
ABSTRACT
TP53 is highly mutated in human cancers, thus targeting this tumor suppressor pathway is highly desirable and will impact many cancer patients.Citation1,2 Therapeutic strategies to reactivate the p53-pathway have been challenging,Citation3,4 and no effective treatment exists.Citation5 We utilized the p53-family members, p63 and p73, which are not frequently mutated in cancer, to treat p53-defective cancers. The N-terminal splice variants of p63 and p73 are denoted as the TA and ΔN isoforms. We recently demonstrated that deletion of either ΔNp63 or ΔNp73 in p53-deficient mouse tumors results in tumor regression mediated by metabolic programming. Using this strategy, we identified pramlintide, a synthetic analog of amylin, as an effective treatment for p53 deficient and mutant tumors. Here, we show the utility of using pramlintide, as a potential cancer preventive option for p53-deficient tumors in mouse models. Additionally, we found that in vivo inhibition of both ΔNp63 and ΔNp73 in combination accelerates tumor regression and increases survival of p53-deficient mice. We report that inhibition of both ΔNp63 and ΔNp73 in combination results in upregulation of 3 key metabolic regulators, IAPP, GLS2, and TIGAR resulting in an increase in apoptosis and tumor regression in ΔNp63/ΔNp73/p53 deficient thymic lymphomas. These data highlight the value of generating inhibitors that will simultaneously target ΔNp63 and ΔNp73 to treat cancer patients with alterations in p53.
Introduction
p53, p63 and p73 in tumor suppression
TP53 is known as the “guardian of the genome” for its well-documented tumor suppressive functions.Citation6 TP53 functions primarily as a transcription factor activating multiple target genes upon DNA damage or cellular stress.Citation7 Under normal physiological conditions, p53 protects the genome by directing the damaged cells to undergo cell death or cell cycle arrestCitation8; however, p53 is highly mutated resulting in p53-loss of function or gain of function phenotypes (TCGA database, ). In the context of cancer, p53 functional status is critical as inactivation of p53 function exposes the genome to multiple insults further accelerating tumor progression.Citation1 Restoration of p53-activity has proven successful in mouse modelsCitation4,9,10; however, therapeutic efforts to reactivate p53 in human cancers have remained challenging. While restoration of p53 in p53 nullizygous mice leads to tumor regression, such a restoration in tumors with a missense mutation in p53, mutations that are present in a wide variety of human tumors, leads primarily to tumor stasis.Citation11 This lack of tumor regression is likely due to the complexity of the p53 family and interactions between family members for its tumor suppressive function.Citation12-15 These studies have necessitated the need to identify alternative approaches to treat human cancers with p53 alterations.
Figure 1. Mutational status of p53, p63 and p73 in human cancers. Graphical representation of the frequency of p53 (red), p63 (blue) and p73 (red) mutations in human cancer. Mutation data was obtained from cbioportal, which represents multiple sequencing datasets from human cancers.
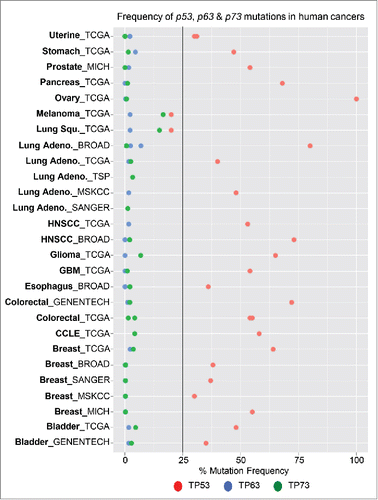
p63 and p73 are ancestors of p53; however, they were identified 20 years after the discovery of p53.Citation16 Based on data from knock out mouse models, the roles of p63 and p73 in developmental processes became very clear.Citation16 Important in vivo knockout mouse models of p63 revealed severe defects in skin and limb developmentCitation17; while, those lacking p73 exhibit defects in the central nervous system (CNS) and immune system.Citation18 Unlike p53, p63 and p73 are not frequently mutated in human cancers (); however, similar to p53, p63 and p73 have tumors suppressive activities through their ability to induce transcriptional target genes to induce apoptosis and other cellular processes involved in tumor suppression.Citation16 The lack of mutations in p63 and p73 in human cancer and their anti-tumorigenic activities make them attractive genes to manipulate for therapeutic intervention of the p53 pathway.
Multiple human cancers express elevated levels of ΔNp63 and ΔNp73 suggesting an oncogenic role of these genes in promoting tumorigenesis.Citation19-22 Our data provided in vivo evidence that ΔNp63 and ΔNp73 act as oncogenes using mouse models. We did this through cre-mediated recombination and deletion of ΔNp63 or ΔNp73 in mice deficient for p53. While p53−/− mice develop thymic lymphomas within a few months of life, deletion of either ΔNp63 or ΔNp73 resulted in complete tumor regression through the induction of TAp63 and TAp73 target genes involved in metabolic reprogramming.Citation23 These data highlights the indispensable role of p63 and p73 in tumor suppression.
p63 and p73 isoforms and their function in cancer
Both TP63 and TP73 encode multiple isoforms both at the N-terminus and C-terminus.Citation16 The TP63 and TP73 isoforms are broadly classified into 2 categories, the isoforms that encode an acidic transactivation domain called the TA isoforms, which encode either TAp63 or TAp73, and the isoforms that lack the acidic domain called ΔN isoforms, which produce either ΔNp63 or ΔNp73.Citation16 In vitro experiments expressing TA isoforms of p63 and p73 induced cell death and provided evidence that they function as tumor suppressors.Citation16 The ΔN isoforms of p63 and p73 function as oncogenes by acting in a dominant negative manner against p53, TAp63 and TAp73.Citation22,24 Mouse models with targeted deletions of TAp63 or TAp73 have provided evidence that these isoforms function as bonafide tumor suppressor genes, since both mouse models are highly tumor prone.Citation25,26 The TAp63 knockout mice develop mammary adenocarcinoma, lung adenocarcinoma, and squamous cell carcinoma.Citation25 Interestingly, these tumors are highly metastatic indicating that TAp63 is a metastasis suppressor.Citation22 The TAp73 knockout mice develop lung adenocarcinoma most frequently.Citation26
Manipulating ΔNp63 and ΔNp73 to therapeutically target tumors with alterations in p53
Given the dominant negative effects of ΔNp63 and ΔNp73 on the tumor suppressors, TAp63 and TAp73, we asked whether silencing of ΔNp63 or ΔNp73 would activate TAp63 and TAp73 to compensate for p53 tumor suppressor function. To do this, we intercrossed ΔNp63 or ΔNp73 conditional knock out (ΔNp63fl/fl or ΔNp73fl/fl) mice to p53 nullizygous (p53−/−) mice to create compound mutants (ΔNp63fl/fl;p53−/− and ΔNp63fl/fl;p53−/−). We then acutely deleted ΔNp63 or ΔNp73 using adenovirus-cre delivered intratumorally to thymic lymphomas that had formed by 10 weeks of age. This resulted in a significant increase in the expression of TAp63 and TAp73 with a concomitant tumor reduction and increased survival of p53−/− mice.Citation23 Additionally, ΔNp63Δ/Δ;p53−/− and ΔNp73Δ/Δ;p53−/− thymic lymphomas exhibited increased apoptosis and senescence when compared to p53−/− thymic lymphomas, suggesting that TAp63 and TAp73 compensate for p53-loss by activating downstream apoptosis and cell cycle targets. To identify other pathways regulated by TAp63 and TAp73 in tumor suppression, RNA sequencing analysis of p53−/−,ΔNp63Δ/Δ;p53−/− and ΔNp73Δ/Δ;p53−/− thymic lymphomas revealed a gene signature enriched for metabolic genes including the well known p53 targets, GLS2Citation27 and TIGAR.,Citation28 and a novel gene, IAPPCitation23
We further demonstrated that IAPP, which encodes amylin, a 37-amino acid peptideCitation29 is causal in the tumor regression of the thymic lymphomas in the ΔNp63Δ/Δ;p53−/− and ΔNp73Δ/Δ;p53−/− mice. Tumor regression by IAPP was induced through the inhibition of glucose uptake and hexokinase, the rate-limiting enzyme in the glycolytic pathway, thus inhibiting tumor glycolysis. IAPP mediated inhibition of glycolysis induces metabolic stress due to nutrient deprivation resulting in accumulation of intra-cellular ROS and the induction of apoptosis in p53 deficient tumors and cancer cells. Importantly, a synthetic analog of amylin called pramlintideCitation30 is an FDA approved drug used to treat type I and II diabetic patients. Treatment of p53-deficient tumors and p53 altered human cancer cell lines with pramlintide resulted in glycolytic inhibition within the tumor and marker tumor regression.Citation23 These results provide evidence that pramlintide can be used to therapeutically treat p53-defective human cancers. We identified that IAPP/amylin and pramlintide mediate their anti-cancer function through 2 critical receptors, RAMP3 and calcitonin receptor (CALCR).Citation31 These data provide important molecular markers that can be used to identify patients that may respond to pramlintide treatment. Here, we further investigate these receptors as molecular determinants of pramlintide efficacy. Additionally, we found that pramlintide is a potential cancer prevention agent and that targeting ΔNp63 and ΔNp73 simultaneously has therapeutic benefit over targeting each alone in p53-deficient cancers.
Results
Molecular determinants of pramlintide for the treatment of p53 mutant tumors
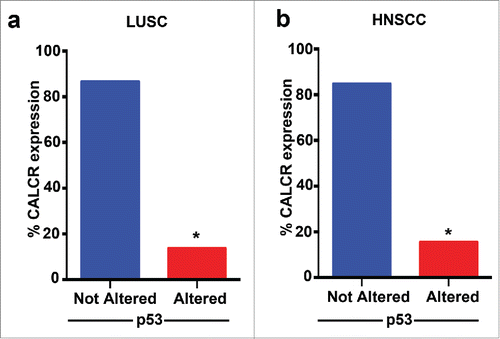
We have demonstrated the pramlintide mediates apoptosis through the inhibition of glycolysis in p53-deficient tumors and human cancer cells. We found that RAMP3 and CALCR are critical for pramlintide function. Inhibition of RAMP3 or CALCR in p53-deficient human cancer cells or in a p53-deficient mouse model of thymic lymphoma resulted in loss of pramlintide efficacy in tumor regression.Citation23 These data suggest that pramlintide efficacy is dependent on the activity of the receptors. We previously analyzed TCGA patient survival data in multiple p53-mutated human cancers and observed increased survival when the expression of RAMP3, CALCR and IAPP were high.Citation23 This suggests that the expression status of the RAMP3 and CALCR receptors could be used as a biomarker to select patients who could be potential responders for pramlintide based therapy. To determine whether patients with p53 alterations are good candidates for pramlintide therapy, we analyzed the TCGA data to find the percentage of patients with alterations in both p53 and CALCR. We focused on lung squamous cell carcinoma and head and neck squamous cell carcinoma, tumors that are frequently p53 mutant. Importantly, only 14% of patients with p53 alterations in lung squamous cell carcinoma () and 15% of head and neck squamous cell carcinoma patients also had low expression of CALCR (). These data indicate that pramlintide based therapy may benefit the majority of patients with p53 mutant tumors. Preclinical studies are underway to assess this.
Figure 2. CALCR expression in patient cohort with p53-mutations. Graphical representation of expression of CALCR in patients with p53-mutations in lung squamous cell carcinoma (LUSC) (a) and head and neck squamous cell carcinoma HNSCC (b).
Targeting both ΔNp63 and ΔNp73 in p53-deficient tumors
Disrupting the expression of either ΔNp63 or ΔNp73 in p53-deficient thymic lymphomas mediates tumor regression and increased survival.Citation23 This tumor shrinkage is mediated by compensation from TAp63 and TAp73, through a common IAPP driven metabolic reprogramming mechanism.Citation23 Additionally, we observed increased expression of known metabolic regulators GLS2 and TIGAR in the ΔNp63Δ/Δ;p53−/− and ΔNp73Δ/Δ;p53−/− tumors. Interestingly, GLS2Citation27 was strongly induced in ΔNp63Δ/Δ;p53−/− while TIGARCitation28 was induced in ΔNp73Δ/Δ;p53−/− tumors. This observation suggests that inhibition of both ΔNp63 and ΔNp73 in p53-deficient tumors may be advantageous to inhibition of ΔNp63 or ΔNp73 individually.
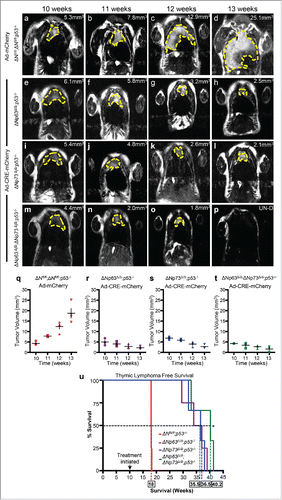
To test whether this is the case, we generated ΔNp63fl/fl;ΔNp73fl/fl;p53−/− mice and deleted both ΔNp63 and ΔNp73 using adenovirus-cre mediated recombination in thymic lymphoma of 10 week old mice (). Administration of adenovirus-CRE resulted in rapid tumor regression by 13 weeks in ΔNp63Δ/Δ;ΔNp73Δ/Δ;p53−/− mice () compared to the ΔNp63fl/fl;ΔNp73fl/fl;p53−/− mice treated with adenovirus-mCherry (). Although, the ΔNp63Δ/Δ;p53−/− () and ΔNp73Δ/Δ;p53−/− () mice also had a significant reduction in thymic lymphomagenesis, deletion of both ΔNp63 and ΔNp73 in p53-deficient tumors had a much greater reduction in tumor burden (). Tumors in some mice were undetectable (). The rapid tumor reduction in ΔNp63Δ/Δ;ΔNp73Δ/Δ;p53−/− correlated with an increased survival time of 1 month compared to p53−/− mice lacking either ΔNp63 (ΔNp63Δ/Δ;p53−/−) or ΔNp73 (ΔNp73Δ/Δ;p53−/−) only and an increased survival time of 5 and a half months over p53−/− mice ().
Figure 3. Combined deletion of ΔNp63 and ΔNp73 accelerated tumor regression in p53-deficient thymic lymphomas. Magnetic Resonance Imaging (MRI) of thymic lymphomas of mice of the indicated genotypes treated with Adenovirus (Ad)-mCherry (a-d) or Ad-Cre-mCherry (e-h, i-l and m-p). Age of mice is indicated across the top of each panel and the tumor volume is indicated (mm3) within each panel. UN-D indicates that the tumor was undetectable. The tumors are indicated by the dashed yellow line. Measurements of thymic lymphomas in mice of the indicated genotypes treated with Ad-mCherry (q) or Ad-Cre-mCherry (r-t), n = 3 to 5 mice per treatment followed over 8 weeks post infection. Kaplan Meier thymic lymphoma free survival curve of ΔNfl/fl;p53−/−, ΔNp63Δ/Δ;p53−/−, ΔNp73Δ/Δ;p53−/− orΔNp63Δ/Δ;ΔNp73Δ/Δ;p53−/− mice treated at 10 weeks with Ad-mCherry or Ad-Cre-mCherry to delete the ΔN isoforms of p63 and p73 (u), n = 3 – 5 mice, p<0.005. Boxed numbers on x-axis represent median time (weeks) of survival for the indicated genotypes.
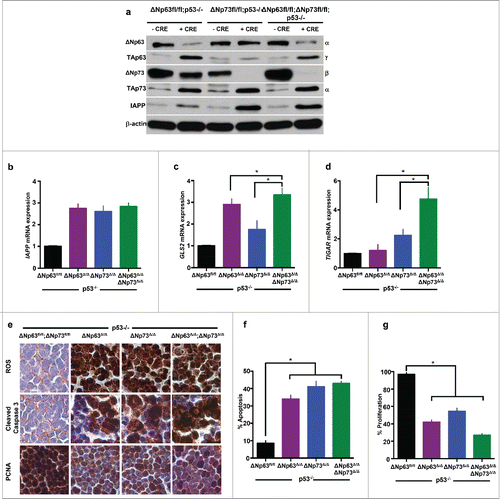
To determine whether there is increased expression of metabolic genes in ΔNp63Δ/Δ;ΔNp73Δ/Δ;p53−/− thymic lymphomas compared to tumors from ΔNp63Δ/Δ;p53−/− or ΔNp73Δ/Δ;p53−/− (), we performed qRT-PCR for IAPP, GLS2, and TIGAR (). We observed a significant increase in the expression of IAPP in all 3 compound mutant thymic lymphomas compared to p53 deficient thymic lymphomas (). Thus, loss of both ΔNp63 and ΔNp73 had no significant effect on the expression of IAPP. In contrast, both GLS2 () and TIGAR () were significantly upregulated in ΔNp63Δ/Δ;ΔNp73Δ/Δ;p53−/− compared to ΔNp63Δ/Δ;p53−/− or ΔNp73Δ/Δ;p53−/− indicating a synergistic effect of deletion of both ΔNp63 and ΔNp73. Lastly, we assayed for levels of ROS, apoptosis, and cell proliferation in compound mutant thymic lymphomas and compared them to p53−/− thymic lymphomas (). We found a modest increase in apoptosis and decrease in cellular proliferation in ΔNp63Δ/Δ;ΔNp73Δ/Δ;p53−/− thymic lymphomas compared to those from ΔNp63Δ/Δ;p53−/− or ΔNp73Δ/Δ;p53−/− mice (). These data also illustrate the synergistic effect of inhibition of both ΔNp63 and ΔNp73 in p53−/− thymic lymphomas. Thus, targeting both ΔNp63 and ΔNp73 may be advantageous to targeting them each individually in the treatment of p53-altered cancers.
Figure 4. ΔNp63 and ΔNp73 deletion mediates upregulation of metabolic regulators resulting in ROS accumulation and apoptosis in p53-deficient thymic lymphomas. Western blot analysis using the indicated antibodies and lysates of mice of the indicated genotypes 48 hours after treatment with Adenovirus (Ad)-mCherry or Ad-Cre-mCherry (a). Quantitative real time (qRT-PCR) of thymic lymphomas treated 48 hours after treatment with Ad-mCherry (ΔNfl/fl;p53−/−) or Ad-Cre-mCherry (ΔNp63Δ/Δ;p53−/− or ΔNp73Δ/Δ;p53−/− orΔNp63Δ/Δ;ΔNp73Δ/Δ;p53−/−) for the indicated target genes (b-d). n = 3, p<0.05. Immunohistochemistry for ROS, cleaved caspase 3 and PCNA in thymic lymphomas 48 hours after treatment with Ad-mCherry (ΔNfl/fl;p53−/−) or Ad-Cre-mCherry (ΔNp63Δ/Δ;p53−/− or ΔNp73Δ/Δ;p53−/− orΔNp63Δ/Δ;ΔNp73Δ/Δ;p53−/−) (e). Bar graph indicating quantification of cleaved caspase 3 (f) and PCNA (g) immunostaining.
Pramlintide in cancer prevention
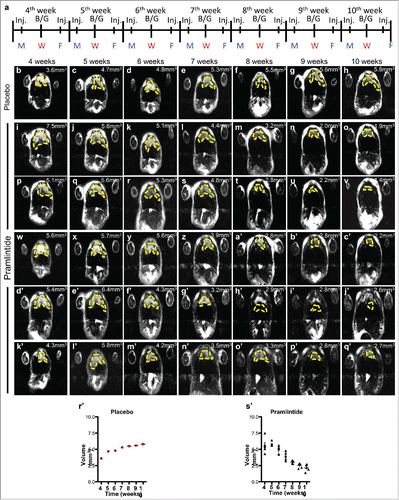
We have shown that pramlintide treatment results in rapid tumor regression in p53−/− mice with thymic lymphomas.Citation23 To determine whether pramlintide can work to prevent the formation of thymic lymphomas, we treated p53−/− mice (n = 6) at 4 weeks of age (before thymic lymphomas form) with 30 μg/kg pramlintide biweekly (). These mice were monitored weekly by MRI for tumor formation. At 10 weeks of age, these mice exhibited no appreciable tumor, and their thymii were the same volume as wild-type mice at 10 weeks of age (˜2.5 mm3) (). These data suggest that pramlintide may be used to prevent tumorigenesis.
Figure 5. Administration of pramlintide prior to thymic lymphoma formation in p53-deficient mice. Schedule of MRI imaging and injection (Inj.) of Pramlintide acetate in mice with p53 deficient thymus. MRI imaging at 10, 11, 12, and 13 weeks after treatment with placebo (b-h) or Pramlintide acetate (i-q'). Tumor volume is indicated (mm3) within each panel. The tumors are indicated by the dashed yellow line. Days of the week are shown as Monday (M), Wednesday (W), Friday (F). Blood glucose (B/G) levels were assessed every Wednesday at the time of MRI.
Conclusions and future directions
TP53 mutations account for ∼50% of all human cancers, and it is known as an undruggable target due to its activity as a transcription factor. Therefore, there is a need to identify effective therapeutic interventions to treat tumors with p53 alterations. p63 and p73 are family members of p53, with independent isoform specific functions.Citation16 We have previously demonstrated that deletion of ΔNp63 or ΔNp73 in p53-deficient cancers mediates tumor regression through metabolic reprogramming induced by the metabolic regulator and tumor glycolysis inhibitor, IAPP.Citation23 IAPP is transcriptionally activated through the TA isoforms of p63 and p73, which are upregulated subsequent to deletion of the ΔN isoforms of p63 or p73.Citation23 Here, we tested whether deletion of both ΔNp63 and ΔNp73 in p53-deficient thymic lymphomas accelerates tumor regression. Indeed, we observed an accelerated reduction in the tumor volume and increased survival upon ablating both ΔNp63 and ΔNp73 in the p53-deficient thymic lymphomas. This correlated with the increased expression of IAPP, GLS2, and TIGAR in the ΔNp63 and ΔNp73 double deficient p53−/− thymic lymphomas. These data suggest that the identification of inhibitors of both ΔNp63 and ΔNp73 would be potentially beneficial to patients with tumors bearing p53 alterations. Moreover, we identified pramlintide as a potential cancer preventative treatment. While exciting and promising, this potentially therapy needs additional testing in other preclinical mouse models of cancer.
The acceleration of tumor regression in the ΔNp63Δ/Δ;ΔNp73Δ/Δ;p53−/− compared to the ΔNp63Δ/Δ;p53−/− and ΔNp73Δ/Δ;p53−/− thymic lymphomas is most likely due to the synergistic effect of the multiple metabolic genes upregulated after the deletion of ΔNp63 and ΔNp73 and subsequent upregulation of TAp63 and TAp73. Further, we observed an increase in apoptosis and decrease in cell proliferation. Although, the metabolic regulator IAPP, which is upregulated in ΔNp63Δ/Δ;ΔNp73Δ/Δ;p53−/− thymic lymphomas is key in mediating the tumor regression,Citation23 we also showed a marked upregulation of GLS2 and TIGAR, key regulators in glutamine metabolism and glycolysis,Citation27,28 in these tumors. Upregulation of these genes has been shown to be important in tumor suppression.Citation27,28 Importantly, we observed that GLS2 was most significantly upregulated in ΔNp63/p53-double deficient compared to ΔNp73/p53 double deficient thymic lymphomas. Conversely, TIGAR was most significantly upregulated in the ΔNp73/p53-double deficient lymphomas. These data indicate that GLS2 and TIGAR are differentially regulated by different p63 and p73 isoforms; hence, upon disrupting both ΔNp63 and ΔNp73 in p53-deficient thymic lymphomas, GLS2, TIGAR and IAPP are upregulated and act in concert to induce rapid tumor regression. Overall, these observations further highlight the importance of targeting both ΔNp63 and ΔNp73 simultaneously to treat p53-deficient and mutant tumors.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by grants to E.R.F. from NCI (R01CA160394 and R01CA134796), CPRIT (RP140271), NCI- Cancer Center Core Grant (CA-16672)(University of Texas M.D. Anderson Cancer Center, a development award from the Lymphoma SPORE (P50CA136411), and the Hildegardo E. and Olga M. Flores Foundation. E.R.F. is a scholar of the Leukemia and Lymphoma Society, the Rita Allen Foundation and the V Foundation for Cancer Research.
Related Research Data
References
- Iwakuma T, Lozano G, Flores ER. Li-Fraumeni syndrome: a p53 family affair. Cell Cycle 2005; 4:865-7; PMID:15917654; http://dx.doi.org/10.4161/cc.4.7.1800
- Muller PA, Vousden KH. p53 mutations in cancer. Nat Cell Biol 2013; 15:2-8; PMID:23263379; http://dx.doi.org/10.1038/ncb2641
- Martins CP, Brown-Swigart L, Evan GI. Modeling the therapeutic efficacy of p53 restoration in tumors. Cell 2006; 127:1323-34; PMID:17182091; http://dx.doi.org/10.1016/j.cell.2006.12.007
- Ventura A. Restoration of p53 function leads to tumour regression in vivo. Nature 2007; 445:661-5; PMID:17251932; http://dx.doi.org/10.1038/nature05541
- Khoo KH, Verma CS, Lane DP. Drugging the p53 pathway: understanding the route to clinical efficacy. Nat Rev Drug Discov 2014; 13:217-36; PMID:24577402; http://dx.doi.org/10.1038/nrd4288
- Brown CJ, Lain S, Verma CS, Fersht AR, Lane DP. Awakening guardian angels: drugging the p53 pathway. Nature Rev Cancer 2009; 9:862-73; http://dx.doi.org/10.1038/nrc2763
- Lane DP. Cancer p53, guardian of the genome. Nature 1992; 358:15-6; PMID:1614522; http://dx.doi.org/10.1038/358015a0
- Brady CA. Distinct p53 transcriptional programs dictate acute DNA-damage responses and tumor suppression. Cell 2011; 145:571-83; PMID:21565614; http://dx.doi.org/10.1016/j.cell.2011.03.035
- Feldser DM, Kostova KK, Winslow MM, Taylor SE, Cashman C, Whittaker CA, Sanchez-Rivera FJ, Resnick R, Bronson R, Hemann MT, et al. Stage-specific sensitivity to p53 restoration during lung cancer progression. Nature 2010; 468:572-5; PMID:21107428; http://dx.doi.org/10.1038/nature09535
- Xue W. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007; 445:656-60; PMID:17251933; http://dx.doi.org/10.1038/nature05529
- Wang Y, Suh YA, Fuller MY, Jackson JG, Xiong S, Terzian T, Quintás-Cardama A, Bankson JA, El-Naggar AK, Lozano G. Restoring expression of wild-type p53 suppresses tumor growth but does not cause tumor regression in mice with a p53 missense mutation. J Clin Invest 2011; 121:893-904; PMID:21285512; http://dx.doi.org/10.1172/JCI44504.
- Lang GA, Iwakuma T, Suh YA, Liu G, Rao VA, Parant JM, Valentin-Vega YA, Terzian T, Caldwell LC, Strong LC, et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell 2004; 119:861-72; PMID:15607981; http://dx.doi.org/10.1016/j.cell.2004.11.006
- Flores ER. p63 and p73 are required for p53-dependent apoptosis in response to DNA damage. Nature 2002; 416:560-4; PMID:11932750; http://dx.doi.org/10.1038/416560a
- Flores ER. Tumor predisposition in mice mutant for p63 and p73: evidence for broader tumor suppressor functions for the p53 family. Cancer Cell 2005; 7:363-73; PMID:15837625; http://dx.doi.org/10.1016/j.ccr.2005.02.019
- Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT, Crowley D, Jacks T. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell 2004; 119:847-60; PMID:15607980; http://dx.doi.org/10.1016/j.cell.2004.11.004
- Su X, Chakravarti D, Flores ER. p63 steps into the limelight: crucial roles in the suppression of tumorigenesis and metastasis. Nat Rev Cancer 2013; 13:136-43; http://dx.doi.org/10.1038/nrc3446
- Yang A. p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature 1999; 398:714-8; PMID:10227294; http://dx.doi.org/10.1038/19539
- Yang A. p73-deficient mice have neurological, pheromonal and inflammatory defects but lack spontaneous tumours. Nature 2000; 404:99-103; PMID:10716451; http://dx.doi.org/10.1038/35003607
- Comperat E. p63 gene expression study and early bladder carcinogenesis. Urology 2007; 70:459-62; PMID:17905096; http://dx.doi.org/10.1016/j.urology.2007.04.030
- Di Como C. J. p63 expression profiles in human normal and tumor tissues. Clin Cancer Res 2002; 8:494-501; PMID:11839669
- Nylander K. Differential expression of p63 isoforms in normal tissues and neoplastic cells. J Pathol 2002; 198:417-27; PMID:12434410; http://dx.doi.org/10.1002/path.1231
- Zaika AI, Slade N, Erster SH, Sansome C, Joseph TW, Pearl M, Chalas E, Moll UM. Np73, A Dominant-Negative Inhibitor of Wild-type p53 and TAp73, Is Up-regulated in Human Tumors. J Exp Med 2002; 196:765-80; PMID:12235210; http://dx.doi.org/10.1084/jem.20020179
- Venkatanarayan A, Raulji P, Norton W, Chakravarti D, Coarfa C, Su X, Sandur SK, Ramirez MS, Lee J, Kingsley CV, et al. IAPP-driven metabolic reprogramming induces regression of p53-deficient tumours in vivo. Nature 2015; 517:626-30; PMID:25409149; http://dx.doi.org/10.1038/nature13910
- Yang A, p63, a p53 homolog at 3q27-29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Mol Cell 1998; 2:305-16; PMID:9774969; http://dx.doi.org/10.1016/S1097-2765(00)80275-0
- Su X. TAp63 suppresses metastasis through coordinate regulation of Dicer and miRNAs. Nature 2010; 467:986-90; PMID:20962848; http://dx.doi.org/10.1038/nature09459
- Tomasini R. TAp73 knockout shows genomic instability with infertility and tumor suppressor functions. Genes Dev 2008; 22:2677-91; PMID:18805989; http://dx.doi.org/10.1101/gad.1695308
- Suzuki S. Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc Natl Acad Sci USA 2010; 107:7461-6; http://dx.doi.org/10.1073/pnas.1002459107
- Bensaad K. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 2006; 126:107-20; PMID:16839880; http://dx.doi.org/10.1016/j.cell.2006.05.036
- Pillay K, Govender P. Amylin uncovered: a review on the polypeptide responsible for type II diabetes. Bio Med Res Int 2013; 2013:826706; PMID:23607096; http://dx.doi.org/10.1155/2013/826706
- Edelman S, Maier H, Wilhelm K. Pramlintide in the treatment of diabetes mellitus. Bio Drugs 2008; 22:375-86; PMID:18998755; http://dx.doi.org/10.2165/0063030-200822060-00004
- Christopoulos G. Multiple amylin receptors arise from receptor activity-modifying protein interaction with the calcitonin receptor gene product. Mol Pharmacol 1999; 56:235-42; PMID:10385705