
ABSTRACT
Families of cyclin-like proteins have emerged that bind and activate cyclin dependent kinases (Cdk)s, directing the phosphorylation of noncanonical Cdk substrates. One of these proteins, Spy1, has demonstrated the unique ability to directly bind and activate both Cdk1 and Cdk2, as well as binding and promoting the degradation of at least one Cdk inhibitor, p27Kip1. Spy1 accelerates somatic cell growth and proliferation and is implicated in a number of human cancers including the breast, brain and liver. Herein we isolate key residues mediating the direct interaction with p27. We use mutants of Spy1 to determine the physiological role of direct interactions with distinct binding partners Cdk2 and p27. We demonstrate that disrupting the direct interaction with either Spy1 binding partner decreased endogenous activity of Cdk2, as well as Spy1-mediated proliferation. However, only the direct interaction with p27 was essential for Spy1-mediated effects on p27 stability. In vivo neither mutation completely prevented tumorigenesis, although each mutation slowed the rate of Spy1-mediated tumorigenesis and decreased overall tumor volumes. This work supports the conclusion that direct interaction with both p27 and Cdk2 contribute to Spy1-mediated effects on cell growth. It is important to elucidate the dynamics of these interactions and to consider these data when assessing functional outcomes.
Introduction
Cell cycle regulation is an intricately controlled process that plays a critical role in all cell fate decisions. The catalytic cyclin dependent kinases (Cdks) are dependent on the production and destruction of their regulatory subunits, the cyclin family of proteins. Cyclin-Cdk complexes undergo a series of well-established post-translational modifications to assume a fully activated state.Citation1,2 The G1/S cyclin-Cdk complexes are also negatively regulated through interactions with the Cip/Kip family of Cdk inhibitors (CKI) which includes p27, p21 and p57. It is crucial that Cdk regulation be tightly controlled to prevent the onset of tumorigenesis; in many forms of cancer elevated Cdk2 kinase activity and decreasing levels of p27 correlate positively with tumorigenic potential and negatively with patient prognosis.Citation3,4 The central requirement for activated Cdks implicates these kinases as potential therapeutic targets, and indeed this approach has shown potential in pre-clinical tumorigenesis models.Citation5,6 However with the exception of hematological malignancies, CKI therapy has been largely disappointing in the clinic.Citation7,8 Targeting metastatic ER+/HER2- patients with elevated Cyclin D and reduced levels of p16 have shown promising response to synthetic CKIs targeting Cdk4/6, this work stresses the need to fully characterize the active Cyclin-Cdk partners in patient populations to optimize CKI therapeuticsCitation8,9 One factor that needs to be considered when stratifying patient populations is the presence of ‘cyclin-like’ proteins capable of activating the Cdks using very unique mechanisms.Citation10,11 Cdks bound to the Speedy/RINGO class of cyclin-like proteins are not dependent on post-translational modifications for activation, and they are less sensitive to inhibition by the CKIs p21 and p27.Citation11,12
One protein within the Speedy/RINGO class, Spy1A1 (gene SPDYA), herein referred to as Spy1, is capable of binding to, and activating, both Cdk1 and Cdk2 leading to increased rates of cell proliferation and inhibition of apoptosis when overexpressed.Citation10,13,14 The region regulating Spy1 interactions with Cdks are within the highly conserved S/R Box and several specific Spy1 residues have been identified to reduce binding affinity with Cdk2 in vitro.Citation12 Unlike any known cyclin proteins, Spy1 has also been shown to directly bind to the CKI p27, ultimately leading to ubiquitin-mediated degradation of the CKI.Citation15 Spy1-p27 and Cdk2 can immunoprecipitate together, supporting the rationale that they can form a trimeric complex, however Spy1 can also interact with each protein directly in vitro.Citation11,16 Data suggest that in a trimeric complex Spy1 preferentially binds p27 to prevent its inhibitory interactions with Cdk2, and that Cdk2 binding to Spy1 stabilizes this interaction and aids in the phosphorylation of p27 on T187, leading to p27 degradation.Citation11,15
Spy1 is elevated in the majority of breast carcinomas and Spy1 overexpression in the mouse mammary gland induces rapid and invasive tumorigenesis.Citation17,18 Defining the physiological role for direct interactions between Spy1-Cdk2 and Spy1-p27 in cell proliferation is an essential step toward resolving the mechanism by which Spy1 promotes mammary tumorigenesis.
This work isolates 4 key amino acids responsible for mediating the binding between Spy1 and p27. Using mutants of these sites we compare effects to a mutant form of Spy1 defective in binding to Cdk2. Our data demonstrate that preventing direct interactions with either Cdk2 or p27 reduce endogenous activity of Cdk2 and subsequently Spy1-mediated proliferation. However, only abrogation of binding between Spy1 and p27 significantly inhibits degradation of the CKI and reduces the proliferative capacity of Spy1 in the presence of exogenously expressed p27. These latter data support the conclusion that Spy1 direct binding to p27 and Cdk2 function via separable mechanisms. Despite this point, we show that direct Spy1 interactions with both p27 and Cdk2 play a role in mediating the rate of tumorigenic onset caused by Spy1. However, disrupting either interaction alone did not completely prevent tumor formation. These data stress that the prevalence and kinetics of these specific interactions in vivo will have significant effects on the functional outcomes of Spy1 expression. These details become very important when considering mechanisms of targeting Spy1, p27 or Cdks for drug therapy.
Methods
Ethics statement
BALB/c mice were maintained following the Canadian Council on Animal Care guidelines at the University of Windsor (protocol #06-19).
Cell culture
Human embryonic kidney cell line, HEK293 (ATCC) were maintained in DMEM medium (Sigma) supplemented with 10% (vol/vol) fetal bovine serum (Hyclone). HC11, a non-transformed, immortalized BALB/c mouse mammary epithelial cell line (provided by Dr. C. Shermanko; University of Calgary) were maintained in RPMI 1640 medium (Hyclone) containing 10% (vol/vol) fetal calf serum (Sigma), supplemented with 5 μg/ml insulin (Sigma), and 10 ng/ml EGF (Calbiochem). Both cell lines were maintained in 2 mM L-glutamine (Sigma), penicillin (Invitrogen), and streptomycin (Invitrogen), and were cultured in a 5% CO2 environment.
Cell counts
Cell counts were conducted with trypan blue exclusion and quantified using a BioRad TC10 Automated Cell Counter.
Plasmid and mutagenesis
Creation of Myc-Spy1 in PCS3 was described previously.Citation10 Spy1-PEIZ was generated by moving Flag-Spy1 from Spy1-PJT0013.Citation11 through EcoR1 and Xba1 restriction sites into the lentiviral PEIZ vector (provided by Dr. B. Welm, University of Utah). Spy1-D90, Spy1-R170 and Spy1-R170-PEIZ were created in Myc-Spy1-PCS3 using Quik Change PCR Multi Site-Directed Mutagenesis (SDM). Spy1-R170 mutation was made using primers # A315 5'- GTTAAGGGACCAGCTCTGGGATGCAATTGACTATGCGGCTATTGTAAGCAGG- 3' and # A316 5'- CCTGCTTACAATAGCCGCATAGTCAATTGCATCCCAGAGCTGGTCCCTTAAC-3';Spy1-D90 was made using primers #A123 5'- GATTTCTTGTGGATGGCATGCTGCTGTAAAATTGC-3' and #A124 5'- GTTAAGGGACCACTGGGATGSAAGACTATGCGGCTATTGTAAGCAGG 3', Spy1-R170-PEIZ lentivirus plasmid was created by engineering an EcoR1 site upstream of Spy1 in Spy1-PCS3 using primers #A532 5’-cttgatttaggtgacactatagaattcaagcttgttctttttg-3’ and #A533 5’-CAAAAAGAACAAGTAGCTTGAATTCTATAGTGTCACCTAAATCAAG-3’. Spy1-R170 was moved to PEIZ lentivirus vector via the EcoR1 and XbaI sites. Successful cloning in all cases was determined by DNA sequencing (Robarts Sequencing Facility; Univ. of Western Ontario).
Immunoblotting (IB) and immunoprecipitation (IP)
Cells were harvested and lysed in 0.1% NP-40 lysis buffer (0.1% NP-40, 20 mM Tris pH 7.5, 5 mM 0.5 M EDTA, 100 mM NaCl in RO water) containing protease inhibitors (100 ug/mL PMSF, 5 ug/mL aprotinin, 2 ug/mL leupeptin) for 30 min on ice. Bradford Reagent was used to determine the protein concentration following the manufacturer's instruction (Sigma). Aliquots of lysates containing 20–30 μg protein were subjected to electrophoresis on denaturing SDS-10% polyacrylamide gels and transferred to PVDF-Plus 0.45 micron transfer membranes (Osmonics Inc.) for 2 hr at 30 V using a wet transfer method. Blots were blocked for 2 hr in TBST containing 3% non-fat dry milk (blocker) at room temperature, primary antibodies were reconstituted in blocker and incubated overnight at 4°C, secondary antibodies were used at a 1:10,000 dilution in blocker for 1 hr at room temperature. Blots were washed 3 times with TBST following incubation with both the primary and secondary antibodies. Washes were 6 min each following the primary antibody and 10 min each following the secondary antibody. Chemilumiminescent Peroxidase Substrate was used for visualization following the manufacturer's instruction (Pierce). Chemiluminescence was quantified on an Alpha Innotech HD2 (Fisher) using AlphaEase FC software.
For IP equal amounts of protein were incubated with primary antisera as indicated overnight at 4°C, followed by the addition of 10 ul protein A–Sepharose (Sigma) and incubated at 4°C with gentle rotation for an additional 2 hr. These complexes were then washed 3 times with 0.1% NP-40 lysis buffer and resolved by 10% SDS-PAGE.
Transfections and infections
Cells were transfected overnight using polyethylenimine (PEI) branched reagent Sigma (408727), 10 μg of DNA was mixed with 50 μL of 150 mM NaCl and 30 ug PEI for 10 min then added to a 10 cm tissue culture plate.
Lentiviral infections were carried out using a modified protocol from Welm et al.Citation19 In brief; PEI transfection of Lenti-XTM 293 producer cell lines (Cat. No. 632180, Clontech, CA) was conducted as described above, 16 hr post-transfection, media containing viral particles was concentrated, titred and stored at −80°C. Cells were screened for ZS-Green using flow cytometry. HC11 infections were optimized using 2 × 105 cells/well in a 6-well plate to determine the multiplicity of infection (MOI). HC11 cells were seeded with media lacking antibiotics for 18 hr, media was removed and transfection media lacking antibiotics and serum added. Viral particles were added in a final concentration of 8*105 transfection units (TU = MOI * cell number), and polybrene was added to a final concentration of 10 ug/ml. Mixture was incubated for 4 hr, followed by a media change to growth media lacking antibiotic.
Antibodies
The generation of Spy1 antibody was previously described.Citation10 Myc antibodies both mouse (9E10) and rabbit (C19), HA (Y11 and F7) and p27 (F8) were purchased from Santa Cruz. Actin antibody (MAB150R) was purchased from Calbiochem. HRP conjugated secondary mouse antibody (A9917) and rabbit antibody (A0545) were purchased from Sigma.
Pulse chase
Cells were transfected with indicated constructs, after 16 hrs serum free media was replaced with Cys-Met free media (D0422; Sigma) supplemented with dialyzed FBS (F0392; Sigma) for 1 hr. S35 was added to a final concentration of 500 μCi for 4 hr followed by 4 washes with 1X PBS and supplementation with growth media containing 2 mM Cys-Met. Cells were lysed at indicated time points, ran on an SDS-PAGE gel. Incorporated sulfur was visualized using a Cyclone storage phosphor system and quantified using OptiQuant software (Perkin Elmer).
Kinase assays
Cells were transfected, cultured in 10% FBS and lysed in 0.1% NP-40 lysis buffer. 18 hr post-transfection IP was carried out as described above and precipitates were washed 4 times prior to the addition of 50 μl of kinase buffer (50 mM Tris-HCl pH 7.5, 10 mM MgCl2, 1 mM DTT, 20 mM EGTA, 50 mM ATP, 10 μCi of [γ-32P]ATP) and 74 μg/ml H1 histone (Boehringer Mannheim). Reactions were incubated for 10 min at 30°C, sample buffer was added to stop the reaction and 50 μl of each sample were analyzed by 10% SDS-PAGE. Incorporated phosphorylation was visualized using a Cyclone storage phosphor system and quantified using OptiQuant software (Perkin Elmer). IPs were subsequently probed on the same membrane.
Fat pad transplants
HC11 cells infected and selected for the relevant lentiviral constructs were injected into the cleared fat pad of the fourth mammary glands in 22-day old mice (250,000 cells per gland) as previously described.Citation17 Tumor incidence was monitored every 2 to 4 d beginning one week after surgery by palpitation of the gland. Glands were allowed to grow for 2 to 4 weeks following surgery. Tumor measurements were recorded using manual calipers and tumor volume was calculated as length (mm) × width (mm) × height (mm). Collected glands were either paraffin embedded for immunohistochemistry or flash frozen for protein, genomic or mRNA analysis.
Results
Resolving Spy1 residues mediating binding with p27
Using a panel of Spy1 deletion mutants previously describedCitation20 we narrowed down the region within the Spy1 protein necessary for p27 binding. Previously, it was determined that truncation of Spy1 at aa 215 retained the ability to bind p27, while truncation at aa 160 in Spy1 disrupted binding to p27Citation15 (; indicated beneath panel). We further tested these results using deletion mutants (DM) of Spy1 where the indicated regions were deleted (). DMA represents a protein devoid of aa 1-57, DMB is devoid of aa 57-83, DMC lacks aa 83-145, DMG lacks aa 147-239 and DMZ lacks aa 241-286. We first determined whether any of these Spy1 deletion mutants would result in abrogation of binding to p27. 293 cells were transfected with wild-type (WT) Spy1 or versions of the Spy1 protein harboring the specified deletions (DMA-DMZ) (). Cells were lysed and equal amounts of protein immunoprecipitated with Myc antibody and analyzed by western blot. DMG failed to bind to p27, demonstrating that aa 145–239 are required for the binding between Spy1 and p27. This was not surprising given the large size of the deletion within DMG, as well as the previous results suggesting the essentiality of this region for binding.Citation15 Although p27 interacts in different ways with known partners, it was previously determined that p27 binding interactions favor positively charged amino acids on the binding partner.Citation21-23 Alignment of the potential p27-binding region within Spy1 from a number of different organisms noted a region of high similarity containing a string of 4 highly conserved positive amino acids (). Previous binding interactions were found to depend on arginine interactions and were disrupted using arginine to alanine substitutions.Citation22,23 Hence, we generated a mutant of Spy1 containing alanine substitutions for arginines 170 and 174 (Spy1-R170) as well as a mutant containing alanine substitutions for arginines 179 and 180 (Spy1-R179) ().
Figure 1. Generation of Spy1 – p27 binding mutants. (A) A schematic diagram of the Spy1 deletion mutants used for screening and their enzyme cut sites. The indicated regions are selectively deleted in each Spy1 mutant construct (ie. DMA lacks region A, DMB lacks region B, DMA lacks region C, DMG lacks region G, DMZ lacks region Z). Dotted line depicts the truncation mutant of Spy1 previously determined to retain the ability to bind p27.Citation15 The solid line reflects the region previously shown to have lost the ability to bind p27. (B) 293 cells were transfected with Myc-Spy1-PCS3 (WT) or the different deletion mutants DMA-DMZ depicted above in the presence of HA-tagged p27 (HA-p27). Transfected cells were treated with MG132 (10μM) for 14 hrs prior to harvest, lysates were immunoprecipitated with Myc antibody and immunoblotted with HA antibody (upper panel) and Myc antibody (lower panel). This is one representative experiment of 3. (C) Alignment of a highly conserved amino acid sequence within the predicted p27 binding region from several species. Conserved positively charged residues to be mutated are noted with a box. Amino acid #s are indicated after the species in brackets. (D) Region G of Spy1 depicting the Arg. (R) residues which were mutated to Alanine (A) to create Spy1-R170 and Spy1-R179 mutations.
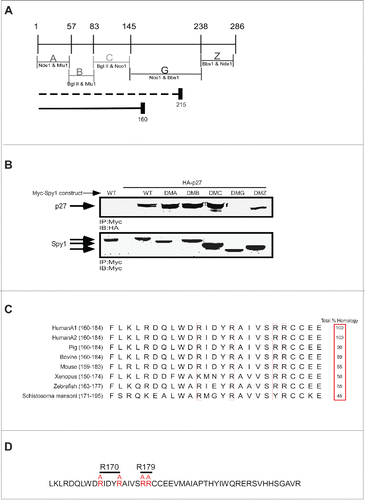
Spy1-R170 and -179 do not bind to p27
To test the necessity of these conserved arginine residues for binding to p27, Myc-tagged Spy1-WT, Spy1-R170, or Spy1-R179 were transfected in combination with HA-tagged p27 in the presence of MG132 (). IP/IB of these lysates demonstrate that Spy1-R170 and Spy1-R179 mutants both reduce binding to p27 as compared to Spy1-WT binding. Reciprocally, IP with Myc antibody isolated p27 when Spy1-WT was overexpressed but not the mutant Spy1 constructs (). It is notable that Cdk2 continued to bind to Spy1 in the presence of both p27-binding mutations (; 3rd panel), which is consistent with previous results demonstrating that Cdk2 binds more efficiently to Spy1 in the presence of overexpressed p27 protein.Citation11 We also examined the interaction between endogenous p27 and Spy1-WT or mutant constructs. Following IP for the Myc-tagged Spy1 proteins, endogenous p27 protein was detected as part of a complex with Spy1-WT but not with either of the Spy1 non-binding mutants (). Collectively, these results demonstrate that mutation of Spy1 at either arginines 170/174 or 179/180 abrogates binding interactions with the CKI p27. These mutants will provide a valuable tool in assessing the specific role for Spy1-p27 interactions in functional experiments.
Figure 2. R170 and R179 mutants of Spy1 abrogate binding to p27. 293 cells were transfected with constructs indicated above the panels and treated with MG132 prior to lysis. Equal amounts of protein was subject to IP/IB as indicated below each panel (upper panels). For each experiment cell lysates were also run and blotted to demonstrate transfection efficiencies (lower panels). (A) Overexpression of all constructs and IP for HA-tagged p27. (B) Overexpression of all constructs and IP for Myc-tagged Spy1 constructs. (C) Transfection of Spy1-WT or mutant constructs and analysis using endogenous p27. Cells were maintained in 2% serum containing media for 14 hr following transfection to elevate endogenous p27 levels. All experiments reflect one representative experiment of 3.
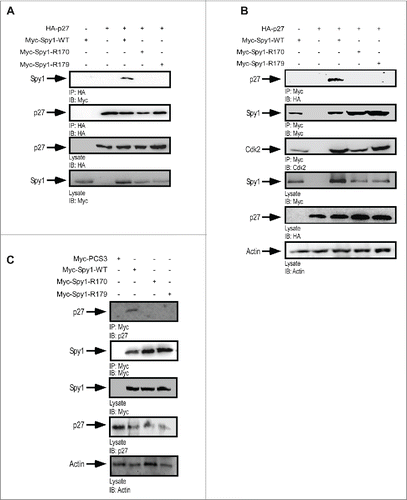
Effect of Spy1 mutants on p27 degradation
Direct binding of Spy1 protein to p27 enhances p27 degradation and subsequently activates Cdk2 kinase activity.Citation11,15 Importantly, Spy1 also directly binds to Cdk2 to activate kinase activity and it is known that Cdk2 phosphorylation of p27 on T187 promotes p27 degradation.Citation24 To determine whether the direct binding of Spy1 to p27 and/or Cdk2 is critical for Spy1-mediated degradation of p27, we utilized 293 cells transfected with either the Spy1-p27 non-binding mutants (Spy1-R170 and Spy1-R179) or the Spy1 mutant previously demonstrated to disrupt interactions with Cdk2 (Spy1-D90).Citation12 Lysates were monitored by IB (; left panel) and results over 3 separate experiments were quantified by densitometry (; right panel). Cells transfected with Spy1-WT and Spy1-D90 resulted in a significant reduction in overall p27 protein levels as compared to cells overexpressing an empty vector control or the Spy1-p27 non-binding mutants. To further confirm these results we used radioactive sulfur (S35) incorporation in a pulse chase assay, IP for p27, followed by IB and phosphor-image analysis to determine relative stability of p27 protein levels over 3 separate experiments (). IgG was used as a control for the IP and lysates were used for control over transfections. Quantification of this data demonstrates that Spy1-WT and Spy1-D90 significantly reduce the stability of p27 protein levels over the empty vector control; however the Spy1-R170/R179 mutants were unable to significantly impact p27 turnover. These data demonstrate that the direct binding between Spy1 and p27 is essential for Spy1-mediated effects on p27 protein turnover. To determine the relative effects of direct interactions with Cdk2 or p27 on Cdk2 kinase activity, a histone kinase assay was performed (). Phosphor-quantification normalized to the Cdk2 IP demonstrated that preventing interactions with either p27 or Cdk2 reduced kinase activity to that of control, showing that each of these interactions is essential for Spy1-mediated activation of Cdk2.
Figure 3. R170 and R179 mutants inhibit p27 down regulation. 293 cells were transfected with HA-tagged p27 in the presence or absence of Myc-tagged empty–vector (PCS3), Spy1-WT, Spy1-R170, Spy1-R179 or Spy1-D90. (A) Lysates were blotted with HA (upper panel), Spy1 (middle panel) and Actin (lower panel) antibodies. Left panel is one representative blot of 3. Right panel reflects densitometry of p27 levels normalized to actin over 3 separate experiments. Error bars represent SEM. *p < 0.05. (B) Pulse chase assay was conducted, followed by IP for HA-tagged p27 and S35 incorporation measured by phosphor image analysis. Immunoblot using IgG antibody was used as a control. Left panel is one representative experiment of 3. Right panel represents quantification using OptiQuant software over 3 separate experiments. Error bars represent SEM, *p < 0.05 (C) Cdk2 IP from transfected lysates. Histone assay was conducted. Left panel is one representative phosphorimage of 2. Right panel represents average phosphor measurements over 2 experiments. Error bars represent SEM.
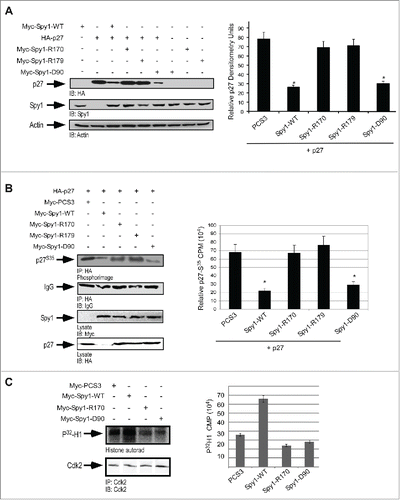
Spy1 is known to override cell cycle inhibition by p27.Citation11 To examine the effect of the Spy1 mutants on p27-induced cell cycle arrest, cells were transfected with the indicated constructs in the presence of p27 and cell number after 24 hr was assessed by trypan blue exclusion (). Cells transfected with the Spy1-p27 non-binding mutants showed a significant decrease in the total cell number as compared to Spy1-WT, being comparable to that of the empty vector control. Importantly, the Spy1-D90 resulted in a significant increase in the total number of cells over the empty vector control, with numbers comparable to that of Spy1-WT. It is important to note that while effects on overall levels of p27 protein were not as distinguishable when p27 was expressed exogenously, Spy1–WT and Spy1-D90A significantly reduced overall levels of exogenous p27 when quantified over multiple trials (Fig. S1). These results demonstrate that the ability of Spy1 to override the inhibitory effects of p27 is dependent on the direct interaction with p27. Measuring cell proliferation in the absence of p27 overexpression demonstrated that both Cdk2 and p27 binding mutants decreased Spy1-proliferative effects, although neither mutant completely rescued the effect of Spy1 overexpression (). These results support the conclusion that direct interactions with both Cdk2 and p27 contribute toward Spy1-medicated proliferative effects.
Figure 4. Direct p27 binding is required to override cell cycle arrest by exogenously expressed p27. 293 cells were transfected with Myc-tagged empty–vector (PCS3), Spy1-WT, Spy1-R170, Spy1-R179 or Spy1-D90 in the presence (A) or absence (B) of HA-p27. Cell numbers were assessed using trypan blue exclusion and quantification over 3 separate experiments. Error bars represent SEM. **p < 0.01; * p< 0.05. Right panels represent one representative blot of 3.
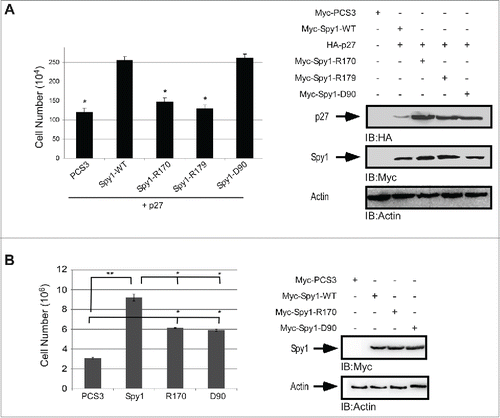
Spy1 direct binding to both p27 and Cdk2 are important for Spy1-mediated tumorigenesis in vivo
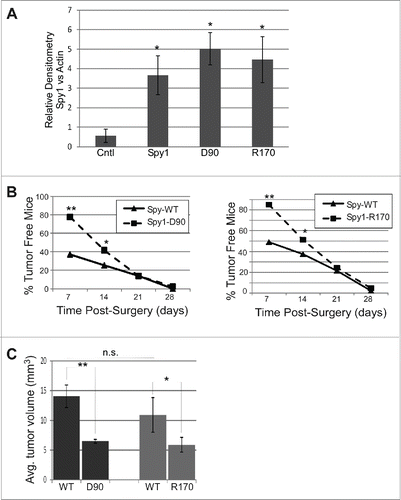
It has previously been shown that overexpression of Spy1 in vivo leads to accelerated rates of mammary tumorigenesis.Citation17 To determine if this is dependent on Spy1s interaction with either Cdk2 or p27, fat pad transplantation was performed on the cleared inguinal mammary gland of Balb/C mice using Spy1-WT expression on one side of the mouse and Spy1 mutants defective for either Cdk2 binding (Spy1-D90) or p27 binding (Spy1-R170) on the opposite side. Relative expression for each construct were statistically similar (). One week following surgery, approximately 50–65% of all Spy1-WT glands had visible tumors while only 20% of all Spy1-D90 glands and 10% of all Spy1-R170 glands had visible tumors (). It is notable that HCll cells alone did not show any signs of tumor formation until ∼30 weeks post-surgery (Fig. S2). At day 21 post-transplant ∼80–90% of all pEIZ-Spy1 transplanted glands had detectible tumors, with no difference in numbers between mutant or WT forms of Spy1. Mice were humanely sacrificed at day 28 post-surgery due to the size and invasiveness of the Spy1-WT glands and glands were removed and studied. There were significant differences in the overall tumor size observed with Spy1-WT glands being much larger than both the Spy1-D90 and Spy1-R170 tumors (). It is notable that at the timepoint of tumor analysis Spy1 expression had been lost in WT and mutant constructs alike (data not shown). Collectively these data indicate that Spy1 direct interactions with both p27 and Cdk2 are important for Spy1-mediated tumorigenesis in vivo. It further suggests that inhibition of both of these functional pathways is required to significantly reduce tumorigenic effects over time.
Figure 5. Direct binding to both p27 and Cdk2 is required for Spy1-mediated tumorigenesis. Cleared mammary fat pads were transplanted with cells expressing Spy1-WT (WT) on the left and either Spy1-R170 (R170) or Spy1-D90 (D90) on the right. (A) Lysates from a subset of cells transplanted were blotted with Spy1 and Actin and quantified by densitometry over 3 individual experiments; Spy1 levels were normalized to actin. Spy1-WT samples were pooled together and results are compared to Control infected HC11 cells (Cntl). Error bars represent SEM. *p < 0.05. (B) Graphic depiction of the percent of mice remaining free from palpable tumors at time points following transplantation with Spy1-WT and Spy1-D90 (right panel) and Spy1-WT and Spy1-R170 (left panel). Treatments occurred in groups of 3 using 3 separate colonies of infected cells. Total sample size = 45. Mann-Whitney, and Wilcoxon Matched Pairs Tests were performed (**p < 0.01; *p < 0.05). (C) Total average tumor volume (length x width x height) over 45 mice transplanted with either R170 or D90 (right gland) and Spy1-WT (left gland) from 3 separate infections are depicted.**p < 0.002; *p < 0.05, n.s = not significant.
Discussion
Spy1 is an atypical Cdk activator known to increase cell proliferation and inhibit apoptosis when overexpressed in mammalian cells.Citation10 In addition to directly binding and activating Cdks, Spy1 binds the CKI p27.Citation11 In vitro and in vivo experiments demonstrate that Spy1 directly binds and co-localizes with nuclear forms of p27, functioning to override p27-mediated cell cycle inhibition.Citation11 Like Cyclin E/Cdk2, Spy1 overexpression is also associated with an increase in phosphorylation of p27 at T187 leading to its proteasomal degradation and enhanced cell cycle progression.Citation15 Whether the effects on p27 degradation are mediated through the direct binding of Spy1 to Cdk2 or p27 was not previously known. To address this question, we mapped 2 critical p27 binding sites on p27 and characterized 2 p27 non-binding mutants (Spy1-R170 and Spy1-R179). These mutants were used along with a Cdk2 binding mutant (Spy1-D90) that has been previously described,Citation16 in proliferation assays as well as in assays to assess p27 protein levels. We determined that direct binding of Spy1 to p27 was required to promote p27 degradation, as well as to override a p27-induced cell cycle arrest. The direct binding of Spy1 to both Cdk2 and p27 were required for Spy1-mediated activation of Cdk2 and overall effects on cell proliferation. These data demonstrate 2 distinct mechanisms by which Spy1 can drive cell proliferation and Cdk2 kinase activity.
Role of Spy1-p27 binding in tumorigenesis
Elevated expression of Spy1 is found in many forms of human cancer.Citation18,25-28 In this present study, we have demonstrated that abrogating direct interactions between Spy1 and either p27 or Cdk2 significantly decreases the rate of tumorigenesis and overall tumor size in vivo. Low levels of p27 protein has been implicated in many human cancers including breastCitation29 and is found to correlate with poor patient outcome.Citation29 Constitutive activation of Cdk2 has also been shown to result in mammary gland hyperplasia, fibrosis, and mammary tumors in a MMTV–cyclin D1–Cdk2 derived cell line.Citation30 Given that Cdk2 targets p27 for degradation,Citation31 it is a valid hypothesis that these proteins are regulating each other; interestingly however, our data suggests that direct interactions with both p27 and Cdk2 are important in driving the tumorigenic activities of Spy1. Details on how these proteins function independently, as well as in a trimeric complex, is required to fully resolve the physiological and pathological roles of these interactions.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Author contributions
Experiments were conceived and designed by MAS, BF, DM, LAP. Experiments were performed by MAS, BF and DM. Data was analyzed by MAS, BF, DM and LAP. Reagents/materials/tools provided by LAP. Manuscript was prepared by MAS, BF and LAP.
KCCY_S_1121327.zip
Download Zip (157.1 KB)Acknowledgments
We thank Dr. C. Shermanko for providing the HCll cell lines, Dr. B. Welm for providing lentivirus vector (PEIZ), Dr. D.J. Donoghue for supplying vectors and the Spy1 antibody and J. Maimaiti and N. Paquette for technical assistance.
Funding
BF was supported by a fellowship funded by the Canadian Breast Cancer Foundation-Ontario Region.
This study was supported by operating funds from the Canadian Cancer Society (CCS)/Canadian Breast Cancer Research Alliance (CBCRA) #020513.
Related Research Data
References
- De Bondt HL, Rosenblatt J, Jancarik J, Jones HD, Morgan DO, Kim SH. Crystal structure of cyclin-dependent kinase 2. Nature 1993; 363:595–602; PMID:8510751; http://dx.doi.org/10.1038/363595a0
- Nurse PM. Nobel Lecture. Cyclin dependent kinases and cell cycle control. Biosci Rep 2002; 22:487-99; PMID:12635846; http://dx.doi.org/10.1023/A:1022017701871
- Said TK, Medina D. Cell cyclins and cyclin-dependent kinase activities in mouse mammary tumor development. Carcinogenesis 1995; 16:823-30; PMID:7728962; http://dx.doi.org/10.1093/carcin/16.4.823
- Alkarain A, Jordan R, Slingerland J. p27 deregulation in breast cancer: prognostic significance and implications for therapy. J Mammary Gland Biol Neoplasia 2004; 9:67-80; PMID:15082919; http://dx.doi.org/10.1023/B:JOMG.0000023589.00994.5e
- Nair BC, Vallabhaneni S, Tekmal RR, Vadlamudi RK. Roscovitine confers tumor suppressive effect on therapy-resistant breast tumor cells. Breast Cancer Res 2011; 13: R80; PMID:21834972; http://dx.doi.org/10.1186/bcr2929
- Appleyard MV, O'Neill MA, Murray KE, Paulin FE, Bray SE, Kernohan NM, Levison DA, Lane DP, Thompson AM. Seliciclib (CYC202, R-roscovitine) enhances the antitumor effect of doxorubicin in vivo in a breast cancer xenograft model. Int J Cancer 2009; 124:465-72; PMID:19003963; http://dx.doi.org/10.1002/ijc.23938
- Dickson MA, Schwartz GK. Development of cell-cycle inhibitors for cancer therapy. Curr Oncol 2009; 16:36-43; PMID:19370178
- Malumbres M, Pevarello P, Barbacid M, Bischoff JR. CDK inhibitors in cancer therapy: what is next? Trends Pharmacol Sci 2008; 29:16-21; PMID:18054800; http://dx.doi.org/10.1016/j.tips.2007.10.012
- Guha M. Blockbuster dreams for Pfizer's CDK inhibitor. Nat Biotechnol 2013; 31:187; PMID:23471056; http://dx.doi.org/10.1038/nbt0313-187a
- Porter LA, Dellinger RW, Tynan JA, Barnes EA, Kong M, Lenormand JL, Donoghue DJ. Human Speedy: a novel cell cycle regulator that enhances proliferation through activation of Cdk2. J Cell Biol 2002; 157:357-66; PMID:11980914; http://dx.doi.org/10.1083/jcb.200109045
- Porter LA, Kong-Beltran M, Donoghue DJ. Spy1 interacts with p27Kip1 to allow G1/S progression. Mol Biol Cell 2003; 14:3664-74; PMID:12972555; http://dx.doi.org/10.1091/mbc.E02-12-0820
- Cheng A, Gerry S, Kaldis P, Solomon MJ. Biochemical characterization of Cdk2-Speedy/Ringo A2 BMC Biochem 2005; 6:19; PMID:16191191; http://dx.doi.org/10.1186/1471-2091-6-19
- Barnes EA, Porter LA, Lenormand JL, Dellinger RW, Donoghue DJ. Human Spy1 promotes survival of mammalian cells following DNA damage. Cancer Res 2003; 63:3701-7; PMID:12839962
- Gastwirt RF, Slavin DA, McAndrew CW, Donoghue DJ. Spy1 expression prevents normal cellular responses to DNA damage: inhibition of apoptosis and checkpoint activation. J Biol Chem 2006; 281:35425-35; PMID:16951407; http://dx.doi.org/10.1074/jbc.M604720200
- McAndrew CW, Gastwirt RF, Meyer AN, Porter LA, Donoghue DJ. Spy1 enhances phosphorylation and degradation of the cell cycle inhibitor p27. Cell Cycle 2007; 6:1937-45; PMID:17671428; http://dx.doi.org/10.4161/cc.6.15.4520
- Cheng A, Xiong W, Ferrell JE Jr., Solomon MJ. Identification and comparative analysis of multiple mammalian Speedy/Ringo proteins. Cell Cycle 2005; 4:155-65; PMID:15611625; http://dx.doi.org/10.4161/cc.4.1.1347
- Golipour A, Myers D, Seagroves T, Murphy D, Evan GI, Donoghue DJ, Moorehead RA, Porter LA. The Spy1/RINGO family represents a novel mechanism regulating mammary growth and tumorigenesis. Cancer Res 2008; 68:3591-600; PMID:18483240; http://dx.doi.org/10.1158/0008-5472.CAN-07-6453
- Al Sorkhy M, Ferraiuolo RM, Jalili E, Malysa A, Fratiloiu AR, Sloane BF, Porter LA. The cyclin-like protein Spy1/RINGO promotes mammary transformation and is elevated in human breast cancer. BMC Cancer 2012; 12:45; PMID:22280365; http://dx.doi.org/10.1186/1471-2407-12-45
- Welm BE, Dijkgraaf GJ, Bledau AS, Welm AL, Werb Z. Lentiviral transduction of mammary stem cells for analysis of gene function during development and cancer. Cell Stem Cell 2008; 2:90-102; PMID:18371425; http://dx.doi.org/10.1016/j.stem.2007.10.002
- Al Sorkhy M, Craig R, Market B, Ard R, Porter LA. The cyclin-dependent kinase activator, Spy1A, is targeted for degradation by the ubiquitin ligase NEDD4. J Biol Chem 2009; 284:2617-27; PMID:19054764; http://dx.doi.org/10.1074/jbc.M804847200
- Wang W, Ungermannova D, Chen L, Liu X. Molecular and biochemical characterization of the Skp2-Cks1 binding interface. J Biol Chem 2004; 279:51362-9; PMID:15452136; http://dx.doi.org/10.1074/jbc.M405944200
- Ungermannova D, Gao Y, Liu X. Ubiquitination of p27Kip1 requires physical interaction with cyclin E and probable phosphate recognition by SKP2. J Biol Chem 2005; 280:30301-9; PMID:15980415; http://dx.doi.org/10.1074/jbc.M411103200
- Sitry D, Seeliger MA, Ko TK, Ganoth D, Breward SE, Itzhaki LS, Pagano M, Hershko A. Three different binding sites of Cks1 are required for p27-ubiquitin ligation. J Biol Chem 2002; 277:42233-40; PMID:12140288; http://dx.doi.org/10.1074/jbc.M205254200
- Montagnoli A, Fiore F, Eytan E, Carrano AC, Draetta GF, Hershko A, Pagano M. Ubiquitination of p27 is regulated by Cdk-dependent phosphorylation and trimeric complex formation. Genes Dev 1999; 13:1181-9; PMID:10323868; http://dx.doi.org/10.1101/gad.13.9.1181
- Zucchi I, Mento E, Kuznetsov VA, Scotti M, Valsecchi V, Simionati B, Vicinanza E, Valle G, Pilotti S, Reinbold R, et al. Gene expression profiles of epithelial cells microscopically isolated from a breast-invasive ductal carcinoma and a nodal metastasis. Proc Natl Acad Sci U S A 2004; 101:18147-52; PMID:15608061; http://dx.doi.org/10.1073/pnas.0408260101
- Ke Q, Ji J, Cheng C, Zhang Y, Lu M, Wang Y, Zhang L, Li P, Cui X, Chen L, et al. Expression and prognostic role of Spy1 as a novel cell cycle protein in hepatocellular carcinoma. Exp Mol Pathol 2009; 87:167-72; PMID:19686732; http://dx.doi.org/10.1016/j.yexmp.2009.07.011
- Hang Q, Fei M, Hou S, Ni Q, Lu C, Zhang G, Gong P, Guan C, Huang X, He S. Expression of Spy1 protein in human Non-Hodgkin's Lymphomas is correlated with phosphorylation of p27(Kip1) on Thr187 and cell proliferation. Med Oncol 2012; PMID:22492278
- Zhang L, Shen A, Ke Q, Zhao W, Yan M, Cheng C. Spy1 is frequently overexpressed in malignant gliomas and critically regulates the proliferation of glioma cells. J Mol Neurosci 2012; 47:485-94; PMID:22447439; http://dx.doi.org/10.1007/s12031-012-9709-5
- Slingerland J, Pagano M. Regulation of the cdk inhibitor p27 and its deregulation in cancer. J Cell Physiol 2000; 183:10-17; PMID:10699961; http://dx.doi.org/10.1002/(SICI)1097-4652(200004)183:110AID-JCP23.0.CO;2-I
- Corsino P, Davis B, Law M, Chytil A, Forrester E, Nørgaard P, Teoh N, Law B. Tumors initiated by constitutive Cdk2 activation exhibit transforming growth factor beta resistance and acquire paracrine mitogenic stimulation during progression. Cancer Res 2007; 67:3135-44; PMID:17409420; http://dx.doi.org/10.1158/0008-5472.CAN-06-3815
- Vlach J, Hennecke S, Amati B. Phosphorylation-dependent degradation of the cyclin-dependent kinase inhibitor p27. Embo J 1997; 16:5334-44; PMID:9311993; http://dx.doi.org/10.1093/emboj/16.17.5334