
ABSTRACT
Previous studies have indicated that inflammatory stimulation represses protein phosphatase 2A (PP2A), a well-known tumor suppressor. However, whether PP2A repression participates in pancreatic cancer progression has not been verified. We used lipopolysaccharide (LPS) and macrophage-conditioned medium (MCM) to establish in vitro inflammation models, and investigated whether inflammatory stimuli affect pancreatic cancer cell growth and invasion PP2A catalytic subunit (PP2Ac)-dependently. Via nude mouse models of orthotopic tumor xenografts and dibutyltin dichloride (DBTC)-induced chronic pancreatitis, we evaluated the effect of an inflammatory microenvironment on PP2Ac expression in vivo. We cloned the PP2Acα and PP2Acβ isoform promoters to investigate the PP2Ac transcriptional regulation mechanisms. MCM accelerated pancreatic cancer cell growth; MCM and LPS promoted cell invasion. DBTC promoted xenograft growth and metastasis, induced tumor-associated macrophage infiltration, promoted angiogenesis, activated the nuclear factor-κB (NF-κB) pathway, and repressed PP2Ac expression. In vitro, LPS and MCM downregulated PP2Ac mRNA and protein. PP2Acα overexpression attenuated JNK, ERK, PKC, and IKK phosphorylation, and impaired LPS/MCM-stimulated cell invasion and MCM-promoted cell growth. LPS and MCM activated the NF-κB pathway in vitro. LPS and MCM induced IKK and IκB phosphorylation, leading to p65/RelA nuclear translocation and transcriptional activation. Overexpression of the dominant negative forms of IKKα attenuated LPS and MCM downregulation of PP2Ac, suggesting inflammatory stimuli repress PP2Ac expression NF-κB pathway–dependently. Luciferase reporter gene assay verified that LPS and MCM downregulated PP2Ac transcription through an NF-κB–dependent pathway. Our study presents a new mechanism in inflammation-driven cancer progression through NF-κB pathway–dependent PP2Ac repression.
Introduction
Pancreatic cancer is one of the most fatal solid malignancies, with a 5-year survival rate of only ˜5%.Citation1 Recently, epidemiological and experimental data have demonstrated a close connection between chronic pancreatitis and pancreatic cancer.Citation2,3 However, the mechanisms involved are still far from defined.
Protein phosphatase 2A (PP2A) is a major serine threonine (Ser/Thr) phosphatase that is involved in the control of many cellular functions including protein synthesis, cell cycle determination, apoptosis, and metabolism.Citation4,5 PP2A modulates several signaling cascades, including that of c-Jun N-terminal kinase (JNK), extracellular signal-related kinase (ERK), p38, Akt and protein kinase C (PKC) among others, most of which can accelerate growth.Citation6 The core PP2A enzyme consists of a catalytic subunit (PP2Ac) and a regulatory subunit termed the A subunit. A third regulatory B subunit is associated with this core structure.Citation6 PP2Ac has α and β isoforms, whose primary sequences share 97% identity.Citation7 Defects in the expression of PP2A components have been linked to cancer.Citation8 PP2A is downregulated in various malignancies, such as lung, breast, colon, and gastric cancer.Citation9,10 Previously, we reported that PP2Ac expression was significantly lower in pancreatic cancer tissues than in adjacent noncancerous tissues. Furthermore, PP2Ac overexpression induced apoptosis and repressed pancreatic cancer cell growth.Citation6,11 These results suggested that PP2A plays a tumor suppressor role and could be a potential target for cancer treatment.
Tumor-associated macrophages (TAMs) are a prominent inflammatory cell population in many tumor types residing in both the perivascular and avascular, hypoxic regions of these tissues. Analysis of TAMs inof human tumor biopsies has shown that they TAMs express a variety of tumor-promoting factors, and evidence the findings from transgenic murine tumor models has have provided unequivocal evidence for the importance of these cells in driving angiogenesis, lymphangiogenesis, immunosuppression, and metastasis.Citation12 We previously demonstrated that macrophage-conditioned medium (MCM) treated breast cancer cells became detached and acquired an epithelial-mesenchymal transition (EMT)-like morphology, which occurs during metastasis.Citation13 However, whether PP2A participates in the TAM- and other inflammatory stimuli-driven pancreatic cancer progression has not been well explored.
Lipopolysaccharide (LPS) is a potent activator of inflammationCitation14 and is widely used as an inflammatory stimulator to establish inflammation models in vitro.Citation14 Previous study proved that LPS treatment represses PP2Ac expression and activity in myocardial cells.Citation15 Considering the tumor suppressive role of PP2A, we speculated that the inflammation-triggered PP2A inhibition participates in the inflammation-driven cancer progression.
Mechanically, LPS is a strong activator of the nuclear factor-κB (NF-κB) pathway, which plays a key role in the inflammation response.Citation16 Moreover, the carcinogenesis effect of TAM has been proved to be related to activation of NF-κB pathway.Citation17 According to the canonical NF-κB pathway cascade,Citation11 the phosphorylated IκB kinase (IKK) can further phosphorylate IκB, which is the cytoplasmic inhibitor of the NF-κB complex. When phosphorylated, IκB undergoes ubiquitination and proteasome-mediated degradation rapidly, resulting in the release and nuclear translocation of the NF-κB complex. The prototypical NF-κB complex is a heterodimer composed of p50 and RelA/p65. Once inside the nucleus, p65 engages the cognate κB enhancers, which contain one or more κB-enhancer consensus sequences (5′-GGGRNYYYCC-3′, where R is a purine, Y is a pyrimidine, and N is any nucleic acid), and regulates the expression of downstream genes.Citation11,18,Citation19 Whether LPS-induced PP2A repression can be mediated in a NF-κB pathway–dependent manner has not been explored.
Therefore, in the present study, we used LPS- and MCM-induced inflammation models, a dibutyltin dichloride (DBTC)-induced mouse chronic pancreatitis model, and a pancreatic orthotopic xenograft mouse model to investigate the relationship between inflammation and PP2Ac expression, both in vitro and in vivo. The involvement of the NF-κB pathway was also investigated.
Results
Inflammatory stimuli promotedpancreatic cancer cell growth and invasion in vitro
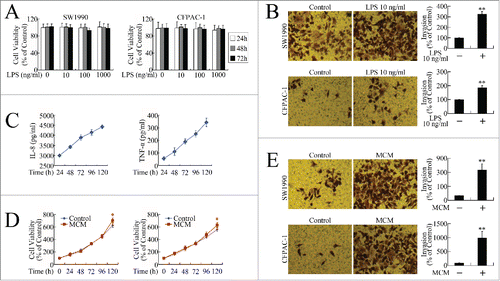
To investigate the effects of inflammatory stimuli on the biological behaviors of the pancreatic cancer cells, methyl thiazolyl tetrazolium (MTT) and invasion assays were performed to evaluate cell growth and metastasis in vitro. Although LPS had no effect on pancreatic cancer cell growth (), it promoted invasion significantly (). Quality control of MCM was performed by evaluating secretion of the cytokines interleukin-8 (IL-8) and tumor necrosis factor-α (TNF-α) ().Citation20,Citation21,22 MCM promoted pancreatic cancer cell growth slightly () and stimulated invasion remarkably (). These in vitro data suggest that inflammation could be a favorable factor for pancreatic cancer progression by accelerating cell growth and invasion.
Figure 1. Inflammatory stimuli promoted growth and invasion of pancreatic cancer cells in vitro. (A) MTT assay of viability of cells incubated with LPS (0, 10, 100, 1000 ng/ml) for 24, 48, and 72 h. (B) Cell invasion assay following 24-h exposure to 10 ng/ml LPS. Cells that had migrated to the lower membranes were photographed under ×400 magnification. **P < 0.01 as compared to control. (C) Quality control of MCM. After 7-day MCSF treatment, IL-8 and TNF-α levels at 24, 48, 72, 96, and 120 h were measured using Milliplex assay. (D) MTT assay of viability of cells incubated with MCM for 24, 48, 72, 96, and 120 h. *P < 0.05 as compared with control. (E) Cell invasion assay following 24-h exposure to MCM. Cells that had migrated to the lower membranes were photographed under ×400 magnification. **P < 0.01 as compared to control.
Inflammatory stimuli promoted pancreatic cancer cell growth and invasion in vivo
To confirm the effects of inflammation on pancreatic cancer progression in vivo, we established an in vivo model using a combination of orthotopic xenograft and DBTC-induced pancreatitis models. The inflammation induction effect of DBTC was first confirmed ()., The segmental glands in DBTC-treated mice were atrophic and associated with chronic inflammatory infiltration of leukocytes, fibroblast proliferation and fibrosis, and large regions of acinar loss (). Using immunohistochemistry and the macrophage marker CD68, we proved that treatment of DBTC induced macrophage infiltration, indicating the successful establishment of the chronic pancreatitis model by DBTC injection. Then, we applied the DBTC treatment to the orthotopic xenograft model. As shown in , mice in the DBTC group developed larger tumors than those in the control group. Moreover, mouse No. Four and 5 in the DBTC group developed visible metastatic cancerous nodes (). Although no visible metastasis was found in other mice, pathological examination identified celiac lymph node metastasis in mouse No. Three and mouse No. Six of the control and DBTC group, respectively. Therefore, consistent with the in vitro data, the in vivo investigation verified that the inflammatory stimuli accelerated growth and metastasis.
Figure 2. Inflammatory stimuli promoted pancreatic cancer cell growth and invasion in vivo. (A) Chronic inflammatory infiltration of leukocytes, fibroblast proliferation and fibrosis, large regions of acinar loss, and infiltration of CD68+ cells in the pancreas of DBTC-treated mice. (B) The effect of DBTC on pancreatic cancer in representative in vivo bioluminescent images and photographs of cancer xenografts. (C) Bioluminescence of pancreatic orthotopic xenografts. **P < 0.01 as compared to control. (D) Tumor weight of pancreatic orthotopic xenografts. **P < 0.01 as compared to control. (E) Pathological examination of metastatic cancerous nodes and celiac lymph nodes. Green arrow: metastatic cancerous node. (F) Pathological and immunohistochemical examination of pancreatic orthotopic xenografts. Immunohistochemistry was performed using antibodies targeting CD68, CD163, Ki67, CD34, p-IKK (phosphorylated IKK), and PP2Ac.
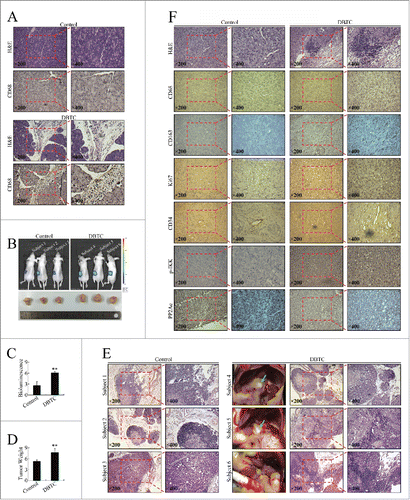
To explore the mechanisms involved, we performed immunohistochemistry examination to evaluate several markers in the orthotopic xenografts. The development of chronic pancreatitis was confirmed by hematoxylin–eosin (H&E) staining (). CD68 is the most widely used macrophage marker, encompassing both the M1 and M2 phenotypes. By using CD68, we confirmed macrophage infiltration in the DBTC-treated group (). To evaluate TAM infiltration, we used an antibody targeting CD163, a more specific marker for identifying M2 macrophages. Similar to CD68-positive cells, there were more CD163-positive cells in the DBTC-treated group (). Therefore, there were more infiltrated macrophages in the DBTC-treated group, consistent with the established inflammatory microenvironment.
Mice in the DBTC-treated group had higher levels of Ki67 (), the well-known proliferation index. Moreover, higher blood vessel density was detected in the DBTC-treated group via the use of an antibody targeting CD34, a vascular endothelial cell marker (), suggesting neoangiogenesis could also be involved in the growth-promoting effect of inflammation through modification of the tumor microenvironment. As inflammatory stimuli promoted pancreatic cancer growth remarkably in vivo, but not in vitro, the involvement of inflammation remodeling of the tumor microenvironment in vivo might be an explanation.
The NF-κB pathway plays a pivotal role in the inflammatory and immune responses and participates in carcinogenesis, including in pancreatic cancer.Citation11 Higher phosphorylation levels of IKK were found in the DBTC-treated group (), suggesting NF-κB pathway activation. As expected, PP2Ac expression was repressed in the DBTC-treated group (), suggesting PP2Ac regulation could be involved in inflammation-driven pancreatic cancer progression.
Inflammatory stimuli promoted in vitro growth and metastasis by repressing PP2Ac expression in pancreatic cancer cells
We performed in vitro studies to investigate the mechanisms involved in PP2Ac repression. The effects of LPS and MCM on PP2Ac expression in pancreatic cancer cells was firstly investigated by real-time PCR. As shown in , the mRNA levels of both PP2Acα and PP2Acβ were markedly decreased after treatment with LPS or MCM treatment, resulting in the down-regulation of total PP2Ac levels. As shown in Figure. 3E and F, the total PP2Ac protein was dramatically decreased after exposure to LPS or MCM exposure.
Figure 3. Inflammatory stimuli promoted in vitro growth and metastasis by repressing PP2Ac expression in pancreatic cancer cells. (A, B) Real-time PCR determination of mRNA levels of PP2Acα, PP2Acβ, and total PP2Ac in SW1990 (A) and CFPAC-1 (B) cells incubated with LPS (0, 10, 100 ng/ml) for 24 h. **P < 0.01 as compared with control. (C, D) Real-time PCR determination of mRNA levels of PP2Acα, PP2Acβ, and total PP2Ac in SW1990 (C) and CFPAC-1 (D) cells incubated with MCM for 24 h. **P < 0.01 as compared with control. (E, F) Western blotting assessment of PP2Ac expression levels of PP2Ac after treatment with LPS (E) or MCM (F). (G) Western blot evaluation of JNK, ERK, PKC and IKK phosphorylation levels in cells transfected with pcDNA3.1(+)-PP2Acα plasmid for 24 h, and then treated with 10 ng/ml LPS for another 12 h. (H) MTT assay of cells transfected with pcDNA3.1(+)-PP2Acα plasmid for 24 h, and then incubated with MCM for an additional 120 h. **P < 0.01 as compared to control. and&P < 0.01 indicates significant differences between fold induction. (I, J) Invasion assay of SW1990 (I) and CFPAC-1 (J) cells transfected with pcDNA3.1(+)-PP2Acα plasmid for 24 h, and then treated with 10 ng/ml LPS or MCM for an additional 24 h. Cells that had migrated to the lower membranes were photographed under ×400 magnification. *P < 0.05, **P < 0.01 as compared to control. @P < 0.05, @@P < 0.01 indicate significant differences between groups. &P < 0.05 indicates significant differences between fold induction.
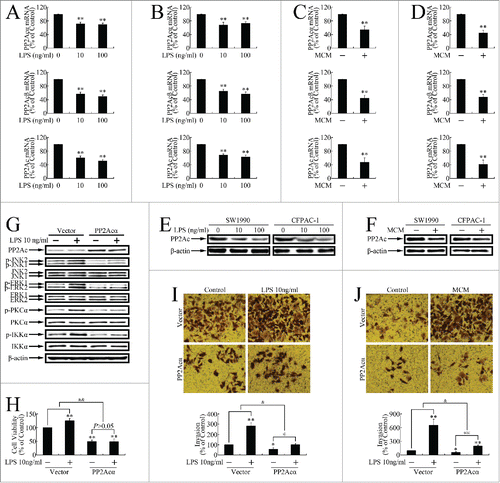
Previous studies have reported JNK, ERK, and PKC activation by LPS.Citation23-26 As all these oncogenic kinases, and IKK, are substrates of PP2Ac, we speculated that PP2Ac repression could result in the comprehensive activation of these kinase pathways. As shown in , JNK, ERK, PKC, and IKK were phosphorylated upon LPS treatment, and PP2Acα overexpression attenuated it, indicating that PP2Ac repression by LPS results in the activation of several oncogenic kinases.Citation27 PP2Acα overexpression also attenuated MCM-induced growth progression (), and impaired LPS or MCM stimulation of invasion (), indicating inflammatory stimuli promoted in vitro pancreatic cancer cell growth and metastasis in a PP2Ac inhibition-dependent manner.
Inflammatory stimuli induced NF-κB pathway activation in pancreatic cancer cells in vitro
As the NF-κB pathway plays a pivotal role in inflammation,Citation11 and as we observed NF-κB pathway activation in vivo, we evaluated the activation of several critical steps in this cascade to confirm LPS and MCM activation of the NF-κB pathway in vitro,. As shown in , LPS and MCM induced IKK phosphorylation, which was blocked by DN-IKKα (S176/180A) overexpression (). LPS and MCM further caused IκBα phosphorylation (), which was attenuated by DN-IKKα (S176/180A) or DN-IκBα (S32/36A) overexpression ().
Figure 4. Inflammatory stimuli induced NF-κB pathway activation in pancreatic cancer cells. (A, B) Milliplex assay evaluation of IKK phosphorylation at Ser176/Ser180 in SW1990 and CFPAC-1 cells treated with 10 ng/ml LPS (A) or MCM (B) for 12 h. (C) DN-IKKα (S176/180A) overexpression repressed LPS-induced IKK phosphorylation in SW1990 (top) and CFPAC-1 (bottom) cells. *P < 0.05, **P < 0.01 as compared with control. &P < 0.05 indicates significant differences between fold induction. (D) DN-IKKα (S176/180A) overexpression repressed MCM-induced IKK phosphorylation in SW1990 cells. *P < 0.05, **P < 0.01 as compared with control. &P < 0.05 indicates significant differences between fold induction. (E, F) Milliplex assay evaluation of IκBα phosphorylation at Ser32 after 12-h treatment with 10 ng/ml LPS (E) or MCM (F). (G) DN-IKKα (S176/180A) or DN-IκBα (S32/36A) overexpression repressed LPS-induced IκBα phosphorylation in SW1990 (top) and CFPAC-1 (bottom) cells. *P < 0.05, **P < 0.01 as compared with control. &P < 0.05, and&P < 0.01 indicate significant differences between fold induction. (H) DN-IKKα (S176/180A) or DN-IκBα (S32/36A) overexpression repressed MCM-induced IκBα phosphorylation in SW1990 cells. *P < 0.05, **P < 0.01 as compared with control. &P < 0.05, and&P < 0.01 indicate significant differences between fold induction. (I) Western blot of p65 expression in cells treated with 10 ng/ml LPS for 12 h, followed by total and nuclear protein extraction. β-actin and histone H1 were the internal controls for the total and nuclear extracts, respectively. (J, K) Luciferase reporter gene assays of cells transiently transfected with pNF-κB-luc before 36-h treatment with 10 ng/ml LPS (J) or MCM (K). **P < 0.01 as compared with control. (L, M) Milliplex assay of c-Myc expression in cells treated with LPS (L) or MCM (M) for 12 h. **P < 0.01 as compared with control.
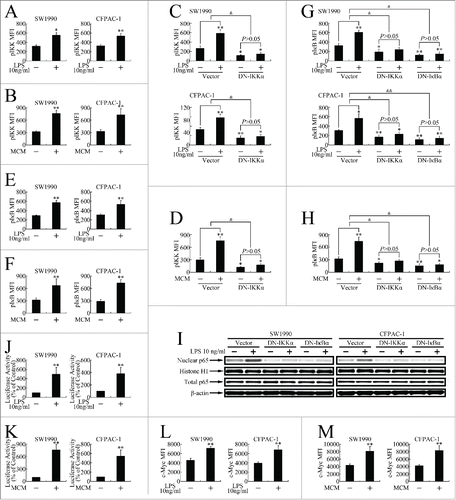
To investigate the nuclear entry of the NF-κB complex, nuclear extracts were prepared and the p65 content was measured by western blotting. LPS induced p65 entry into the nucleus, while DN-IKKα (S176/180A) or DN-IκBα (S32/36A) (), respectively, repressed it.
To investigate the transcriptional regulation of LPS- or MCM-activated p65, cells were transiently transfected with a pNF-κB-luc reporter construct. As shown in and K, LPS- and MCM-treated cells exhibited increased luciferase activity. Using a Milliplex assay, we further confirmed that the expression of c-Myc, a classic target gene downstream of the NF-κB pathway, was elevated upon LPS or MCM treatment ().
Inflammatory stimuli repressed PP2Ac expression in an NF-κB pathway-dependent manner
As shown in , DN-IKKα (S176/180A) or DN-IκBα (S32/36A) overexpression attenuated the LPS-induced downregulation of PP2Ac in pancreatic cancer cells. LPS repression of PP2Ac was also impaired by pretreatment with the IKK inhibitor EF-24 or the IκB phosphorylation inhibitor BAY 11-7082 (). DN-IKKα (S176/180A) or DN-IκBα (S32/36A) overexpression and EF-24 or BAY 11-7082 pretreatment also attenuated the MCM downregulation of PP2Ac (), suggesting inflammatory stimuli repressed PP2Ac expression in an NF-κB pathway-dependent manner.
Figure 5. Inflammatory stimuli repressed PP2Ac expression in a NF-κB pathway–dependent manner. (A, B) Determination of PP2Ac mRNA expression in SW1990 cells pretreated with 20 μM BAY 11-7082 or 5 μM EF-24 2 h before 24-h LPS treatment (A), or transfected with DN-IKKα (S176/180A) or DN-IκBα (S32/36A) 24 h before 24-h LPS treatment (B). *P < 0.05, **P < 0.01 indicate significant differences from the control. @P < 0.05 indicates significant differences between groups. &P < 0.05, and&P < 0.01 indicate significant differences between fold induction. (C, D) Determination of PP2Ac mRNA expression in SW1990 cells pretreated with 20 μM BAY 11-7082 or 5 μM EF-24 2 h before 24-h MCM treatment (C), or transfected with DN-IKKα (S176/180A) or DN-IκBα (S32/36A) 24 h before 24-h MCM treatment (D). *P < 0.05, **P < 0.01 indicate significant differences from the control. @@P < 0.01 indicates significant differences between groups. &P < 0.05 indicates significant differences between fold induction. (E, F) Luciferase gene reporter assay of SW1990 cells transfected with pGL3-Basic-PP2Acα or pGL3-Basic-PP2Acβ plasmids and treated with 20 μM BAY 11-7082 or 5 μM EF-24 for 2 h, followed by 36-h LPS (E) or MCM (F) treatment. *P < 0.05, **P < 0.01 indicate significant differences from the control. @P < 0.05 indicates significant differences between groups. &P < 0.05, and&P < 0.01 indicate significant differences between fold induction. (G) Illustration of NF-κB binding sites or NF-κB downstream genes in the 5′upstream regions of PP2Acα and PP2Acβ.
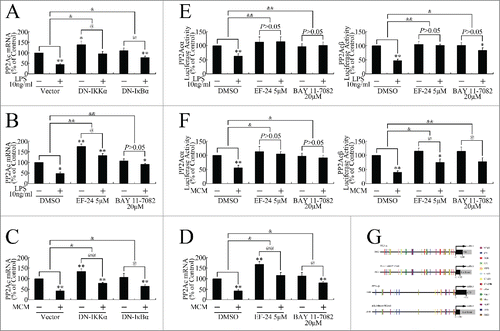
To further investigate whether PP2Ac down-regulation occurred through transcriptional repression, we cloned the 5′ upstream regions of PP2Acα and PP2Acβ respectively. Using the luciferase reporter gene assay, we found that LPS or MCM treatment induced transcriptional repression of PP2Acα and PP2Acβ, which EF-24 or BAY 11-7082 attenuated (), suggesting LPS repressed PP2Ac transcription in an NF-κB pathway-dependent manner.
To investigate the detailed mechanism involved in the repression of PP2Acα and PP2Acβ transcription, we analyzed the potential binding sites of NF-κB or NF-κB downstream genesCitation28-30 in the 5′ upstream regions of PP2Acα and PP2Acβ by using Genomatix software.Citation31 In addition to NF-κB itself, the potential binding sites of several transcription factors, which have been proven to be the target genes of the NF-κB pathway, were identified (). For example, the binding sites of c-Myc, which is overexpressed as reported above, were located at the upstream regions of both PP2Acα and PP2Acβ.
Discussion
Inflammation is a vital defensive response that plays critical roles in a variety of physiological situations, and when dysregulated, can contribute to the pathogenesis of many diseases, including cancer. The possible mechanism is that the normal inflammatory response is activated on an acute basis and is auto-regulated, whereas, chronic inflammation leads to failure in the regulatory mechanism. Chronic inflammation is a well-documented risk for carcinogenesis, particularly in the pancreas and gastrointestinal tract. Chronic pancreatitis involves long-standing inflammation of the pancreas associated with an increased risk (˜20 folds) for pancreatic cancer.Citation32
PP2A is a key Ser/Thr phosphatase responsible for dephosphorylating of a wide array of signaling molecules and it responds to inflammatory stimulation. Transforming growth factor-β (TGF-β), a major profibrotic cytokine implicated in scleroderma fibrosis, down-regulates PP2Ac expression in fibroblasts, resulting in ERK phosphorylation.Citation33 In addition to reducing its expression levels, prostaglandin E2 (PGE2), TGF-β,Citation34 and H2O2 Citation35 repress PP2A activity. More than an inflammatory responder, PP2A is widely accepted as a cancer repressor. It remains unclear whether inflammatory stimulation represses PP2A expression and/or activity in cancer cells. The response of PP2A to inflammation and PP2A participation in cancer evolution inspired our investigation of whether PP2A could mediate inflammation-driven cancer progression.
In recent years, many efforts have been made to identify the underlying mechanisms that contribute to inflammation-driven tumor progress. IL-1 (interleukin-1) and TGF-β1 are elevated in the serum of patients with pancreatic cancer.Citation36,37 IL-1α promotes proliferation, adhesion, and migration in pancreatic cancer cell lines, associating with the activation of Ras and the downstream ERK signaling pathway.Citation38 Another study demonstrated that IL-1α activates the NF-κB signaling pathway and up-regulates several anti-apoptotic genes, that for Bcl-xL and BfL-139. TGF-β upregulates matrix metalloproteinase 2 (MMP-2) and the urokinase-type plasminogen activator (uPA) system and consequently mediates invasive behavior.Citation40 IL-8 and IL-6 activate the mitogen-activated protein kinase (MAPK) pathway, leading to the production of vascular endothelial growth factor ( VEGF ) and neuropilin-2,Citation41,42 both of which correlate with angiogenesis and the metastatic behavior of cancer cells.Citation41-43 These investigations suggest that multiple cytokines and mechanisms could be involved in inflammation-driven cancer progression.
As a single cytokine can not mimic the inflammatory environment, we used LPS and MCM to establish non-specific inflammation models in vitro. LPS itself is not intrinsically harmful. Instead, it induces myeloid and/or non-myeloid cells to produce proinflammatory cytokines, such as tumor necrosis factor (TNF), interleukins, inducible nitric oxide synthase, and cyclooxygenase-2.Citation14 Macrophages are classified into 2 categories: type 1 or classically activated (M1), and type 2 or alternatively activated (M2). M1 macrophages are characterized by a proinflammatory phenotype and display microbicidal activity that results in tumor suppression, whereas M2 macrophages promote tissue repair, matrix remodeling, and angiogenesis supporting tumorigenesis. M2 macrophages, also termed TAMs, produce cytokines, chemokines, and growth factors involved in tumor invasion, metastasis, angiogenesis, and immunosuppression. These soluble stimuli include IL-8, IL-10, adrenomedullin (ADM), TNF-α, TGF-β, VEGFs, uPA, colony stimulating factor 1 (CSF-1), and chemokine (C-C motif) ligand 2 (CCL2).Citation21,22,44 In the present study, we proved that LPS facilitates pancreatic cancer cell invasion, while MCM promotes both pancreatic cancer cell growth and invasion. Both LPS and MCM repressed PP2Ac expression in pancreatic cancer cells in vitro, suggesting PP2Ac regulation could be involved in inflammation-driven pancreatic cancer progression.
To reveal the relationship between inflammation and PP2Ac in vivo, we established a pancreatic orthotopic xenograft model of inflammation using DBTC. DBTC-induced pancreatitis has a complex pathogenesis, involving a biliary and a hematogenic component. Merkord and colleagues reported that, in rats, this model appeared suitable for investigations of the process leading to pancreatic fibrosis.Citation45 However, to the best of our knowledge, the DBTC-induced pancreatitis model has not been well developed in mice. It was noteworthy that we successfully established a pancreatitis model by DBTC injection in nude mice. Xenografts grown based on the inflammation were not only bigger, but also tended to metastasize. Using this model, we confirmed that inflammation induced TAM infiltration, upregulated the proliferation index, and promoted angiogenesis. Moreover, inflammation repressed PP2Ac expression in the pancreatic orthotopic xenografts in vivo, consistent with the in vitro data.
Many studies have demonstrated that PP2A disruption and dysfunction are requirements for malignant transformation.Citation46 As the present study reveals the inflammation-triggered repression of PP2A in pancreatic cancer cells, we investigated whether PP2A repression could promote cancer progression. Using protein gel blotting, we confirmed that LPS induced phosphorylation of the PP2A substrate kinases, including ERK, JNK, PKC, and IKK, most of which accelerate cancer progression.Citation27 PP2Ac overexpression attenuated the activation of these oncogenic cascades. Furthermore, PP2Ac overexpression impaired the inflammatory stimuli promotion of cell growth and invasion. Therefore, our present study suggests a new theory; that a PP2A-dependent mechanism might be involved in the inflammation-promoted cancer elevation.
Our present study also reveals the cell signaling transduction mechanisms involved in PP2Ac downregulation by inflammatory stimuli. The role of the NF-κB pathway in inflammation has been established. There are 2 distinct NF-κB signaling cascades: canonical and non-canonical pathway. The canonical pathway, involved in innate immunity, is activated by pro-inflammatory stimuli, including TNF-α, various interleukins, microbes, and virus-related ligands.Citation47 Based on its close structural similarity with the viral oncoprotein v-Rel, the link between the NF-κB pathway and cancer was first suspected in 1990.Citation48 In the past decade, an increasing number of studies have shown that many cancers have constitutively active NF-κB, including lung, colon, pancreatic, and ovarian cancers.Citation49-51 In addition, NF-κB down-regulation renders cancer cells more sensitive to treatments, including chemo- and radiation therapies.Citation52 Therefore, the NF-κB pathway plays critical roles both in the inflammatory response and in tumors, indicating NF-κB pathway participation in inflammation-driven cancer progression. In the present study, we showed that LPS and MCM markedly increased IKK and IκB phosphorylation of, and p65 nuclear translocation and transcriptional activation, followed by up-regulation of the downstream oncogenic c-Myc. The activation of the NF-κB pathway further repressed PP2Ac transcription and expression of, stimulating pancreatic cancer cell invasion. Interestingly, IKK presents both upstream and as a substrate of PP2A, suggesting the involvement of a positive feedback mechanism. Here, we here present a potential mechanism involved in the inflammation-driven cancer progression mediated by the NF-κB/PP2Ac pathway.
Taking together, our present study demonstrates that that PP2Ac responds to inflammatory stimuli both in vitro and in vivo. The molecular mechanism involved in PP2Ac inhibition by inflammation appears to be mediated by the NF-κB pathway. Moreover, the inflammation-stimulated NF-κB/PP2Ac pathway promoted JNK, ERK, PKC and IKK phosphorylation, and accelerated pancreatic cancer cell invasion in vitro. Previous studies have porven that multiple kinase pathways are overactivited in inflammation and cancer. However, the mechanisms involved in this extensive and comprehensive activation have not been well explored. Considering the multi-substrate characteristic of PP2A, our investigation suggests a potential mechanism in the extensive kinase activation and inflammation-driven cancer progression: repression on PP2A induced by inflammation results in extensive and comprehensive activation of oncogenic kinases, leading to cancer progression. Moreover, the extensive activation of kinases in cancer always renders single-targeting therapeutics inefficient, while toxic and side effects always restrict multi-target therapeutics. We now present a potential indirect multi-targeting therapeutic that targets PP2A, which may shed new light on pancreatic cancer treatment.
Materials and methods
Cells culture
The human pancreatic cancer cell lines SW1990, CFPAC-1 and PANC-1 were purchased from the American Type Culture Collection (ATCC; Manassas, VA, USA). Cells were maintained in Dulbecco's modified essential medium (DMEM; Gibco, Grand Island, NY, USA). Medium was supplemented with 10% fetal calf serum (FBS; Gibco), 100 U/ml penicillin, and 100 mg/ml streptomycin at 37˚C in a 5% CO2 incubator with a humidified atmosphere. The cells were passaged every 2-3 d to maintain exponential growth.
Reagents
Lipopolysaccharide (LPS) was purchased from Enzo Life Sciences International (farmingdale, NY, USA). BAY11-7082 and EF-24 were purchased from Sigma (St. Louis, MO, USA).
MTT assay
Cell growth was evaluated using a methyl thiazolyl tetrazolium (MTT) assay.Citation53 Briefly, 5 × 104 cells per well were seeded onto 24-well plates. Following treatments for various durations of time, MTT (Sigma) was added to each well at a final concentration of 0.5 mg/ml. The mixture was incubated at 37°C for 4 h. The medium was then removed, and then 800μl of dimethyl sulfoxide (DMSO) was added to each well. The absorbance was measured at 490 nm using a microplate reader (Bio-Rad Laboratories). The relative cell viability was calculated as follows: relative cell viability = (mean absorbance of the test wells/mean absorbance of the control wells) × 100%.
Invasion assay
Matrigel (100 μl, 1:30 dilution in serum-free DMEM) was added to Transwell polycarbonate filters (8-μm pore size; Corning) and incubated with the filters at 37°C for 6 h. Cells were trypsinized and washed 3 times with DMEM containing 1% FBS, followed by resuspension in DMEM containing 1% FBS at a density of 2 × 106 cells/ml. The cell suspensions (100 μl) were seeded into the top chambers, and 600μl DMEM containing 10% FBS was added to the bottom chambers. Cells (2 × 105/well) were allowed to invade for 24 h, and the membranes were then stained with 1% methylrosanilinium chloride. Cells that had migrated to the underside of the filter were counted using a light microscope in 5 randomly selected fields.
Macrophage preparation and culture
MCM was performed as previously described.Citation13,Citation54 Buffy coats containing mononuclear cells were collected from the blood of healthy individual donors at the First Affiliated Hospital of Soochow University with an approved Institutional Review Board application (the Committee on Medical Ethics, the First Affiliated Hospital of Soochow University). Primary blood monocytes were isolated by density-gradient centrifugation through Ficol/Hypaque (Amersham Bioscience, Piscataway, NJ), suspended (8×106 cells/ml) in DMEM medium (Gibco) with 10% heat-inactivated human serum (Sigma, St. Louis, MO), and seeded in culture dishes. After 2-h incubation at 37°C, adherent cells were cultured in medium supplemented with 40 ng/ml human macrophage colony-stimulating-factor (MCSF, PeproTech Inc., Rocky Hill, NJ). Cells were allowed to differentiate for 7 d in the presence of MCSF. On day 7, fresh medium without MCSF was added to the cells, and the cells were cultured for anothere 24-120 h. The culture medium was collected, centrifuged, stored in aliquots at -80°C, and defined as MCM.
Milliplex assay
Multiplex biometric immunoassay kits containing fluorescent dyed microbeads (Milliplex NF-κB Signaling Magnetic Bead kit; Cat. No. 48-630MAG, and Human Cytokine MAGNETIC Kit; Cat. No. HCYTOMAG-60K, Millipore Corp, St Charles, MO) were used for measuring phosphorylated IKKα/β (Ser176/Ser180), phosphorylated IκBα (Ser32), c-Myc, IL-8, and TNF-α. Mean fluorescence intensity (MFI) was calculated using Luminex technology (Bio-Plex Workstation; Bio-Rad Laboratories, Hercules, CA, USA). Data were analyzed by using the Milliplex® Analyst 5.1 software (Bio-Rad Laboratories).
Animal
Four-week-old female BALB/c athymic nude mice (SLAC Laboratory Animal Co. Ltd., Shanghai, China) were maintained in a climate-controlled room with foods and water on a 12-h light/dark cycle (light on at 8:00 AM). Mice were allowed to acclimate to the housing facilities for 7 d before any experimental procedures were begun. All protocols were approved by the animal use and care committee of Soochow University.
Chronic pancreatitis model
Chronic pancreatitis was produced by injection of DBTC (Sigma) dissolved firstly in 100% ethanol (2 parts), and then mixed with glycerol (3 parts).Citation55 DBTC (2 mg/kg) in 50 μl solvent was injected into the tail vein. Control mice only received the equivalent volume of solvent. The animals were sacrificed 28 d after DBTC treatment. The pancreas was removed, formalin-fixed, and paraffin-embedded.
Orthotopic xenograft nude mouse model and treatment
The pancreatic cancer cell line PANC-1 expressing firefly luciferase was used. Orthotopic cell implantation was performed as previously described.Citation56 Cells (5 × 106) in 100 ml Matrigel (Becton Dickinson) were injected into the body-tail of the pancreas of nude mice. One week after orthotopic implantation, the mice were randomized into 2 groups: control and DBTC-treated. DBTC injection was administered according to the chronic pancreatitis model establishment protocol. Twenty-eight days after DBTC treatment, the mice were anaesthetized and given D-luciferin in phosphate-buffered saline (PBS). At 20 min after injection, bioluminescence was imaged with a charge-coupled device camera (IVIS; Lumina II, PerkinElmer). Then, the pancreas and tumor tissue were stripped, formalin-fixed, and paraffin-embedded.
Immunohistochemistry
All resection specimens were fixed in 10% buffered formalin and paraffin-embedded by routine processing. Sections were obtained at 4-μm thickness, heated at 60°C for 30 min, deparaffinized, and hydrated through a series of xylene and alcohol baths before staining. The slides were microwaved with antigen retrieval solution (citrate buffer, pH 6.0, containing 0.3% trisodium citrate and 0.04% citric acid) for 5 min. After replenishment of the solution, the slides were microwaved again for 5 min and then allowed to cool for 20 min. The sections were then rinsed in PBS and immersed in 3% H2O2 for 15 min to block endogenous peroxidase. Thereafter, the sections were incubated with 10% bull serum albumin (BSA) at room temperature for 60 min to block nonspecific antibodies. Immunohistochemical staining was performed with mouse anti-CD68 antibody (ab49777; Abcam), rabbit anti-CD163 antibody (ab87099; Abcam), rabbit anti-CD34 antibody (ab81289; Abcam), rabbit anti-PP2Ac antibody (ab32141; Abcam), or rabbit anti–phosphorylated IKK (S176, ab138426; Abcam) at room temperature for 2 h. After 20-min incubation with the corresponding secondary antibodies, the bound complex was visualized using a SuperPicture Polymer Detection kit (No. 87-8963; Invitrogen).
Real-time PCR
Total RNA was extracted using TRIzol reagent (Invitrogen,Valencia, California, USA) according to the manufacturer's protocol. After spectrophotometric quantification, 1 μg of total RNA in a final volume of 20 μl was used for reverse transcription with a PrimeScript RT Reagent Kit (TAKARA, Otsu, Shiga, Japan) according to the manufacturer's protocol. Aliquots of cDNA (cDNA) corresponding to equal amounts of RNA were used for mRNA quantification via real-time PCR using a LightCycler 96 Real-Time Quantitative PCR System (Roche, Indianapolis, IN, USA). The reaction system (25 µl) contained the corresponding cDNA, forward and reverse primers, and SYBR-Green PCR Master Mix (Roche). All data were analyzed using β-actin gene expression as the internal standard. The specific primers were as follows: (1) PP2Acα (Homo): forward, 5′-CGCCAGAAGTACACGAGGAAC-3′, reverse, 5′-CGTTGGATTCTTTTGTCAGGATTT-3′, product, 240 bp; (2) PP2Acβ (Homo): forward, 5′-GGGAAACCTGCCTTTGTAT-3′, reverse, 5′-CATCATTAGTATGGCACATTTGGTC-3′, product, 156 bp; (3) PP2Ac (Homo): forward, 5′- GTTCACCAAGGAGCTGGACCA-3′, reverse, 5′- CATGCACATCTAAACAGACAGTAAC-3′, product, 164 bp; (4) β-actin (Homo): forward, 5′-TCATGAAGTGTGACGTGGACAT-3′, reverse, 5′-CTCAGGAGGAGCAATGATCTTG-3′, product, 158 bp.
Western blot analysis
Total protein was extracted using a lysis buffer containing 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1% Triton X-100, 0.1% sodium dodecyl sulfate (SDS), 1 mM EDTA, protease inhibitors (10 mg/ml leupeptin, 10 mg/ml aprotinin, 10 mg/ml pepstatin A, 1 mM 4-[2-aminoethyl] benzenesulfonyl fluoride), and phosphatase inhibitors (1 mM NaF, 1 mM Na3VO4). Cytosolic and nuclear extracts were prepared using NE-PER Nuclear and Cytoplasmic Extraction Reagents (Pierce Biotechnology) supplemented with a protease inhibitor cocktail kit (Pierce Biotechnology) according to the manufacturer's instructions. The protein extracts were separated by 10% SDS–PAGE (PAGE) and were transferred to polyvinylidene fluoride (PVDF) membranes (Millipore). After 1-h blocking in 5% non-fat milk, the membranes were incubated overnight with primary antibodies at 4°C. The primary mouse antibodies were anti-PP2Ac, anti–phosphorylated PP2Ac (Tyr307), and anti–β-actin antibodies (Santa Cruz Biotechnologies). Rabbit anti-histone H1 antibodies were purchased from Proteintech Group. Protein expression was determined using horseradish peroxidase–conjugated antibodies followed by enhanced chemiluminescence) (ECL) detection (Millipore). Protein band analysis was performed using Quantity One 4.6.2 software (Bio-Rad Laboratories). β-actin and histone H1 were used as internal controls for the total and nuclear extracts, respectively.
Plasmids construction and transient transfection
The pcDNA3.1(+)-PP2Acα, pcDNA3.1(+)-DN-IKKα, and pcDNA3.1(+)-DN-IκBα plasmids were constructed as previously described.Citation11 The dominant negative (DN) mutant form of IKKα (DN-IKKα) was PCR-amplified from wild-type (WT) IKKα using site-directed mutagenesis to change Ser176 and Ser180 to Ala.Citation11 The DN mutant form of IκB (DN-IκB) was amplified from WT IκBα to change Ser32 and Ser36 to Ala.Citation11 The luciferase reporter plasmid, pNF-κB-luc, containing the κB-enhancer consensus sequences [(5′-TGGGGACTTTCCGC-3′)×5] and NF-κB-dependent firefly luciferase gene was purchased from Stratagene (La Jolla, CA, USA). The internal control plasmid, pRL-SV40, containing the Renilla luciferase gene was obtained from Promega (Madison, WI, USA). The reporter plasmids, pGL3-Basic-PP2Acα and pGL3-Basic-PP2Acβ, were constructed to measure the transcription of these 2 genes. PGL3-Basic-PP2Acα contained a 3,278-bp DNA fragment (from -3,093 to 185) upstream from the transcription initiation site of the PP2Acα gene, and pGL3-Basic-PP2Acβ containeds a 3,594-bp DNA fragment (from -3,404 to 190) upstream from the transcription initiation site of the PP2Acβ gene. These fragments were PCR-amplified from homo genomic DNA using PrimerSTAR HS DNA polymerase (TAKARA) with the following primers: (1) PP2Acα: forward, 5′-TGTCTCAGTAAGTTTAGAAGTCAGGG-3′, reverse, 5′-TACTTCTGGCGGCTGTTGAGG-3′; (2) PP2Acβ: forward, 5′-AAGGATTGGAGGTTGATGGTTA-3′, reverse, 5′-TGAAGGACGCGGTGAGGT-3′. The PCR-amplified fragments were firstly cloned into TA-clone by using a pMD19-T Vector Cloning Kit (TAKARA) and followed by subcloning into pGL3-Basic vector (Promega). Cells were transfected with plasmids by Lipofectamine 3000 (Invitrogen, Valencia, California, USA) according to manufacturer's protocol.
Luciferase reporter gene assay
Cells were transiently co-transfected with the reporter plasmid (500 ng/well) and the internal control plasmid pRL-SV40 (100 ng/well) using Lipofectamine 3000 (Invitrogen), according to the manufacturer's protocol. After transfection the medium was renewed, and the treatments were initiated. Subsequently, cell lysates were subjected to dual luciferase reporter assay (Promega) according to the manufacturer's recommendations. Luciferase activity was measured using a GloMax-20/20 luminometer (Promega). The results are expressed as relative luciferase activity (the ratio of firefly luciferase activity to Renilla luciferase activity).
Statistical analysis
Each experiment was performed at least in triplicate. Results are expressed as the mean value ± standard deviation (SD). Statistical analysis was performed using an unpaired Student's t-test. A P value less than 0.05 was considered significant.
Abbreviations
| LPS | = | lipopolysaccharide |
| TAM | = | tumor-associated macrophage |
| MCM | = | macrophage-conditioned medium |
| PP2A | = | protein phosphatase 2A |
| PP2Ac | = | PP2A catalytic subunit |
| IκB kinase | = | IKK |
| JNK | = | c-Jun N-terminal kinase |
| ERK | = | extracellular signal-related kinase |
| PKC | = | protein kinase C |
| DBTC | = | dibutyltin dichloride |
| PBS | = | phosphate buffered saline |
| MFI | = | Mean fluorescence intensity |
| MTT | = | methyl thiazolyl tetrazolium |
| DMSO | = | dimethyl sulfoxide |
| PVDF | = | polyvinylidene fluoride |
| PGE2 | = | prostaglandin E2 |
| IL-1 | = | interleukin-1 |
| TGF-β | = | transforming growth factor β |
| MMP-2 | = | matrix metalloproteinase 2 |
| MAPK | = | Mitogen-activated protein kinase |
| VEGF | = | vascular endothelial growth factor |
| TNF | = | tumor necrosis factor |
| ADM | = | adrenomedullin |
| uPA | = | urokinase-type plasminogen activator, CSF-1, colony stimulating factor 1 |
| CCL2 | = | chemokine (C-C motif) ligand 2 |
| BSA | = | bull serum albumin |
| cDNA | = | cDNA |
| SDS | = | sodium dodecyl sulfate |
| PAGE | = | polyacrylamide gel electrophoresis |
| PVDF | = | polyvinylidene fluoride |
| ECL | = | enhanced chemiluminescence |
| DN | = | dominant negative |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by grants from the National Natural Science Foundation of China [Nos. 81472296, 81101867, 81272542, 81200369, 81572992 and 81501970], the CSPAC-Celgene Foundation, the China International Medical Foundation (grant no. CIMFFH001057), the Special Foundation of Clinical Medicine of Jiangsu Provincial Bureau of Science and Technology (grant no. BL2014039), the Scientific Research Project of Jiangsu Provincial Bureau of Traditional Chinese Medicine (grant no. L213236), the Medical Scientific Research Project of Jiangsu Provincial Bureau of Health (grant no. Z201206), the Special Foundation of Wu Jieping Medical Foundation for Clinical Scientific Research (grant nos. 320.6753.1225 and 320.6750.12242), the Science and Education for Health Foundation of Suzhou for Youth (grant nos. SWKQ1003 and SWKQ1011), the Science and Technology Project Foundation of Suzhou (grant nos. SYS201112, SYSD2012137, SYSD201464 and SYS201335).
References
- Wolfgang CL, Herman JM, Laheru DA, Klein AP, Erdek MA, Fishman EK, Hruban RH. Recent progress in pancreatic cancer. CA Cancer J Clin 2013; 63:318-48; PMID:23856911
- Guerra C, Schuhmacher AJ, Canamero M, Grippo PJ, Verdaguer L, Perez-Gallego L, Dubus P, Sandgren EP, Barbacid M. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell 2007; 11:291-302; PMID:17349585; http://dx.doi.org/10.1016/j.ccr.2007.01.012
- Lowenfels AB, Maisonneuve P, Cavallini G, Ammann RW, Lankisch PG, Andersen JR, Dimagno EP, Andren-Sandberg A, Domellof L. Pancreatitis and the risk of pancreatic cancer. International Pancreatitis Study Group. N Engl J Med 1993; 328:1433-7; PMID:8479461; http://dx.doi.org/10.1056/NEJM199305203282001
- Janssens V, Goris J. Protein phosphatase 2A: a highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem J 2001; 353:417-39; PMID:11171037; http://dx.doi.org/10.1042/bj3530417
- Virshup DM. Protein phosphatase 2A: a panoply of enzymes. Curr Opin Cell Biol 2000; 12:180-5; PMID:10712915; http://dx.doi.org/10.1016/S0955-0674(99)00074-5
- Li W, Chen Z, Gong FR, Zong Y, Chen K, Li DM, Yin H, Duan WM, Miao Y, Tao M, et al. Growth of the pancreatic cancer cell line PANC-1 is inhibited by protein phosphatase 2A inhibitors through overactivation of the c-Jun N-terminal kinase pathway. Eur J Cancer 2011; 47:2654-64; PMID:21958460; http://dx.doi.org/10.1016/j.ejca.2011.08.014
- Arino J, Woon CW, Brautigan DL, Miller TB, Jr., Johnson GL. Human liver phosphatase 2A: cDNA and amino acid sequence of two catalytic subunit isotypes. Proc Natl Acad Sci U S A 1988; 85:4252-6; PMID:2837763; http://dx.doi.org/10.1073/pnas.85.12.4252
- Mumby M. PP2A: unveiling a reluctant tumor suppressor. Cell 2007; 130:21-4; PMID:17632053; http://dx.doi.org/10.1016/j.cell.2007.06.034
- Westermarck J, Hahn WC. Multiple pathways regulated by the tumor suppressor PP2A in transformation. Trends Mol Med 2008; 14:152-60; PMID:18329957; http://dx.doi.org/10.1016/j.molmed.2008.02.001
- Li J, Sheng C, Li W, Zheng JH. Protein phosphatase-2A is down-regulated in patients within clear cell renal cell carcinoma. Int J Clin Exp Pathol 2014; 7:1147-53; PMID:24696731
- Li W, Chen Z, Zong Y, Gong F, Zhu Y, Lv J, Zhang J, Xie L, Sun Y, Miao Y, et al. PP2A inhibitors induce apoptosis in pancreatic cancer cell line PANC-1 through persistent phosphorylation of IKKalpha and sustained activation of the NF-kappaB pathway. Cancer Lett 2011; 304:117-27; PMID:21376459; http://dx.doi.org/10.1016/j.canlet.2011.02.009
- Coffelt SB, Hughes R, Lewis CE. Tumor-associated macrophages: effectors of angiogenesis and tumor progression. Biochim Biophys Acta 2009; 1796:11-8; PMID:19269310
- Wu Y, Deng J, Rychahou PG, Qiu S, Evers BM, Zhou BP. Stabilization of snail by NF-kappaB is required for inflammation-induced cell migration and invasion. Cancer Cell 2009; 15:416-28; PMID:19411070; http://dx.doi.org/10.1016/j.ccr.2009.03.016
- Rhee SH. Lipopolysaccharide: basic biochemistry, intracellular signaling, and physiological impacts in the gut. Intest Res 2014; 12:90-5; PMID:25349574; http://dx.doi.org/10.5217/ir.2014.12.2.90
- Marshall M, Anilkumar N, Layland J, Walker SJ, Kentish JC, Shah AM, Cave AC. Protein phosphatase 2A contributes to the cardiac dysfunction induced by endotoxemia. Cardiovasc Res 2009; 82:67-76; PMID:19201758; http://dx.doi.org/10.1093/cvr/cvp037
- Kawai T, Akira S. Signaling to NF-kappaB by Toll-like receptors. Trends Mol Med 2007; 13:460-9; PMID:18029230; http://dx.doi.org/10.1016/j.molmed.2007.09.002
- Feng F, Wu Y, Zhang S, Liu Y, Qin L, Yan Z, Wu W. Macrophages facilitate coal tar pitch extract-induced tumorigenic transformation of human bronchial epithelial cells mediated by NF-kappaB. PloS One 2012; 7:e51690; PMID:23227270; http://dx.doi.org/10.1371/journal.pone.0051690
- Gupta SC, Singh R, Pochampally R, Watabe K, Mo YY. Acidosis promotes invasiveness of breast cancer cells through ROS-AKT-NF-kappaB pathway. Oncotarget 2014; 5:12070-82; PMID:25504433; http://dx.doi.org/10.18632/oncotarget.2514
- Sun Y, Ai X, Shen S, Lu S. NF-kappaB-mediated miR-124 suppresses metastasis of non-small-cell lung cancer by targeting MYO10. Oncotarget 2015; 6:8244-54; PMID:25749519; http://dx.doi.org/10.18632/oncotarget.3135
- Krausgruber T, Blazek K, Smallie T, Alzabin S, Lockstone H, Sahgal N, Hussell T, Feldmann M, Udalova IA. IRF5 promotes inflammatory macrophage polarization and TH1-TH17 responses. Nat Immunol 2011; 12:231-8; PMID:21240265; http://dx.doi.org/10.1038/ni.1990
- Zumsteg A, Christofori G. Corrupt policemen: inflammatory cells promote tumor angiogenesis. Curr Opin Oncol 2009; 21:60-70; PMID:19125020; http://dx.doi.org/10.1097/CCO.0b013e32831bed7e
- Riabov V, Gudima A, Wang N, Mickley A, Orekhov A, Kzhyshkowska J. Role of tumor associated macrophages in tumor angiogenesis and lymphangiogenesis. Front Physiol 2014; 5:75; PMID:24634660; http://dx.doi.org/10.3389/fphys.2014.00075
- Kim JK, Lee SM, Suk K, Lee WH. A novel pathway responsible for lipopolysaccharide-induced translational regulation of TNF-α and IL-6 expression involves protein kinase C and fascin. J Immunol 2011; 187:6327-34; PMID:22102721; http://dx.doi.org/10.4049/jimmunol.1100612
- Boncoeur E, Bouvet GF, Migneault F, Tardif V, Ferraro P, Radzioch D, de Sanctis JB, Eidelman D, Govindaraju K, Dagenais A, et al. Induction of nitric oxide synthase expression by lipopolysaccharide is mediated by calcium-dependent PKCalpha-beta1 in alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol 2013; 305:L175-84; PMID:23686852; http://dx.doi.org/10.1152/ajplung.00295.2012
- Liu SH, Lu TH, Su CC, Lay IS, Lin HY, Fang KM, Ho TJ, Chen KL, Su YC, Chiang WC, et al. Lotus leaf (Nelumbo nucifera) and its active constituents prevent inflammatory responses in macrophages via JNK/NF-kappaB signaling pathway. Am J Chin Med 2014; 42:869-89; PMID:25004880; http://dx.doi.org/10.1142/S0192415X14500554
- Zhao LL, Hu GC, Zhu SS, Li JF, Liu GJ. Propofol pretreatment attenuates lipopolysaccharide-induced acute lung injury in rats by activating the phosphoinositide-3-kinase/Akt pathway. Brazilian journal of medical and biological research = Revista brasileira de pesquisas medicas e biologicas / Sociedade Brasileira de Biofisica [et al] 2014; 47:1062-7
- Li W, Xie L, Chen Z, Zhu Y, Sun Y, Miao Y, Xu Z, Han X. Cantharidin, a potent and selective PP2A inhibitor, induces an oxidative stress-independent growth inhibition of pancreatic cancer cells through G2/M cell-cycle arrest and apoptosis. Cancer Sci 2010; 101:1226-33; PMID:20331621; http://dx.doi.org/10.1111/j.1349-7006.2010.01523.x
- Israel A. The IKK complex: an integrator of all signals that activate NF-kappaB? Trends Cell Biol 2000; 10:129-33; PMID:10740266; http://dx.doi.org/10.1016/S0962-8924(00)01729-3
- Oeckinghaus A, Ghosh S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol 2009; 1:a000034; PMID:20066092; http://dx.doi.org/10.1101/cshperspect.a000034
- Bunting K, Rao S, Hardy K, Woltring D, Denyer GS, Wang J, Gerondakis S, Shannon MF. Genome-wide analysis of gene expression in T cells to identify targets of the NF-kappa B transcription factor c-Rel. J Immunol 2007; 178:7097-109; PMID:17513759; http://dx.doi.org/10.4049/jimmunol.178.11.7097
- Gong F, Sun L, Wang Z, Shi J, Li W, Wang S, Han X, Sun Y. The BCL2 gene is regulated by a special AT-rich sequence binding protein 1-mediated long range chromosomal interaction between the promoter and the distal element located within the 3′-UTR. Nucleic Acids Res 2011; 39:4640-52; PMID:21310710; http://dx.doi.org/10.1093/nar/gkr023
- Liu ST, Pham H, Pandol SJ, Ptasznik A. Src as the link between inflammation and cancer. Front Physiol 2013; 4:416; PMID:24474940
- Samuel GH, Bujor AM, Nakerakanti SS, Hant FN, Trojanowska M. Autocrine transforming growth factor β signaling regulates extracellular signal-regulated kinase 1/2 phosphorylation via modulation of protein phosphatase 2A expression in scleroderma fibroblasts. Fibrogenesis Tissue Repair 2010; 3:25; PMID:21134273; http://dx.doi.org/10.1186/1755-1536-3-25
- Young MR. Tumor-derived prostaglandin E2 and transforming growth factor-β stimulate endothelial cell motility through inhibition of protein phosphatase-2A and involvement of PTEN and phosphatidylinositide 3-kinase. Angiogenesis 2004; 7:123-31; PMID:15516833; http://dx.doi.org/10.1007/s10456-004-1027-2
- Sommer D, Coleman S, Swanson SA, Stemmer PM. Differential susceptibilities of serine/threonine phosphatases to oxidative and nitrosative stress. Arch Biochem Biophys 2002; 404:271-8; PMID:12147265; http://dx.doi.org/10.1016/S0003-9861(02)00242-4
- Fukumura Y, Kumasaka T, Mitani K, Karita K, Suda K. Expression of transforming growth factor beta1, beta2, and beta3 in chronic, cancer-associated, obstructive pancreatitis. Arch Pathol Lab Med 2006; 130:356-61; PMID:16519564
- Ren C, Chen Y, Han C, Fu D, Chen H. Plasma interleukin-11 (IL-11) levels have diagnostic and prognostic roles in patients with pancreatic cancer. Tumour Biol 2014; 35(11):11467-72
- Mori A, Moser C, Lang SA, Hackl C, Gottfried E, Kreutz M, Schlitt HJ, Geissler EK, Stoeltzing O. Upregulation of Kruppel-like factor 5 in pancreatic cancer is promoted by interleukin-1beta signaling and hypoxia-inducible factor-1alpha. Mol Cancer Res 2009; 7:1390-8; PMID:19671674; http://dx.doi.org/10.1158/1541-7786.MCR-08-0525
- Melisi D, Niu J, Chang Z, Xia Q, Peng B, Ishiyama S, Evans DB, Chiao PJ. Secreted interleukin-1alpha induces a metastatic phenotype in pancreatic cancer by sustaining a constitutive activation of nuclear factor-kappaB. Mol Cancer Res 2009; 7:624-33; PMID:19435817; http://dx.doi.org/10.1158/1541-7786.MCR-08-0201
- Ellenrieder V, Hendler SF, Ruhland C, Boeck W, Adler G, Gress TM. TGF-β-induced invasiveness of pancreatic cancer cells is mediated by matrix metalloproteinase-2 and the urokinase plasminogen activator system. Int J Cancer 2001; 93:204-11; PMID:11410867; http://dx.doi.org/10.1002/ijc.1330
- Feurino LW, Zhang Y, Bharadwaj U, Zhang R, Li F, Fisher WE, Brunicardi FC, Chen C, Yao Q, Min L. IL-6 stimulates Th2 type cytokine secretion and upregulates VEGF and NRP-1 expression in pancreatic cancer cells. Cancer Biol Ther 2007; 6:1096-100; PMID:17568185; http://dx.doi.org/10.4161/cbt.6.7.4328
- Li M, Zhang Y, Feurino LW, Wang H, Fisher WE, Brunicardi FC, Chen C, Yao Q. Interleukin-8 increases vascular endothelial growth factor and neuropilin expression and stimulates ERK activation in human pancreatic cancer. Cancer Sci 2008; 99:733-7; PMID:18307536; http://dx.doi.org/10.1111/j.1349-7006.2008.00740.x
- Holmer R, Goumas FA, Waetzig GH, Rose-John S, Kalthoff H. Interleukin-6: a villain in the drama of pancreatic cancer development and progression. Hepatobiliary Pancreat Dis Int 2014; 13:371-80; http://dx.doi.org/10.1016/S1499-3872(14)60259-9
- Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, Kaiser EA, Snyder LA, Pollard JW. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 2011; 475:222-5; PMID:21654748; http://dx.doi.org/10.1038/nature10138
- Merkord J, Jonas L, Weber H, Kroning G, Nizze H, Hennighausen G. Acute interstitial pancreatitis in rats induced by dibutyltin dichloride (DBTC): pathogenesis and natural course of lesions. Pancreas 1997; 15:392-401; PMID:9361094; http://dx.doi.org/10.1097/00006676-199711000-00010
- Chen W, Wang Z, Jiang C, Ding Y. PP2A-Mediated Anticancer Therapy. Gastroenterol Res Pract 2013; 2013:675429; PMID:24307892
- Smits HH, van Beelen AJ, Hessle C, Westland R, de Jong E, Soeteman E, Wold A, Wierenga EA, Kapsenberg ML. Commensal Gram-negative bacteria prime human dendritic cells for enhanced IL-23 and IL-27 expression and enhanced Th1 development. Eur J Immunol 2004; 34:1371-80; PMID:15114670; http://dx.doi.org/10.1002/eji.200324815
- Ghosh S, Gifford AM, Riviere LR, Tempst P, Nolan GP, Baltimore D. Cloning of the p50 DNA binding subunit of NF-kappa B: homology to rel and dorsal. Cell 1990; 62:1019-29; PMID:2203532; http://dx.doi.org/10.1016/0092-8674(90)90276-K
- Tsao SM, Hsia TC, Yin MC. Protocatechuic Acid Inhibits Lung Cancer Cells by Modulating FAK, MAPK, and NF-kappaB Pathways. Nutr Cancer 2014; 66:1331-41; PMID:25356681; http://dx.doi.org/10.1080/01635581.2014.956259
- Keshk WA, Zineldeen DH, Wasfy RE, El-Khadrawy OH. Fatty acid synthase/oxidized low-density lipoprotein as metabolic oncogenes linking obesity to colon cancer via NF-kappa B in Egyptians. Med Oncol 2014; 31:192; PMID:25173531; http://dx.doi.org/10.1007/s12032-014-0192-4
- Yang MH, Kim HT, Lee KT, Yang S, Lee JK, Lee KH, Rhee JC. KML001 inhibits cell proliferation and invasion in pancreatic cancer cells through suppression of NF-kappaB and VEGF-C. Anticancer Res 2014; 34:3469-74; PMID:24982355
- Staudt LM. Oncogenic activation of NF-kappaB. Cold Spring Harb Perspect Biol 2010; 2:a000109; PMID:20516126; http://dx.doi.org/10.1101/cshperspect.a000109
- Carmichael J, DeGraff WG, Gazdar AF, Minna JD, Mitchell JB. Evaluation of a tetrazolium-based semiautomated colorimetric assay: assessment of chemosensitivity testing. Cancer Res 1987; 47:936-42; PMID:3802100
- Li W, Liang RR, Zhou C, Wu MY, Lian L, Yuan GF, Wang MY, Xie X, Shou LM, Gong FR, et al. The association between expressions of Ras and CD68 in the angiogenesis of breast cancers. Cancer cell Int 2015; 15:17; PMID:25685069; http://dx.doi.org/10.1186/s12935-015-0169-1
- Sparmann G, Jaschke A, Loehr M, Liebe S, Emmrich J. Tissue homogenization as a key step in extracting RNA from human and rat pancreatic tissue. Biotechniques 1997; 22:408-10, 12; PMID:9067010
- Hudson SV, Huang JS, Yin W, Albeituni S, Rush J, Khanal A, Yan J, Ceresa BP, Frieboes HB, McNally LR. Targeted noninvasive imaging of EGFR-expressing orthotopic pancreatic cancer using multispectral optoacoustic tomography. Cancer Res 2014; 74:6271-9; PMID:25217521; http://dx.doi.org/10.1158/0008-5472.CAN-14-1656