
ABSTRACT
Recent studies have found BCL10 can localize to the nucleus and that this is linked to tumor aggression and poorer prognosis. These studies suggest that BCL10 localization plays a novel role in the nucleus that may contribute to cellular transformation and carcinogenesis. In this study, we show that BCL10 functions as part of the DNA damage response (DDR). We found that BCL10 facilitates the rapid recruitment of RPA, BRCA1 and RAD51 to sites of DNA damage. Furthermore, we also found that ATM phosphorylates BCL10 in response to DNA damage. Functionally, BCL10 promoted DNA double-strand breaks repair, enhancing cell survival after DNA damage. Taken together our results suggest a novel role for BCL10 in the repair of DNA lesions.
Introduction
B cell lymphoma 10 (BCL10) was originally identified as a target of recurrent chromosomal translocation in mucosa-associated lymphoid tissue lymphomas.1,2 In lymphoid cells BCL10 plays a critical role in the activation of the transcription factor nuclear factor kappa B (NF-κB) downstream of a variety of immune receptors including the TCR, B cell receptor, NK-cell receptors, C type family lectin receptors, Ig family receptors and G protein-coupled receptors.3-5 In these cells activation of NF-κB through receptor-mediated pathways requires BCL10 to form a complex in the cytoplasm with the paracaspase MALT1.6 Receptor engagement leads to recruitment of BCL10-MALT1 to the CARD-containing adaptor protein CARMA1 forming a trimeric complex, the CARMA, BCL10, MALT1 (CBM) signalosome. Although the precise contributions of the components have yet to be differentiated, the CBM signalosome appears necessary for the ubiquitylation and degradation of IKKβ, part of the heterotrimeric NF-κB regulatory IKK complex, thereby enabling NF-κB to enter the nucleus and induce transcription.7 CARMA1 is found predominantly in hematopoietic tissues whereas CARMA3 shares extensive homology with CARMA1 and is widely expressed outside the immune system.8 BCL10 and MALT1 may therefore interact with CARMA proteins in a variety of cell types to activate NF-κB. Knockdown of CBM proteins has been shown to impede NF-κB activation in ovarian cancers and to inhibit NF-κB mediated ovarian cancer cell invasion in-vitro, suggesting that CBM proteins regulate oncogenesis.9 Recently it was demonstrated that invasion by head and neck squamous cell carcinomas induced by the chemokine SDF-1, requires activation of NF-κB by the CBM complex further implicating CBM proteins in cancer.10 In breast cancer activation of NF-κB downstream of EGF receptors was recently shown to require CARMA3 suggesting that the CBM complex may also regulate NF-κB activation in breast epithelia.11 However, although CBM proteins are implicated in cancer progression the function of BCL10 in carcinomas is poorly understood.
In this report, we describe the participation of BCL10 in the regulation of DNA double-strand breaks (DSBs) repair. We found that BCL10 is recruited to sites of DNA damage where it co-localizes with several DNA repair proteins. We demonstrate that the DSB kinase ATM phosphorylates BCL10 but this phosphorylation is not required for its recruitment to sites of DNA damage. Rather, BCL10 recruitment to DNA breaks requires the E3 ubiquitin ligases RNF8/RNF168 and together they regulate the formation of ubiquitin conjugates at the sites of DNA damage. Finally, we show that knockdown of BCL10 reduces the ability of cells to repair DSBs and renders cells sensitive to radiation.
Material and methods
Cell transfection, immunoblotting and immunostaining
U2OS and T47D breast carcinoma cells were obtained from the American Type Culture Collection. These cells were cultured in DMEM medium containing 10% fetal calf serum (FCS) at 37°C and 5% CO2. DR-GFP U2OS and EJ5GFP cells (a gift from Dr. Jeremy Stark) were grown in Mycos 5A media without pyruvate supplemented with 10% FCS. Unless otherwise stated, cells were irradiated in ambient air using a model CS-600 137Cs irradiator (Picker, Glendale, CA) at a dose rate of 2 Gy/minute. We used the following BRCA1 siRNAs: GGAACCUGUCUCCACAAAG-dTdT. KU-55933 (ATM Inhibitor), KU-57788 (DNA-Pkcs inhibitor) and Wortmannin were purchase from Selleckchem, dissolved in DMSO and kept in aliquots at -80C. Cell transfections with plasmid DNA or siRNA duplexes were performed by using Lipofectamine 3000 and Lipofectamine RNAiMax (Invitrogen), respectively, following the manufacturer's instructions. To prepare aggregated IgG, mouse IgG (100 μg/mL, Sigma) were heat-aggregated by an incubation at 63°C in a borate-buffer saline pH 8.0 for 30 min. Immunofluorescence staining, cell fractionation and immunoblot analysis were performed as described previously.12 A panel of commercially available primary antibodies γ-H2AX (Millipore), BCL10 (Sigma), BRCA1 (Santa Cruz inc.), Ubiquitin (FK2, Enzo), p-BCL10 Antibody (6D3) (Santa Cruz inc.), RAD51 (Santa Cruz inc.), p-ATM-S1981 (Active Motif), CtIP (Active Motif) and RPA32 (Abcam) were used. Western blot and immunoprecipitation experiments were done as previously described.12
DNA repair and cell survival assays
HR and NHEJ assays were performed as described previously using the GFP reporter cell lines.13 Cell survival was performed as described previously.12
Results
BCL10 is recruited to sites of DNA damage
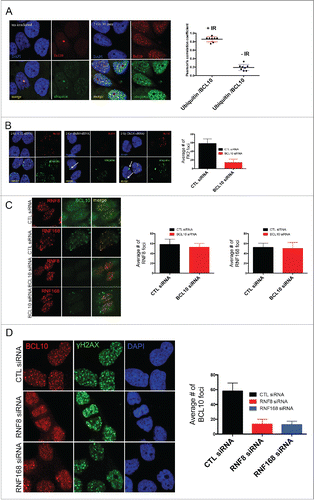
Using indirect immunofluorescence staining, we investigated the sub-cellular localization of BCL10 in breast cancer cells.14 In contrast to lymphoid cells, in which BCL10 is largely cytoplasmic, we found in the breast cancer cell line T47D that BCL10 was concentrated in large nuclear foci (). In examining BCL10 nuclear domains, we noted similarities with the cryptogenic γ-H2AX (the phosphorylated form of histone H2AX) foci that are previously shown in unirradiated cells ().12,15 This raises the possibility that BCL10 is involved in the DDR. To evaluate the possible involvement of BCL10 in DNA repair, we examined its immunofluorescence (IF) staining pattern relative to that of γ-H2AX foci in T47D cells. BCL10 concentrated in several large domains that were positive for γ-H2AX staining, despite the absence of any treatment to induce DNA damage. Previous reports showed that these large γ-H2AX foci represent persistent DSBs that may include eroded telomeres.16-18 To investigate whether BCL10 is required for formation of constitutive γ-H2AX foci in cells we knocked down BCL10 with siRNA (). BCL10 knockdown significantly reduced the number of constitutive γ-H2AX foci () indicating BCL10 may play a role in the maintenance or function of these foci. Knockdown of BCL10 leads to loss of BCL10 IF signal, demonstrating the specificity of the antibody (). This suggested a possible role for BCL10 in the DDR. To test this hypothesis, we exposed cells to IR and examined the localization of BCL10 relative to γ-H2AX. Upon DNA damage, BCL10 localized to a large number of foci that co-localized with the DSB marker γ-H2AX (). When the breast cancer cell line T47D was irradiated with 2 Gy γ-H2AX and Phosphorylation of serine 1981 in endogenous ATM (p-ATM) foci became detectable by 5 minutes, peaked within 30 to 60 minutes and became less numerous by 480 to 960 minutes () indicating that most of the lesions had been successfully repaired. BCL10 co-localized extensively with irradiation-induced foci, the degree of co-localization becoming greater with time after irradiation suggesting BCL10 could contribute to their function. Interestingly, large (and presumably constitutive) BCL10 foci were not observable by 30 minutes after irradiation suggesting BCL10 preferentially associates with the newer lesions. When T47D cells were exposed to 5 Gy ionizing irradiation, BCL10 was recruited to γ-H2AX and P-ATM foci with the association again being most apparent at later time intervals post-irradiation ().
Figure 1. BCL10 is recruited to sites of DNA damage. (A) Immunodetection of BCL10 and γ-H2AX in T47D cells transfected with control siRNA (CTL siRNA) or BCL10 siRNA (BCL10 siRNA). Quantification of the average number of cryptic γ-H2AX foci per cells is shown on the right. Efficiency of BCL10 knockdown was shown by an immunoblot on the top. (B) Immunodetection of BCL10 and γ-H2AX 60 min after IR (2 Gy) irradiation. The right panel shows Pearson's correlation coefficient values for co-localization of γ-H2AX and BCL10 with and without IR.
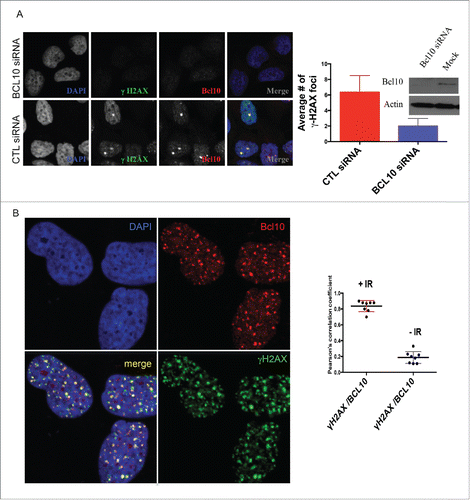
Figure 2. Kinetics of DNA damage-induced BCL10 foci formation. Immunodetection of BCL10 and γ-H2AX or p-ATM (S1981) at the indicated time points after IR (2 Gy) irradiation. Quantification of the average number of IR-induced BCL10/γ-H2AX or BCL10/p-ATM foci per cells is shown on the bottom. (A) BCL10/γ-H2AX. (B) BCL10/p-ATM.
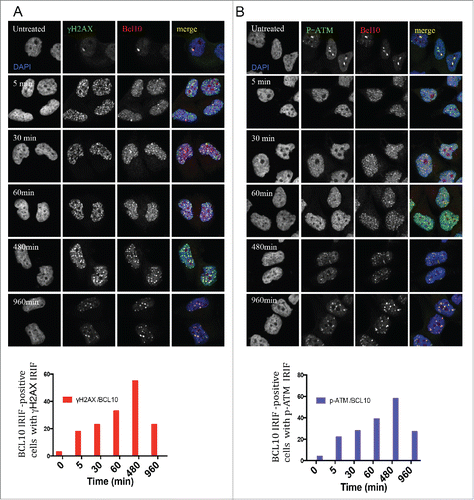
BCL10 forms a complex with MALT1 that is required for the activation of NF-κB in the cytoplasm.6 We next determined if MALT1, similar to BCL10, is also recruited to sites of DNA damage. We observed that MALT1 co-localized with γ-H2AX in constitutive foci in the nucleus of T47D cells (). However, in contrast to knockdown of BCL10, knockdown of MALT1 did not impede formation of γ-H2AX constitutive foci (). This indicates that BCL10-dependent maintenance of constitutive foci does not require MALT1 and presumably operates independently of activation of NF-κB.
Figure 3. MALT1 is dispensable for γ-H2AX cryptic foci formation. (A) Immunodetection of BCL10 and γ-H2AX in T47D cells transfected with control siRNA (CTL siRNA), or MALT1 siRNA (MALT1 siRNA). Quantification of the average number of cryptic γ-H2AX foci per cells is shown on the right. MALT1 knocking down efficiency was shown by an immunoblot on the top. (B) The effect of BCL10 knock down on NFκB activation in response to DNA damage. NFκB activation was measured by the translocation of p65 into the nucleus in response to DNA damage with or without BCL10 depletion. Quantification of the p65 nuclear translocation was measured.
BCL10 is dispensable for NF-κB activation upon DNA damage
Since ionizing radiation is reported to activate NF-κB we wondered whether BCL10 knockdown would influence translocation of the NF-κB subunit p65 to the nucleus, an indicator of NF-κB activation. We observed that the average nuclear intensity of p65 within the nucleus of T47D cells did increase after irradiation (5 Gy ionizing radiation) (). BCL10 knockdown had very little effect on p65 translocation to the nucleus after irradiation (). These data suggest that the novel BCL10 function in the DDR, reported here, is distinct from the irradiation-induced NF-κB activation. In line with these data, we found that pretreatment of cells with NF-κB activators, lipopolysaccharide (LPS) and aggregated IgG, before exposure to radiation did not significantly change the conjugated ubiquitin (FK2) foci formation (Fig. S1). Similarly, pretreatment of cells with LPS and aggregated IgG did not change cellular response to radiation in BCL10-defcient background (Fig. S1). In control experiments, we showed that LPS and aggregated IgG activated NF-κB pathway (Fig. S2).
BCL10 localization to DNA breaks is independent of ATM activity
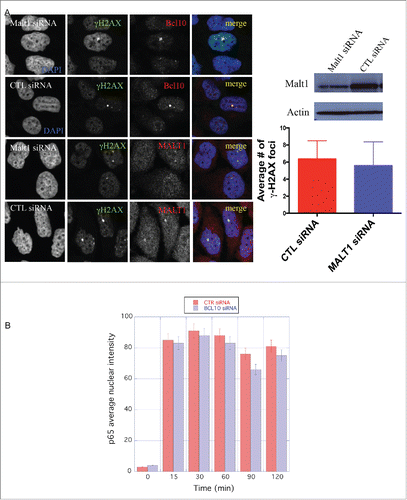
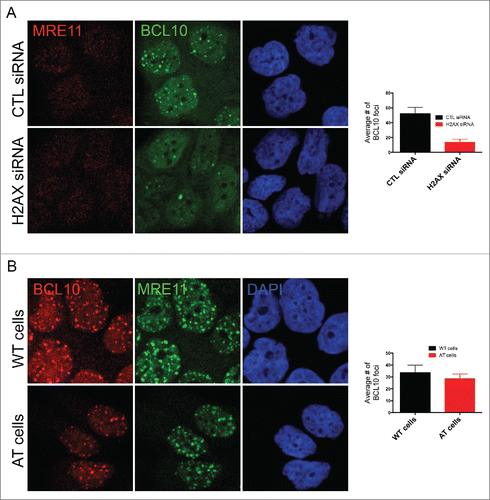
We next sought to determine the mechanism of BCL10 accumulation at sites of DNA damage. Initially, we focused on the potential involvement of H2AX phosphorylation (γ-H2AX) because this histone modification has previously been implicated in the recruitment of several DDR factors to damaged chromatin (reviewed in 19). Interestingly, BCL10 accumulation at DNA damage sites was impaired in H2AX deficient cells () indicating that H2AX is required for BCL10 foci formation. H2AX is phosphorylated at serine 139 in response to DNA damage in ATM dependent manner. We then tested the potential contribution of ATM activity in BCL10 accumulation at the sites of DNA damage. The use of AT fibroblasts (ATM deficient cells) revealed that ATM activity was dispensable for BCL10 recruitment to damage sites ().
Figure 4. H2AX-dependent but ATM-independent recruitment of BCL10 to the DNA damage sites. (A) Immunodetection of BCL10 and MRE11 in T47D cells transfected with control siRNA (CTL siRNA) or H2AX siRNA (H2AX siRNA) in response to IR (2 Gy). Quantification of the average number of DNA damage-induced BCL10 foci is shown on the right. (B) Immunodetection of BCL10 and MRE11 in ATM proficient (WT) and ATM deficient (AT) cells in response to IR (2 Gy). Quantification of the average number of DNA damage-induced BCL10 foci per cell is shown on the right.
BCL10 regulates ubiquitin conjugates formation at the sites of DNA damage
In the cytoplasm, BCL10 is thought to activate NF-κB in part by contributing to the ubiquitination and degradation of inhibitors of NF-κB. In resting T47D cells, we observed that BCL10 colocalized with ubiquitin in constitutive nuclear foci (). When cells were exposed to 2 Gy ionizing radiation, we observed that BCL10 co-localized with ubiquitin in multiple foci at early, as well as at later, time points suggesting BCL10 could play a role in the ubiquitination of proteins associated with DNA repair (). BCL10 knockdown with siRNA was associated with loss of radiation-induced foci accompanied by accumulation of ubiquitin in the perinuclear region (arrowed) (). DNA damage induced ubiquitin foci are mainly regulated by the E3 ubiquitin ligases RNF8/RNF168. The previous results could be explained if RNF8/RNF168 are not recruited to sites of DNA damage upon siRNA mediated BCL10 depletion. To test this hypothesis, cells were transfected with with BCL10 siRNA and the recruitment of RNF8 and RNF168 to form foci was monitored. We found that BCL10 knockdown did not impact RNF8/RNF168 recruitment to foci (). We also examined if BCL10 is downstream of RNF8/RNF168 dependent ubiquitin signaling events. To this aim, we asked whether or not BCL10 retention at sites of DNA damage is affected by RNF8/RNF168 depletion. In contrast to the previous results, we found that RNF8/RNF168 ubiquitin ligases are required for BCL10 retention at sites of DNA damage (). Moreover, we found that the proteasome inhibitor MG132 inhibited the recruitment of BCL10 to DNA damage foci (Fig. S3). MG132 treatment depletes free nuclear ubiquitin and consequently inhibits ubiquitylation at sites of DNA damage. Taken together, our data suggest that BCL10 is recruited to the sites of DNA damage via RNF8/RNF168 mediated ubiquitylation events.
Figure 5. BCL10 regulates ubiquitylation at the sites of DNA damage. (A) Immunodetection of BCL10 and ubiquitin (FK2) in T47D cells exposed or not exposed to IR. Quantification of the average number of cryptic γ-H2AX foci per cells is shown on the right. The right panel shows Pearson's correlation coefficient values for co-localization of ubiquitin and BCL10 with and without IR. (B) Immunodetection of BCL10 and ubiquitin (FK2) in T47D cells transfected with control siRNA (CTL siRNA) or BCL10 siRNA (BCL10 siRNA) and exposed to IR (2 Gy). Arrows indicate the accumulation of ubiquitin in the cytoplasm upon BCL10 depletion. Quantification of the average number of ubiquitin (FK2) foci per cell is shown on the right. (C) Immunodetection of BCL10 and RNF8 or RNF168 in T47D cells transfected with control siRNA (CTL siRNA) or BCL10 siRNA (BCL10 siRNA). Quantification of the average number of RNF8 and RNF168 foci per cell is shown on the right. (D) Immunodetection of BCL10 foci in T47D cells transfected with control siRNA (CTL siRNA), RNF8 siRNA (RNF8 siRNA) or RNF168 siRNA (RNF168 siRNA) and exposed to IR (2 Gy). Quantification of the average number of ubiquitin BCL10 foci per cell is shown on the right.
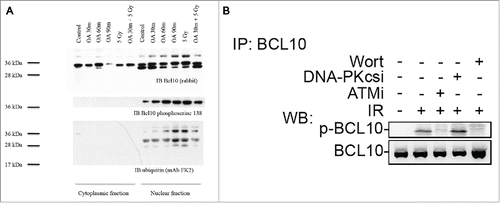
BCL10 is phosphorylated upon DNA damage in an ATM-dependent manner
A hallmark of many DDR proteins is their post-translational modification in response to DNA damage.20 Examining BCL10 amino acid sequence revealed that BCL10 has multiple phosphorylation sites. In lymphoid cells, serine 138 is a key target for BCL10 phosphorylation.21 To investigate whether BCL10 is phosphorylated in the nucleus of T47D cells, we separated cytoplasmic and nuclear fractions and immunoblotted with antibody to BCL10 following exposure to the serine/threonine phosphatase inhibitor okadaic acid (). BCL10 was identifiable as a 30 kDa band in both the cytoplasm and the nucleus. Exposure to okadaic acid for 30 to 90 minutes resulted in appearance of a slower migrating band of 38 kDa that was present in the nucleus but not in the cytoplasm. Irradiating cells with 5 Gy also resulted in appearance of a 38 kDa band in the nucleus (). To determine whether the slowly migrating band was phosphorylated, we immunoblotted using an antibody directed against BCL10 phosphoserine 138. BCL10 phosphoserine 138 was specifically detected in the nucleus confirming that the nucleus is an important site of BCL10 phosphorylation. Phosphoserine 138 was detectable in resting cells and the signal was amplified by exposure to okadaic acid or by 5 Gy ionizing radiation (). Furthermore, this BCL10 phosphorylation was prevented when cells were incubated with wortmannin or the ATM inhibitor KU-55933 but was not inhibited when cells were treated with DNA-PKcs inhibitor (DNA-PKcsi) (). Consistent with these data, proteomic screens in human cells have identified BCL10 as a target for the DDR kinases, ATM and ATR.20 These results suggest that BCL10 is phosphorylated at serine 138 in response to DNA damage in an ATM dependent manner. The requirement for RNF8 and ubiquitylation suggest that BCL10 may itself be ubiquitylated. Indeed, when we blotted with the ubiquitin-specific monoclonal antibody FK2 we identified a band of 38 kDa that was present only in the nucleus (). FK2 antibody detects mono- and poly-ubiquitylated conjugates. The relative intensity of that band was enhanced in the nucleus of cells exposed to okadaic acid or to 5 Gy suggesting that BCL10 is targeted by serine/threonine kinases when cells are irradiated. Since the ubiquitylated protein band had an identical mobility to phosphorylated BCL10, phosphorylation may be an upstream requirement for ubiquitination.22,23
Figure 6. ATM phosphorylates BCL10 is in response to DNA damage. (A) Cells were treated with the indicated drugs and cytoplasmic and nuclear fractions were isolated. BCL10 immunoprecipitation was conducted using BCL10 antibodies. Immunoblots were performed as indicated. (B) Cells were incubated with the indicated inhibitors (20μM Wort., Wortmannin; 2μM DNA-PKi, DNA-PK inhibitor; 10 μM ATMi, ATM inhibitor) for 1 hr and then exposed to IR (10 Gy). Nuclear extracts were isolated and subjected to immunoprecipitated using BCL10 antibody. Western blot analysis was conducted with BCL10 and p-BCL10 antibodies.
BCL10 mediates DSB repair via HR pathway
DSB repair involves 2 major pathways, non-homologous end-joining (NHEJ) and homologous recombination (HR). BCL10 is known to interact with Ubc13, a key player in DSB repair by HR.24,25 We therefore decided to examine the role of BCL10 in HR repair. We used DR-GFP U2OS cells containing a single-copy integrated tandem GFP-based reporter of HR 13 after siRNA-mediated knockdown of BCL10 (). BCL10 knockdown by siRNA resulted in 60% reduction in the proportion of cells that successfully repaired by HR to become GFP positive (). BRCA1 is an important factor for HR repair and hence served as a positive control.Citation26 We found that siRNA mediated BRCA1 knockdown also results in a low frequency of GFP-positive cells (approximate 80%; ). However, the effect on DSB repair by NHEJ did not reach significance, suggesting that BCL10 may be less important for NHEJ repair (). In contrast, Ku80 knockdown results in a low frequency of GFP-positive cells and served as a positive control.
Figure 7. BCL10 promotes DSB repair and cellular sensitivity to DNA damage. Efficiency of BCL10 cellular depletion on DSB repair in cells transfected with control siRNA (CTL siRNA) or BCL10-specific siRNA (BCL10 siRNA). (A) Cells contain an integrated tandem GFP reporter of HR. The percentage of GFP positive was measured by flow cytometer. Each data point represents the mean ± the SEM of 2 separate experiments. (B) Cells contain an integrated tandem GFP reporter of NHEJ. The percentage of GFP positive was measured as in A. (C) Effect of BCL10 depletion on HR proteins. Cells were treated as in A, laser micro-irradiated and allowed to recover for 1 hr and immunostained with different antibodies as indicated. Representative pictures were shown on the right. The percentage of cells with γ-H2AX/BRCA1, γ-H2AX/RPA, γ-H2AX/RAD51 and γ-H2AX/ CtIP were shown on the right. (D and E) Sensitivity of BCL10 knock down cells to different DNA damaging agents (IR and CPT). Survival of U2OS cells upon BCL10 knockdown (BCL10 siRNA) compared with control (CTL siRNA) in response to (D) IR or (E) CPT. Error bars indicate SEM from 2 independent experiments. The results are normalized to plating efficiency.
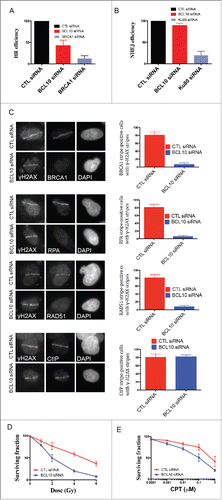
As BCL10 was required for effective HR, we investigated whether BCL10 depletion influenced the accumulation of the HR components RAD51 and BRCA1 at DNA damage sites. IR-induced focus formation of HR proteins such as RAD51 and BRCA1 was reduced in BCL10 knockdown cells (). The effect of BCL10 depletion on HR components is specific because BCL10 did not inhibit γ-H2AX foci formation ().
Resection of DSB ends is one of the critical initial reactions in the HR repair pathway, and this resection step can be visualized by RPA and CtIP recruitment to sites of DNA damage.Citation27 We observed that the relocation of RPA to DNA damage sites was reduced in BCL10 knockdown cells (). In contrast, BCL10 knockdown did not alter CtIP recruitment to the sites of DNA breaks. We conclude that BCL10 might participate in HR directly, perhaps by facilitating the initial recruitment/accumulation of key HR proteins such as RPA, RAD51 and BRCA1 to DSB sites.
BCL10 promotes cell survival after DNA damage
We next determined whether or not cellular depletion of BCL10 would render cells sensitive to ionizing radiation (IR) and Camptothecin (CPT). CPT is a topoisomerase-I inhibitor that prevents repair of the topoisomerase-I-induced SSB and during DNA replication collision with the replication forks, converts these cleavage complexes into DSB.Citation28 We used the colony formation assay to determine the survival of cells after IR and CPT. siRNA mediated BCL10 cellular depletion reduced cell survival to both IR and CPT (). This reduction in survival was statistically significant (P = 0.0003). The effect of BCL10 knockdown on cellular IR sensitivity was more pronounced (4 fold) at higher doses (). These data indicate that depletion of BCL10 levels in U2OS increased the sensitivity of the cells to IR and CPT.
Discussion
BCL10 together with CARMA1 and MALT1 forms the CBM complex. This complex links T cell receptor signaling to the canonical IκB kinase (IKK)/NF-κB pathway. Our study uncovers a novel function of BCL10 that is independent of the CBM complex, and operates in the DDR. We found that BCL10 regulates RNF8/RNF168-mediated ubiquitylation and subsequently the recruitment of BRCA1. DNA damage induces phosphorylation of BCL10 in an ATM dependent manner. BCL10 knock down therefore modulates the cellular sensitivity of cells to radiation and regulates DSB repair through HR repair.
The DNA-PK subunits Ku70 and Ku80 are upregulated by NF-κB.Citation29 Therefore, it is possible that BCL10 regulates DSB repair through NHEJ. To the contrary, we did not observe any contribution of BCL10 in NHEJ repair as measured by an NHEJ GFP-based reporter assay. On the other hand, BCL10 knock down inhibited DSB repair by HR. In agreement with the function of BCL10 in HR repair, we found that BCL10 facilitates the recruitment of key HR repair proteins RAD51, BRCA1 and RPA. Moreover, BCL10 did not have a noticeable effect on the early DNA end resection protein CtIP.
We also showed that BCL10 regulates ubiquitin turnover at the sites of DNA damage. The formation of these ubiquitin conjugates at the sites of DNA damage is dependent on RNF8/RNF168 activity.Citation30–34 These data, together with our data showing the requirement for RNF8/RNF168 for the accumulation of BCL10 at sites of DNA damage, suggest that BCL10 collaborates with RNF8/RNF168 to regulate ubiquitin signaling at sites of DNA damage. It remains unclear how BCL10 regulates RNF8/RNF168 mediated ubiquitin signaling. One possibility is that the recruitment of RNF8/RNF168 is dependent on BCL10. Our data showing normal accumulation of these ubiquitin ligases argue against such a hypothesis. Alternatively, it is possible that BCL10 facilitates the interaction of Ubc13, an E2 enzyme required for RNF8/RNF168 mediated ubiquitin signaling, with RNF8/RNF168. In agreement with this notion, a recent report suggests that the RNF8/UBC13-containing complex ubiquitylates BCL10.Citation35 Ubiquitylated BCL10 could subsequently bind to the UBDs of RNF168 and present UBC13 to RNF168. This could confer E3 ligase activity on RNF168 for the mono-ubiquitylation of γ-H2AX, and possibly other substrates, and subsequently the RNF8/RNF168 ligase activity for poly-ubiquitylation of γ-H2AX.Citation35 Ubiquitylated γ-H2AX around the DSBs serve as a platform for the recruitment of DSB signaling and repair factors.
It is not clear how BCL10 is activated in response to DNA damage. It is possible that BCL10 is post-tanslationally modified in response to DNA damage. Consistent with this notion and previous data (ref. Citation20), we found that ATM phosphorylates BCL10 in irradiated cells.Citation20 Although the physiological significance of this phosphorylation is unclear, it is possible that ATM-mediated phosphorylation is required for interaction with RNF8/RNF168 or for binding to damaged chromatin. The latter does not appear to be the case, however, as BCL10 recruitment to sites of DNA damage is independent of ATM activity. It is also possible that ATM-mediated phosphorylation is required for RNF8/RNF168 dependent ubiquitylation signaling events. In agreement with this, we found that pharmacological inhibition of ATM inhibits DNA damage induced ubiquitylation as measured by FK2 antibody. It is possible, therefore, that ATM mediated ubiquitylation is mediated, at least in part, through BCL10.
In the cytosol, BCL10 is part of CBM complex that promotes NEMO ubiquitination by TRAF6, leading to NFκB activation.Citation25 Despite the function of BCL10 in DNA repair, the constitutive partner of BCL10, MALT1, does not seem to play a role in the DDR, because depletion of MALT1 did not have an impact on the localization of DSB repair proteins at the damage sites and it did not localize to de-novo sites of DNA damage. Therefore, it would be interesting to determine if any of canonical BCL10-dependent events such as NEMO mono-ubiquitination and subsequent translocation from the nucleus to cytosol are dependent on ATM initiated BCL10 phosphorylation.
In the cytosol, BCL10 forms a complex with MALT1 and CARMA1 that functions to stimulate the immune response triggered by T/B-cell antigen receptors.Citation36 However, several research groups have reported that BCL10 nuclear expression correlates with a significant decrease in patient survival in primary cutaneous marginal zone B-cell lymphomas and oral squamous cell carcinomas. We now report that BCL10 regulates HR repair through mediating RNF8/RNF168-mediated ubiquitylation events. It is reasonable to speculate that BCL10 may enhance DSB repair capacity of tumor cells and thus confers resistance to chemotherapeutic agents, contributing to poor overall survival in patients.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
KCCY_S_1121322.zip
Download Zip (10.4 MB)Acknowledgments
We thank Dr. Jeremy Stark for providing valuable reagents. We also thank the cellular imaging facility and flow cytometry units at the Cross cancer institute for the use of microscopes and flow cytometers.
Funding
This work was supported by an operating grant from the Canadian Institute of Health Research (CIHR) and by operating grants from the Alberta Cancer Foundation (ACF) and the Canadian Breast Cancer Foundation (CBCF).
Related Research Data
References
- Isaacson P, Wright DH. Malignant lymphoma of mucosa-associated lymphoid tissue. A distinctive type of B-cell lymphoma. Cancer 1983; 52:1410-6; PMID:6193858; http://dx.doi.org/10.1002/1097-0142(19831015)52:8%3c1410::AID-CNCR2820520813%3e3.0.CO;2-3
- Rosebeck S, Rehman AO, Lucas PC, McAllister-Lucas LM. From MALT lymphoma to the CBM signalosome: three decades of discovery. Cell Cycle 2011; 10:2485-96; PMID:21750409; http://dx.doi.org/10.4161/cc.10.15.16923
- Ruland J, Duncan GS, Elia A, del Barco Barrantes I, Nguyen L, Plyte S, Millar DG, Bouchard D, Wakeham A, Ohashi PS, et al. Bcl10 is a positive regulator of antigen receptor-induced activation of NF-kappaB and neural tube closure. Cell 2001; 104:33-42; PMID:11163238; http://dx.doi.org/10.1016/S0092-8674(01)00189-1
- Rueda D, Gaide O, Ho L, Lewkowicz E, Niedergang F, Hailfinger S, Rebeaud F, Guzzardi M, Conne B, Thelen M, et al. Bcl10 controls TCR- and FcgammaR-induced actin polymerization. J Immunol 2007; 178:4373-84; http://dx.doi.org/10.4049/jimmunol.178.7.4373
- Xue L, Morris SW, Orihuela C, Tuomanen E, Cui X, Wen R, Wang D. Defective development and function of Bcl10-deficient follicular, marginal zone and B1 B cells. Nat Immunol 2003; 4:857-65; PMID:12910267; http://dx.doi.org/10.1038/ni963
- Thome M, Charton JE, Pelzer C, Hailfinger S. Antigen receptor signaling to NF-kappaB via CARMA1, BCL10, and MALT1. Cold Spring Harbor perspectives in biology 2010; 2:a003004; PMID:20685844; http://dx.doi.org/10.1101/cshperspect.a003004
- Israel A. The IKK complex, a central regulator of NF-kappaB activation. Cold Spring Harb Perspect Bio 2010; 2:a000158; PMID:20300203; http://dx.doi.org/10.1101/cshperspect.a000158
- Jiang C, Lin X. Regulation of NF-kappaB by the CARD proteins. Immunol Rev 2012; 246:141-53; PMID:22435552; http://dx.doi.org/10.1111/j.1600-065X.2012.01110.x
- Nestle FO, Banchereau J, Hart D. Dendritic cells: On the move from bench to bedside. Nat Med 2001; 7:761-5; PMID:11433329; http://dx.doi.org/10.1038/89863
- Rehman AO, Wang CY. SDF-1alpha promotes invasion of head and neck squamous cell carcinoma by activating NF-kappaB. J Biol Chem 2008; 283:19888-94; PMID:18448428; http://dx.doi.org/10.1074/jbc.M710432200
- Jiang T, Grabiner B, Zhu Y, Jiang C, Li H, You Y, Lang J, Hung MC, Lin X. CARMA3 is crucial for EGFR-Induced activation of NF-kappaB and tumor progression. Cancer Res 2011; 71:2183-92; PMID:21406399; http://dx.doi.org/10.1158/0008-5472.CAN-10-3626
- Ismail IH, Andrin C, McDonald D, Hendzel MJ. BMI1-mediated histone ubiquitylation promotes DNA double-strand break repair. J Cell Biol 2010; 191:45-60; PMID:20921134; http://dx.doi.org/10.1083/jcb.201003034
- Bennardo N, Cheng A, Huang N, Stark JM. Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Gen 2008; 4:e1000110; PMID:18584027; http://dx.doi.org/10.1371/journal.pgen.1000110
- Dronyk A. New roles for B cell lymphoma 10 in the nucleus. MSc thesis. Edmonton, Alberta, Canada: Alberta, 2011:165.
- McManus KJ, Hendzel MJ. ATM-dependent DNA damage-independent mitotic phosphorylation of H2AX in normally growing mammalian cells. Mol Biol Cell 2005; 16:5013-25; PMID:16030261; http://dx.doi.org/10.1091/mbc.E05-01-0065
- Hao LY, Strong MA, Greider CW. Phosphorylation of H2AX at short telomeres in T cells and fibroblasts. J Biol Chem 2004; 279:45148-54; PMID:15322096; http://dx.doi.org/10.1074/jbc.M403924200
- Lukas C, Savic V, Bekker-Jensen S, Doil C, Neumann B, Pedersen RS, Grofte M, Chan KL, Hickson ID, Bartek J, et al. 53BP1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nat Cell Biol 2011; 13:243-53; PMID:21317883; http://dx.doi.org/10.1038/ncb2201
- Harrigan JA, Belotserkovskaya R, Coates J, Dimitrova DS, Polo SE, Bradshaw CR, Fraser P, Jackson SP. Replication stress induces 53BP1-containing OPT domains in G1 cells. J Cell Biol 2011; 193:97-108; PMID:21444690; http://dx.doi.org/10.1083/jcb.201011083
- Yuan J, Adamski R, Chen J. Focus on histone variant H2AX: to be or not to be. FEBS Lett 2010; 584:3717-24; PMID:20493860; http://dx.doi.org/10.1016/j.febslet.2010.05.021
- Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER, 3rd, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini N, Lerenthal Y, et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007; 316:1160-6; PMID:17525332; http://dx.doi.org/10.1126/science.1140321
- Zeng H, Di L, Fu G, Chen Y, Gao X, Xu L, Lin X, Wen R. Phosphorylation of Bcl10 negatively regulates T-cell receptor-mediated NF-kappaB activation. Mol Cell Biol 2007; 27:5235-45; PMID:17502353; http://dx.doi.org/10.1128/MCB.01645-06
- Woelk T, Sigismund S, Penengo L, Polo S. The ubiquitination code: a signalling problem. Cell Div 2007; 2:11; PMID:17355622; http://dx.doi.org/10.1186/1747-1028-2-11
- Nguyen LK, Kolch W, Kholodenko BN. When ubiquitination meets phosphorylation: a systems biology perspective of EGFR/MAPK signalling. Cell Commun Signal 2013; 11:52; PMID:23902637; http://dx.doi.org/10.1186/1478-811X-11-52
- Zhou H, Wertz I, O'Rourke K, Ultsch M, Seshagiri S, Eby M, Xiao W, Dixit VM. Bcl10 activates the NF-kappaB pathway through ubiquitination of NEMO. Nature 2004; 427:167-71; PMID:14695475; http://dx.doi.org/10.1038/nature02273
- Sun L, Deng L, Ea CK, Xia ZP, Chen ZJ. The TRAF6 ubiquitin ligase and TAK1 kinase mediate IKK activation by BCL10 and MALT1 in T lymphocytes. Mol Cell 2004; 14:289-301; PMID:15125833; http://dx.doi.org/10.1016/S1097-2765(04)00236-9
- Powell SN, Kachnic LA. Roles of BRCA1 and BRCA2 in homologous recombination, DNA replication fidelity and the cellular response to ionizing radiation. Oncogene 2003; 22:5784-91; PMID:12947386; http://dx.doi.org/10.1038/sj.onc.1206678
- Huertas P. DNA resection in eukaryotes: deciding how to fix the break. Nat Struct Mol Biol 2010; 17:11-6; PMID:20051983; http://dx.doi.org/10.1038/nsmb.1710
- Pommier Y. Topoisomerase I inhibitors: camptothecins and beyond. Nat Rev Cancer 2006; 6:789-802; PMID:16990856; http://dx.doi.org/10.1038/nrc1977
- Volcic M, Karl S, Baumann B, Salles D, Daniel P, Fulda S, Wiesmuller L. NF-kappaB regulates DNA double-strand break repair in conjunction with BRCA1-CtIP complexes. Nucleic Acids Res 2012; 40:181-95; PMID:21908405; http://dx.doi.org/10.1093/nar/gkr687
- Doil C, Mailand N, Bekker-Jensen S, Menard P, Larsen DH, Pepperkok R, Ellenberg J, Panier S, Durocher D, Bartek J, et al. RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell 2009; 136:435-46; PMID:19203579; http://dx.doi.org/10.1016/j.cell.2008.12.041
- Stewart GS, Panier S, Townsend K, Al-Hakim AK, Kolas NK, Miller ES, Nakada S, Ylanko J, Olivarius S, Mendez M, et al. The RIDDLE syndrome protein mediates a ubiquitin-dependent signaling cascade at sites of DNA damage. Cell 2009; 136:420-34; PMID:19203578; http://dx.doi.org/10.1016/j.cell.2008.12.042
- Kolas NK, Chapman JR, Nakada S, Ylanko J, Chahwan R, Sweeney FD, Panier S, Mendez M, Wildenhain J, Thomson TM, et al. Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science 2007; 318:1637-40; PMID:18006705; http://dx.doi.org/10.1126/science.1150034
- Huen MS, Grant R, Manke I, Minn K, Yu X, Yaffe MB, Chen J. RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell 2007; 131:901-14; PMID:18001825; http://dx.doi.org/10.1016/j.cell.2007.09.041
- Mailand N, Bekker-Jensen S, Faustrup H, Melander F, Bartek J, Lukas C, Lukas J. RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell 2007; 131:887-900; PMID:18001824; http://dx.doi.org/10.1016/j.cell.2007.09.040
- Zhao H, Zhu M, Dou G, Zhao H, Zhu B, Li J, Liao J, Xu X. BCL10 regulates RNF8/RNF168-mediated ubiquitination in the DNA damage response. Cell cycle 2014; 13:1777-87; PMID:24732096; http://dx.doi.org/10.4161/cc.28707
- Thome M. CARMA1, BCL-10 and MALT1 in lymphocyte development and activation. Nat Rev Immunol 2004; 4:348-59; PMID:15122200; http://dx.doi.org/10.1038/nri1352