase type M2 and its role in tumor growth and spreading. Semin Cancer Biol 2005; 15:300–8; PMID:15908230; http://dx.doi.org/10.1016/j.semcancer.2005.04.009
- Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, Fleming MD, Schreiber SL, Cantley LC. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 2008; 452:230–3; PMID:18337823; http://dx.doi.org/10.1038/nature06734
- Mazurek S. Pyruvate kinase type M2: A key regulator of the metabolic budget system in tumor cells. Int J Biochem Cell Biol 2011; 43:969–80; PMID:20156581; http://dx.doi.org/10.1016/j.biocel.2010.02.005
- Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 2009; 324:1029–33; PMID:19460998; http://dx.doi.org/10.1126/science.1160809
- Hensley CT, Wasti AT, DeBerardinis RJ. Glutamine and cancer: cell biology, physiology, and clinical opportunities. J Clin Invest 2013; 123:3678–84; PMID:23999442; http://dx.doi.org/10.1172/JCI69600
- Mullen AR, Wheaton WW, Jin ES, Chen PH, Sullivan LB, Cheng T, Yang Y, Linehan WM, Chandel NS, DeBerardinis RJ. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 2012; 481:385–8
- David CJ, Chen M, Assanah M, Canoll P, Manley JL. HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature 2010; 463:364–8; PMID:20010808; http://dx.doi.org/10.1038/nature08697
- Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK, Nissim I, Daikhin E, Yudkoff M, McMahon SB, Thompson CB. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci U S A 2008; 105:18782–7; PMID:19033189; http://dx.doi.org/10.1073/pnas.0810199105
- Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, Zeller KI, De Marzo AM, Van Eyk JE, Mendell JT, Dang CV. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009; 458:762–5; PMID:19219026; http://dx.doi.org/10.1038/nature07823
- DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, Thompson CB. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci U S A 2007; 104:19345–50; PMID:18032601; http://dx.doi.org/10.1073/pnas.0709747104
- Sonveaux P, Vegran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, De Saedeleer CJ, Kennedy KM, Diepart C, Jordan BF, et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J Clin Invest 2008; 118:3930–42; PMID:19033663
- Van Hee VF, Perez-Escuredo J, Cacace A, Copetti T, Sonveaux P. Lactate does not activate NF-kB in oxidative tumor cells. Front Pharmacol 2015; 6:228; PMID:26528183; http://dx.doi.org/10.3389/fphar.2015.00228
- Bonuccelli G, Tsirigos A, Whitaker-Menezes D, Pavlides S, Pestell RG, Chiavarina B, Frank PG, Flomenberg N, Howell A, Martinez-Outschoorn UE, et al. Ketones and lactate "fuel" tumor growth and metastasis: Evidence that epithelial cancer cells use oxidative mitochondrial metabolism. Cell Cycle 2010; 9:3506–14; PMID:20818174; http://dx.doi.org/10.4161/cc.9.17.12731
- Lu H, Forbes RA, Verma A. Hypoxia-inducible factor 1 activation by aerobic glycolysis implicates the Warburg effect in carcinogenesis. J Biol Chem 2002; 277:23111–5; PMID:11943784; http://dx.doi.org/10.1074/jbc.M202487200
- Lu H, Dalgard CL, Mohyeldin A, McFate T, Tait AS, Verma A. Reversible inactivation of HIF-1 prolyl hydroxylases allows cell metabolism to control basal HIF-1. J Biol Chem 2005; 280:41928–39; PMID:16223732; http://dx.doi.org/10.1074/jbc.M508718200
- De Saedeleer CJ, Copetti T, Porporato PE, Verrax J, Feron O, Sonveaux P. Lactate activates HIF-1 in oxidative but not in Warburg-phenotype human tumor cells. PLoS ONE 2012; 7:e46571; PMID:23082126; http://dx.doi.org/10.1371/journal.pone.0046571
- DeBerardinis RJ, Cheng T. Q's next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene 2010; 29:313–24; PMID:19881548; http://dx.doi.org/10.1038/onc.2009.358
- Leite TC, Coelho RG, Da Silva D, Coelho WS, Marinho-Carvalho MM, Sola-Penna M. Lactate downregulates the glycolytic enzymes hexokinase and phosphofructokinase in diverse tissues from mice. FEBS Lett 2011; 585:92–8; PMID:21074528; http://dx.doi.org/10.1016/j.febslet.2010.11.009
- Halestrap AP, Wilson MC. The monocarboxylate transporter family–role and regulation. IUBMB Life 2012; 64:109–19; PMID:22162139; http://dx.doi.org/10.1002/iub.572
- Invernizzi F, D'Amato I, Jensen PB, Ravaglia S, Zeviani M, Tiranti V. Microscale oxygraphy reveals OXPHOS impairment in MRC mutant cells. Mitochondrion 2012; 12:328–35; PMID:22310368; http://dx.doi.org/10.1016/j.mito.2012.01.001
- Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis?. Nat Rev Cancer 2004; 4:891–9; PMID:15516961; http://dx.doi.org/10.1038/nrc1478
- De Saedeleer CJ, Porporato PE, Copetti T, Perez-Escuredo J, Payen VL, Brisson L, Feron O, Sonveaux P. Glucose deprivation increases monocarboxylate transporter 1 (MCT1) expression and MCT1-dependent tumor cell migration. Oncogene 2014; 33:4060–8; PMID:24166504; http://dx.doi.org/10.1038/onc.2013.454
- Katt WP, Cerione RA. Glutaminase regulation in cancer cells: a druggable chain of events. Drug Discov Today 2014; 19:450–7; PMID:24140288; http://dx.doi.org/10.1016/j.drudis.2013.10.008
- Pappalardi MB, McNulty DE, Martin JD, Fisher KE, Jiang Y, Burns MC, Zhao H, Ho T, Sweitzer S, Schwartz B, et al. Biochemical characterization of human HIF hydroxylases using HIF protein substrates that contain all three hydroxylation sites. Biochem J 2011; 436:363–9; PMID:21410436; http://dx.doi.org/10.1042/BJ20101201
- Loboda A, Jozkowicz A, Dulak J. HIF-1 and HIF-2 transcription factors–similar but not identical. Mol Cells 2010; 29:435–42; PMID:20396958; http://dx.doi.org/10.1007/s10059-010-0067-2
- Corn PG, Ricci MS, Scata KA, Arsham AM, Simon MC, Dicker DT, El-Deiry WS. Mxi1 is induced by hypoxia in a HIF-1-dependent manner and protects cells from c-Myc-induced apoptosis. Cancer Biol Ther 2005; 4:1285–94; PMID:16319523; http://dx.doi.org/10.4161/cbt.4.11.2299
- Zhang H, Gao P, Fukuda R, Kumar G, Krishnamachary B, Zeller KI, Dang CV, Semenza GL. HIF-1 inhibits mitochondrial biogenesis and cellular respiration in VHL-deficient renal cell carcinoma by repression of C-MYC activity. Cancer Cell 2007; 11:407–20; PMID:17482131; http://dx.doi.org/10.1016/j.ccr.2007.04.001
- Walenta S, Schroeder T, Mueller-Klieser W. Lactate in solid malignant tumors: potential basis of a metabolic classification in clinical oncology. Curr Med Chem 2004; 11:2195–204; PMID:15279558; http://dx.doi.org/10.2174/0929867043364711
- Walenta S, Mueller-Klieser WF. Lactate: mirror and motor of tumor malignancy. Semin Radiat Oncol 2004; 14:267–74; PMID:15254870; http://dx.doi.org/10.1016/j.semradonc.2004.04.004
- Kennedy KM, Scarbrough PM, Ribeiro A, Richardson R, Yuan H, Sonveaux P, Landon CD, Chi JT, Pizzo S, Schroeder T, Dewhirst MW. Catabolism of exogenous lactate reveals it as a legitimate metabolic substrate in breast cancer. PLoS ONE 2013; 8:e75154; PMID:24069390; http://dx.doi.org/10.1371/journal.pone.0075154
- Busk M, Walenta S, Mueller-Klieser W, Steiniche T, Jakobsen S, Horsman MR, Overgaard J. Inhibition of tumor lactate oxidation: Consequences for the tumor microenvironment. Radiother Oncol 2011; 99:404–11; PMID:21704401; http://dx.doi.org/10.1016/j.radonc.2011.05.053
- Nakajima EC, Van HB. Metabolic symbiosis in cancer: refocusing the Warburg lens. Mol Carcinog 2013; 52:329–37; PMID:22228080; http://dx.doi.org/10.1002/mc.21863
- Guillaumond F, Leca J, Olivares O, Lavaut MN, Vidal N, Berthezene P, Dusetti NJ, Loncle C, Calvo E, Turrini O, et al. Strengthened glycolysis under hypoxia supports tumor symbiosis and hexosamine biosynthesis in pancreatic adenocarcinoma. Proc Natl Acad Sci U S A 2013; 110:3919–24; PMID:23407165; http://dx.doi.org/10.1073/pnas.1219555110
- McGillen JB, Kelly CJ, Martinez-Gonzalez A, Martin NK, Gaffney EA, Maini PK, Perez-Garcia VM. Glucose-lactate metabolic cooperation in cancer: Insights from a spatial mathematical model and implications for targeted therapy. J Theor Biol 2014; 361:190–203; PMID:25264268; http://dx.doi.org/10.1016/j.jtbi.2014.09.018
- Kianercy A, Veltri R, Pienta KJ. Critical transitions in a game theoretic model of tumour metabolism. Interface Focus 2014; 4:20140014; PMID:25097747; http://dx.doi.org/10.1098/rsfs.2014.0014
- Sonveaux P, Copetti T, De Saedeleer CJ, Vegran F, Verrax J, Kennedy KM, Moon EJ, Dhup S, Danhier P, Frerart F, et al. Targeting the lactate transporter MCT1 in endothelial cells inhibits lactate-induced HIF-1 activation and tumor angiogenesis. PLoS ONE 2012; 7:e33418; PMID:22428047; http://dx.doi.org/10.1371/journal.pone.0033418
- Vegran F, Boidot R, Michiels C, Sonveaux P, Feron O. Lactate influx through the endothelial cell monocarboxylate transporter MCT1 supports an NF-kappaB/IL-8 pathway that drives tumor angiogenesis. Cancer Res 2011; 71:2550–60; PMID:21300765; http://dx.doi.org/10.1158/0008-5472.CAN-10-2828
- Roland CL, Arumugam T, Deng D, Liu SH, Philip B, Gomez S, Burns WR, Ramachandran V, Wang H, Cruz-Monserrate Z, et al. Cell surface lactate receptor GPR81 is crucial for cancer cell survival. Cancer Res 2014; 74:5301–10; PMID:24928781; http://dx.doi.org/10.1158/0008-5472.CAN-14-0319
- Appelhoff RJ, Tian YM, Raval RR, Turley H, Harris AL, Pugh CW, Ratcliffe PJ, Gleadle JM. Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor. J Biol Chem 2004; 279:38458–65; PMID:15247232; http://dx.doi.org/10.1074/jbc.M406026200
- DeBoer LW, Bekx PA, Han L, Steinke L. Pyruvate enhances recovery of rat hearts after ischemia and reperfusion by preventing free radical generation. Am J Physiol 1993; 265:H1571–6; PMID:8238569
- Russell RR, III, Taegtmeyer H. Pyruvate carboxylation prevents the decline in contractile function of rat hearts oxidizing acetoacetate. Am J Physiol 1991; 261:H1756–62; PMID:1750532
- Corbet C, Draoui N, Polet F, Pinto A, Drozak X, Riant O, Feron O. The SIRT1/HIF2alpha Axis Drives Reductive Glutamine Metabolism under Chronic Acidosis and Alters Tumor Response to Therapy. Cancer Res 2014; 74:5507–19; PMID:25085245; http://dx.doi.org/10.1158/0008-5472.CAN-14-0705
- Reitzer LJ, Wice BM, Kennell D. Evidence that glutamine, not sugar, is the major energy source for cultured HeLa cells. J Biol Chem 1979; 254:2669–76; PMID:429309
- Cardaci S, Rizza S, Filomeni G, Bernardini R, Bertocchi F, Mattei M, Paci M, Rotilio G, Ciriolo MR. Glutamine deprivation enhances antitumor activity of 3-bromopyruvate through the stabilization of monocarboxylate transporter-1. Cancer Res 2012; 72:4526–36; PMID:22773663; http://dx.doi.org/10.1158/0008-5472.CAN-12-1741
- Doherty JR, Yang C, Scott KE, Cameron MD, Fallahi M, Li W, Hall MA, Amelio AL, Mishra JK, Li F, et al. Blocking lactate export by inhibiting the Myc target MCT1 Disables glycolysis and glutathione synthesis. Cancer Res 2014; 74:908–20; PMID:24285728; http://dx.doi.org/10.1158/0008-5472.CAN-13-2034
- Soderberg O, Gullberg M, Jarvius M, Ridderstrale K, Leuchowius KJ, Jarvius J, Wester K, Hydbring P, Bahram F, Larsson LG, et al. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat Methods 2006; 3:995–1000; PMID:17072308; http://dx.doi.org/10.1038/nmeth947