
Oncogenic activation of NOTCH1 signaling as result of gain of function mutations in the NOTCH1 gene plays a central role in the pathogenesis of T-ALL.Citation1 Accordingly, γ-secretase inhibitors (GSI), which block a transmembrane proteolytic cleavage required for NOTCH1 receptor activation have been proposed as a potential targeted therapy in T-ALL.Citation1 The therapeutic relevance of this approach is supported by in vitro studies demonstrating a rapid clearance of intracellular activated NOTCH1 in T-ALL cell lines and induction of cell cycle arrest upon GSI treatment.Citation1 In addition, GSIs impair the engraftment of primary T-ALL cells in mice and induce significant antileukemic responses in mouse NOTCH1-induced leukemia and in patient-derived primary T-ALL xenografts.Citation2 However, and despite some exceptional responders, GSIs have shown limited therapeutic activity in early clinical trials.Citation3 This lack of therapeutic efficacy may reflect weak oncogene addiction to NOTCH1 in T-ALL, perhaps due to activation of parallel signaling pathways or adaptive epigenetic responses. Thus, mutational loss of PTEN is associated with GSI resistance in human T-ALL cell lines.Citation4 Moreover, MYC can overcome the antileukemic effects of NOTCH1 inhibition in some tumors, and mutations in FBXW7, which encodes a negative regulator of NOTCH1, MYC and mTOR, are also highly prevalent in GSI-resistant cell lines. Notably, these different proposed mechanisms could be interrelated as NOTCH1, PI3K signaling and MYC are closely interconnected. Moreover, some T-ALL cell lines contain dynamic population of GSI-tolerant ‘persister’ cells, supporting a potential role for epigenetic mechanisms of adaptation to NOTCH1 inhibition in GSI resistance.Citation5 However, the significance of these in vitro models is limited as they primarily rely on results from immortalized cell lines and fall short of evaluating clinical resistance, which is best defined as disease progression under treatment in vivo. Finally, the downstream effector mechanism mediating the response to NOTCH1 inhibition and the effector pathways driving resistance remain to be elucidated.
These key questions have now been formally addressed via phenotypic, transcriptional and metabolomic profiling of the in vivo response to GSI therapy in isogenic Pten-positive and Pten-deleted mouse NOTCH1-induced leukemias.Citation2 In this model genetic loss of Pten induced resistance to GSI treatment in vivo despite effective inhibition of NOTCH1 signaling. Mechanistically, NOTCH1 inhibition induced transcriptional downregulation of anabolic pathway genes with concomitant upregulation of catabolic pathways and increased autophagy in Pten-positive tumors, but not in Pten-deleted cells, supporting a potential major role for metabolic dowregulation in the response to GSI treatment. Metabolomic profiling of these tumors showed a prominent role for glutaminolysis in Pten-positive NOTCH1 induced T-ALL and revealed that NOTCH1 inhibition results in decreased levels of glycolysis and glutaminolysis in ALL lymphoblast cells. Notably, isogenic loss of Pten in this model increased the glycolytic efficiency of leukemic cells and rescued the inhibitory effects of GSI treatment in leukemia cell metabolism (). Of note Myc was effectively downregulated upon NOTCH inhibition in both Pten-positive and Pten-deleted T-ALLs. A key role for inhibition of cellular metabolism in the response to NOTCH inhibition is further supported by the reversal of antileukemic effects induced by GSI in PTEN positive T-ALL cell lines upon treatment with membrane soluble forms of pyruvate and α-ketoglutarate or by forced expression of key enzymes driving glycolysis (PKM2) and glutaminolysis (GLS). A corollary of these results is that inhibition of autophagy, which may serve as salvage pathway partially sustaining cell growth in Pten-positive ALL cells upon inhibition of NOTCH signaling, or further suppression of cellular metabolism by direct inhibition of glutaminolysis, could accentuate the antileukemic effects of GSI treatment. Consistently, genetic abrogation of autophagy in NOTCH1 induced leukemias with conditional inducible loss of Atg7, a key mediator of autophagy, enhanced the antileukemic effects of GSI treatment. Moreover, genetic and pharmacologic inhibition of glutaminase was highly synergistic with inhibition of NOTCH1 signaling in T-ALL cell lines, mouse NOTCH1-induced tumors and human patient-derived T-ALL xenografts ().
Figure 1. Metabolic effects of NOTCH1 inhibition and Pten loss in T-ALL. NOTCH1 drives glycolysis and glutaminolysis and NOTCH1 inhibition promotes an anabolic block, forcing lymphoblasts to use autophagy to survive. Consistently, inhibition of autophagy or glutaminolysis enhances the antileukemic effects of GSIs. Conversely, loss of Pten promotes a hyperglycolytic switch that overrides the metabolic effects of GSIs, driving resistance to therapy.
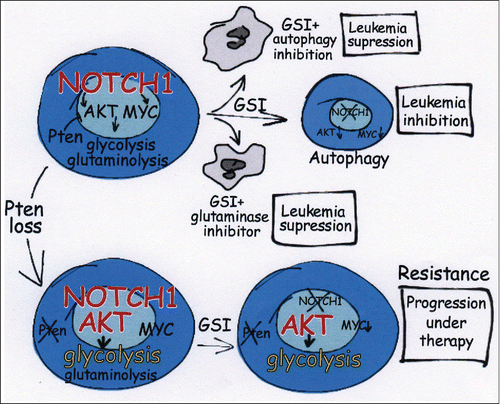
Overall these results underline the critical importance of carbon metabolism in mediating the antileukemic effects of NOTCH1 inhibition and highlight glutaminolysis and autophagy as new therapeutic targets in T-ALL. More broadly, the interplay between NOTCH1 and Pten in the regulation of cancer metabolism and response to NOTCH1 inhibition underscore the importance of metabolic rewiring in the context of resistance to targeted therapies and demonstrates that secondary mutations can greatly impact on the metabolic profile of tumors.
Still, the discovery of the acute metabolic effects of NOTCH1 inhibition, and their abrogation by loss of Pten, opens new relevant questions. First, the mechanisms by which NOTCH1 regulates glycolysis and glutaminolysis in T-ALL remain to be fully elucidated. Moreover, the transcriptional control of gene expression programs related to anabolic and catabolic pathways by NOTCH1 and Pten is yet to be linked with specific transcription factors and epigenetic regulators. Finally, the lack of robust antitumor effects to GSI treatment and glutaminase inhibition in Pten negative NOTCH1 induced tumors highlights the need to identify new therapeutic strategies for the treatment of these most refractory leukemias.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- Weng AP, et al. Science 2004; 306:269-271; PMID:15472075; http://dx.doi.org/10.1126/science.1102160
- Herranz D, et al. Nat Med 2015; 21:1182-1189; PMID:26390244; http://dx.doi.org/10.1038/nm.3955
- Takebe N, et al. Pharmacol Therapeutics 2014; 141:140-149; PMID:24076266; http://dx.doi.org/10.1016/j.pharmthera.2013.09.005
- Palomero T, et al. Nat Med 2007; 13:1203-1210; PMID:17873882; http://dx.doi.org/10.1038/nm1636
- Knoechel B, et al. Nat Genet 2014; 46:364-370; PMID:24584072; http://dx.doi.org/10.1038/ng.2913