
ABSTRACT
Multiple myeloma (MM) is a B-cell malignancy characterized by an accumulation of abnormal clonal plasma cells in the bone marrow. Introduction of the proteasome-inhibitor bortezomib has improved MM prognosis and survival; however hypoxia-induced or acquired bortezomib resistance remains a clinical problem. This study highlighted the role of thioredoxin reductase 1 (TrxR1) in the hypoxia-induced and acquired bortezomib resistance in MM. Higher TrxR1 gene expression correlated with high-risk disease, adverse overall survival, and poor prognosis in myeloma patients. We demonstrated that hypoxia induced bortezomib resistance in myeloma cells and increased TrxR1 protein levels. Inhibition of TrxR1 using auranofin overcame hypoxia-induced bortezomib resistance and restored the sensitivity of hypoxic-myeloma cells to bortezomib. Hypoxia increased NF-кβ subunit p65 nuclear protein levels and TrxR1 inhibition decreased hypoxia-induced NF-кβ p65 protein levels in the nucleus and reduced the expression of NF-кβ-regulated genes. In addition, higher TrxR1 protein levels were observed in bortezomib-resistant myeloma cells compared to the naïve cells, and its inhibition using either auranofin or TrxR1-specific siRNAs reversed bortezomib resistance. TrxR1 inhibition reduced p65 mRNA and protein expression in bortezomib-resistant myeloma cells, and also decreased the expression of NF-кβ-regulated anti-apoptotic and proliferative genes. Thus, TrxR1 inhibition overcomes both hypoxia-induced and acquired bortezomib resistance by inhibiting the NF-кβ signaling pathway. Our findings demonstrate that elevated TrxR1 levels correlate with the acquisition of bortezomib resistance in MM. We propose considering TrxR1-inhibiting drugs, such as auranofin, either for single agent or combination therapy to circumvent bortezomib-resistance and improve survival outcomes of MM patients.
Introduction
Multiple myeloma (MM) is an incurable B-cell malignancy characterized by the accumulation of abnormal clonal plasma cells in the bone marrow (BM). In the past decade, many effective anti-myeloma agents have been developed including bortezomib, thalidomide, and lenalidomide.Citation1-3 The introduction of bortezomib as an anti-myeloma agent has greatly improved overall survival in MM patients.Citation4 Despite such advancements, relapse following prolonged bortezomib treatment is inevitable due to either genetic modificationsCitation5 or emergence of bortezomib-resistant plasma cell sub-clones.Citation6 Once myeloma cells acquire bortezomib resistance, they can no longer be treated with bortezomib. Clinical data shows that roughly half of initially bortezomib-sensitive MM patients do not respond to bortezomib once the disease is relapsed.Citation7 Therefore, new therapeutic strategies are needed to treat such bortezomib-resistant or relapsed MM patients.
A crucial microenvironmental factor that plays an important role in the acquisition of drug resistance and MM disease progression is hypoxia. Hypoxia is an imbalance between the supply of oxygen and its consumption that deprives cells of sufficient oxygen. Recent studies have shown that myeloma cells reside in an extensively hypoxic BM microenvironment.Citation8 Hypoxia induces myeloma cell dedifferentiation, acquisition of quiescent state by reducing cell proliferation and inducing G1-cell cycle arrest, and enhances tumor progression. Hypoxia also induces drug resistance to proteasome inhibitors including bortezomib, and carfilzomib.Citation9 However, the exact molecular mechanism by which hypoxia induces such resistance remains to be elucidated. In recent years, therapies including the hypoxia-activated prodrug TH-302, which targets the hypoxic myeloma cells have been developed.Citation8 Together TH-302 and bortezomib synergistically induce apoptosis in myeloma cells growing under hypoxic conditions.Citation10 Thus, development of therapies that target not only the cell growing under normal oxygen conditions but also under hypoxia, either alone or in combination with conventional anti-myeloma drugs, may provide a better therapeutic approach to treat MM.
Acquisition of bortezomib resistance has been attributed to different mechanisms including increased growth factor expression,Citation11 a mutated proteasome subunit PSMβ5 and overexpression of PSMβ5 protein,Citation5 an upregulated NF-кβ signaling pathway,Citation12,13 and overexpressed antioxidant molecules.Citation14,15 The pro-survival NF-кβ signaling pathway and its members are upregulated in MM patients compared to healthy individuals.Citation12,16 Bortezomib has been shown to inhibit NF-кβ expression in myeloma cells by inhibiting the 20S proteasome β5 subunit.Citation17 However, bortezomib also increases constitutive NF-кβ expression levels by activating IKKβ, leading to NF-кβ nuclear translocation and the transcription of multiple NF-кβ-regulated genes including Survivin, and Cyclin D1, which are associated with multi-drug resistance in MM.Citation18-20 This upregulation of NF-кβ expression and its signaling pathway has been associated with acquisition of bortezomib resistance due to prolonged drug exposure in MM.Citation13,18 However, the role of the NF-кβ signaling pathway in hypoxia-induced bortezomib resistance in MM is unknown.
The thioredoxin (Trx) system is one of the major antioxidant systems in the body and is comprised of thioredoxin 1 (Trx1), thioredoxin reductase 1 (TrxR1) and NADPH. Both Trx1 and TrxR1 have been shown to be upregulated in many human cancer types including MM, and correlated with cancer cell proliferation, survival, and chemoresistance.Citation14,21-23 We have previously shown that bortezomib-resistant cells contained higher Trx1 protein levels compared to parent myeloma cells and its inhibition induced apoptosis in bortezomib-resistant myeloma cells.Citation14 TrxR1 expression has been shown to be upregulated in drug-resistant cancer cells and its inhibition induced apoptosis in those cells.Citation24,25 However, there are no previous reports on the therapeutic significance of TrxR1 in bortezomib-resistant myeloma cells.
The present study was designed to characterize the role of TrxR1 in the acquisition of bortezomib resistance in myeloma cells, to investigate whether TrxR1 inhibition overcomes the hypoxia-induced and acquired bortezomib resistance in myeloma cells, and to elucidate the involvement of the NF-кβ signaling pathway. Our results show that TrxR1 inhibition overcomes the hypoxia-induced and acquired bortezomib resistance in MM by inhibiting the NF-κβ signaling pathway. These data provide justification for considering TrxR1 as an effective therapeutic target to treat bortezomib-resistant MM patients. These results also establish the possibility of using TrxR1-inhibiting drugs, such as auranofin, in conjunction with bortezomib for therapeutic intervention of relapsed/bortezomib-resistant MM patients in the clinic.
Results
TrxR1 upregulation decreases myeloma patient survival and responsiveness to the treatment
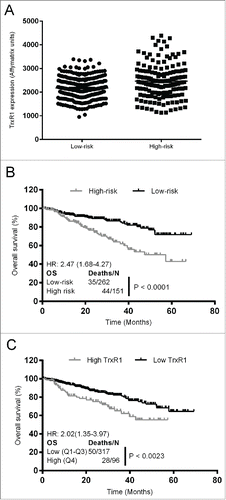
We first aimed to examine the effect of TrxR1 on myeloma patient survival and responsiveness to the given treatment. By using the gene expression dataset (GSE6477), we previously showed that TrxR1 is expressed at significantly higher levels in the newly diagnosed and relapsed MM patients compared to the healthy individuals.Citation14 To analyze the role of TrxR1 in patient survival upon treatment, we used the gene expression data set (GSE4581) in which myeloma patients were subsequently treated with the Total Therapies (as described in the method section). TrxR1 gene expression was analyzed in the high-risk and low-risk group of MM patients and the data showed that high-risk patients exhibited approximately 1.2-fold increased TrxR1 expression relative to low-risk patients (). Next, survival analysis methods were used to analyze the effect of TrxR1 expression on the overall survival (OS) for MM patients. The OS was significantly decreased in the high-risk group of patients compared to low-risk patients (HR = 2.47, log-rank P < 0.0001) (). Moreover, patients having higher TrxR1 expression were at a higher death risk compared to the patients with low TrxR1 expression (HR = 2.02, log-rank P < 0.0023) (). Overall, retrospective analysis of primary samples showed that increased TrxR1 expression decreases MM patient survival in response to the treatment and portends poor prognosis.
Figure 1. Increased TrxR1 expression in MM patients correlates with decreased overall survival and poor prognosis. (A) TrxR1 expression in high-risk and low-risk MM patients was determined from the gene expression profiling data deposited in the gene expression omnibus database (GSE4581). Unpaired student t test was performed. P < 0.05 (compared to low-risk patients) (B) Overall survival of high-risk and low-risk patients receiving Total Therapy 2 and 3 was estimated by generating the Kaplan-Meier curve. (C) Overall survival was estimated in the patients with high and low TrxR1 expression levels receiving Total Therapy 2 and 3 by generating the Kaplan-Meier curve.
TrxR1 is upregulated in hypoxic myeloma cells and its inhibition sensitizes hypoxic myeloma cells to bortezomib
Hypoxia is a critical microenvironmental factor that promotes myeloma cell metastasis, disease progression, and drug resistance.Citation9,10,26 We firstly investigated the effect of hypoxia on myeloma cell proliferation in the presence of bortezomib. RPMI8226 and U266 cells were cultured under hypoxic (Hx, 1% O2) and normoxic (Nrx, 21% O2) conditions for 24 hours, and subsequently treated with or without bortezomib (0–40 nM) for 24 hours, and cell proliferation was analyzed. In line with another study,Citation9 our data showed that hypoxia induced bortezomib resistance in myeloma cells, whereas normoxic myeloma cells were sensitive to bortezomib (). Western blot analysis showed that culturing RPMI8226 and U266 cells under hypoxia for different time-periods markedly increased HIF-1α protein expression indicating that myeloma cells have become hypoxic (). Moreover, hypoxia increased TrxR1 protein expression in both RPMI8226 and U266 cells in a time-dependent manner ().
Figure 2. TrxR1 is upregulated in hypoxic myeloma cells and its inhibition overcomes hypoxia-induced bortezomib-resistance in myeloma cells. (A, B) RPMI8226 (A) and U266 (B) cells were cultured under normoxia (Nrx, 21% O2) and hypoxia (Hx, 1% O2) for 24 hours. Both cell lines were treated with bortezomib (0–40 nM) for 24 hours under normoxia and hypoxia and cell proliferation was analyzed by CellTiter blue assays. (C, D) RPMI8226 and U266 cells were cultured under hypoxia for indicated time periods. Total cell extracts were prepared, and HIF-1α and TrxR1 protein expression was analyzed by western blot analysis. β-tubulin was used as a loading control. Western blots are the representative of 3 independent experiments. (E) Hx-RPMI8226 and Hx-U266 cells were treated with auranofin (0–4 μM) for 24 hours under hypoxia (1% O2) and protein was extracted. The TrxR1 activity was analyzed by measuring the NADPH-dependent reduction of DTNB by TrxR1 enzyme and normalizing against the protein concentration. (F, G) RPMI8226 (F) and U266 (G) cells were cultured under normoxia (21% O2) and hypoxia (1% O2) for 24 hours. Both cell lines were treated with auranofin (0–8 μM) for 24 hours under normoxia and hypoxia and cell proliferation was analyzed by CellTiter blue assay. (H) Hx-RPMI8226 and Hx-U266 cells were treated with auranofin (2 μM) and bortezomib (10 nM) either alone or in combination for 24 hours under hypoxia. Cell proliferation was analyzed by CellTiter Blue assays. Values indicate mean ± SEM (n = 3). One-way ANOVA followed by Tukey's post-test were employed. *, P < 0.05.
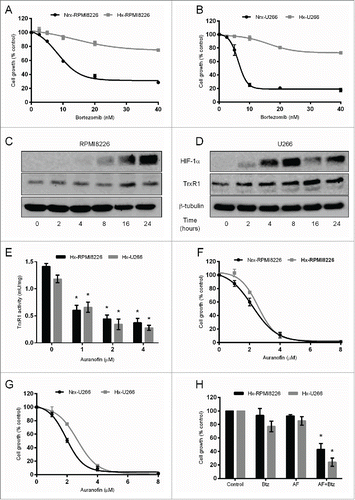
Based on these results, we hypothesized that upregulated TrxR1 levels may be responsible for hypoxia-induced bortezomib resistance in myeloma cells. We aimed to investigate whether TrxR1 inhibition reduces proliferation and sensitizes hypoxic myeloma cells to bortezomib. RPMI8226 and U266 cells were cultured under normoxic and hypoxic conditions for 24 hours and subsequently treated with specified auranofin concentrations for 24 hours, and TrxR1 activity and cell proliferation were analyzed. Auranofin treatment significantly inhibited TrxR1 activity in both RPMI8226 and U266 cells (). Unlike bortezomib treatment, auranofin treatment exerted a similar anti-proliferative effect on the hypoxic as well as normoxic RPMI8226 in a concentration-dependent manner (). While auranofin reduced proliferation of both normoxic and hypoxic U266 cells in a concentration-dependent manner, hypoxic U266 cells were observed to be more resistant to auranofin compared to normoxic U266 cells (). We then analyzed whether TrxR1 inhibition sensitizes hypoxic myeloma cells to the cytotoxic effect of bortezomib. Hypoxic RPMI8226 and U266 cells were treated with a sub-lethal concentration of auranofin (2 μM) and bortezomib (10 nM) either alone or in combination for 24 hours under hypoxia, and cell proliferation was measured. Both auranofin and bortezomib alone had no effect on RPMI8226 and U266 cell proliferation, whereas auranofin and bortezomib in combination significantly reduced RPMI8226 and U266 () cell proliferation by approximately 50% and 75%, respectively, under hypoxic conditions.
TrxR1 inhibition overcomes hypoxia-induced bortezomib resistance by inhibiting the NF-кβ signaling pathway
Hypoxia has been shown to activate the NF-кβ signaling pathway and hypoxia-induced NF-кβ activation has been associated with drug resistance in cancer cells.Citation27-29 Moreover, increased NF-кβ expression has been linked to bortezomib resistance in myeloma cells.Citation13 Therefore, we first aimed to examine the effect of hypoxia on NF-кβ p65 expression in myeloma cells. U266 cells were cultured under hypoxia for different time-periods (0–24 hours) and NF-кβ p65 protein levels were analyzed by western blot analysis. Results showed that 2 to 8 hours exposure of U266 cells to hypoxia markedly increased NF-кβ p65 protein levels in the nucleus ().
Figure 3. TrxR1 inhibition overcomes hypoxia-induced bortezomib resistance by inhibiting NF-кβ signaling pathway. (A) U266 cells were cultured under hypoxia (1% O2) for indicated time periods. NF-κβ subunit p65 protein levels were analyzed by western blot analysis. (B) U266 cells were cultured under hypoxia for 24 hours and subsequently treated with auranofin (4 μM) for 4 hours, and cell proliferation was analyzed. (C) U266 cells were cultured under hypoxia for 0, 2, and 4 hours with or without auranofin (4 μM) treatment. NF-кβ subunit p65 protein levels were analyzed by western blot analysis. Lamin B1 (nuclear fraction) and β-tubulin (cytosolic fraction) were used as loading controls. Western blots are the representative of 3 independent experiments. (D) Hypoxic U266 cells were treated with 4 μM AF for 4 hours under hypoxia (1% O2). Expression of indicated NF-κβ-regulated genes was analyzed by RT-qPCR and normalized against L32. Values indicate mean ± SEM (n = 3). Unpaired student t test was employed. *, P < 0.05.
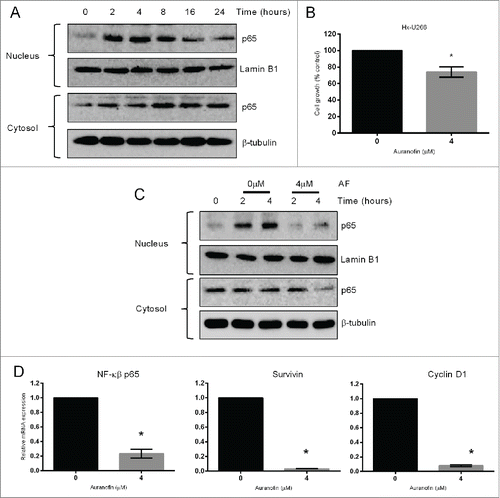
Based on these results, we hypothesized that an activated NF-кβ pathway may be responsible for the acquisition of hypoxia-induced bortezomib resistance in myeloma cells. We then investigated the effect of TrxR1 inhibition on the NF-кβ signaling pathway in hypoxic myeloma cells. U266 cells were treated with or without auranofin (4 μM) and cultured under hypoxia for 0, 2, and 4 hours. Although 4 μM auranofin significantly reduced the proliferation of hypoxic-U266 cells by approximately 85% after 24 hours treatment (), only 25% reduction in cell proliferation was observed after 4 hours treatment (). Thus, this concentration of auranofin was selected to analyze the effect on the NF-κβ signaling pathway. Western blot analysis showed that auranofin treatment inhibited hypoxia-induced nuclear accumulation of NF-кβ p65 (). Auranofin treatment also reduced NF-кβ p65 protein levels in the cytosol after 4 hours exposure to hypoxia (). Hypoxic U266 cells were treated with or without auranofin (4 μM) for 4 hours under hypoxia and the expression of the downstream NF-кβ-regulated genes were analyzed. RT-qPCR results showed a significant decrease in the mRNA levels of NF-кβ p65 and its target genes, Survivin and Cyclin D1, in auranofin-treated hypoxic U266 cells compared to the untreated cells ().
Basal TrxR1 protein expression is increased in bortezomib-resistant myeloma cells
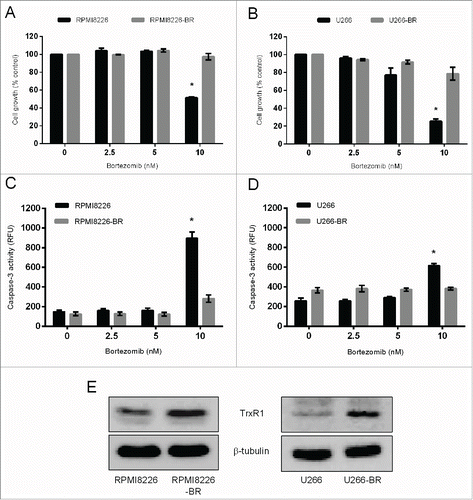
In addition to hypoxia-induced bortezomib resistance, we also aimed to determine the role of TrxR1 in the bortezomib resistance acquired due to prolonged drug exposure in MM. To investigate the role of TrxR1 in the acquired bortezomib resistance in myeloma cells we first generated bortezomib-resistant (BR) cell lines (RPMI8226-BR and U266-BR) from the highly sensitive parent RPMI8226 and U266 cell lines. Bortezomib resistance was confirmed by exposing both RPMI8226-BR and U266-BR cell lines to bortezomib (0–10 nM) for 24 hours. Results showed that 10 nM bortezomib significantly reduced the proliferation of parent RPMI8226 and U266, whereas no significant effect was observed on RPMI8226-BR and U266-BR cell proliferation (). In addition, we analyzed apoptosis by measuring caspase-3 activity in the parent and bortezomib-resistant cell lines following bortezomib exposure. Caspase-3 activity was significantly increased in the parent RPMI8226 and U266 cells after exposure to 10 nM bortezomib whereas, no significant increase in caspase-3 activity was observed in RPMI8226-BR and U266-BR cells (). Western blot analysis showed higher TrxR1 protein levels in both bortezomib-resistant cell lines compared to the parent cell lines ().
Figure 4. TrxR1 protein expression in the parent and bortezomib-resistant myeloma cells. (A-D)Parent and bortezomib-resistant RPMI8226 and U266 cells (RPMI8226-BR and U266-BR) were treated with indicated concentrations of bortezomib for 24 hours and cell proliferation (A, B) and caspase-3 activity (C, D) were measured. One-way ANOVA followed by Tukey's post-test was employed. *, P < 0.05 (compared to the 0nM bortezomib treatment) (E) TrxR1 protein levels in the parent and bortezomib-resistant myeloma cells were analyzed by western blot. β-tubulin was used as a loading control. Western blots are the representative of 3 independent experiments.
TrxR1 inhibition induces apoptosis in bortezomib-resistant myeloma cells
We have previously shown that TrxR1 expression is increased in myeloma cells compared to normal cells and its inhibition leads to apoptosis in the parent myeloma cells.Citation14 Increased TrxR1 expression has been correlated with drug resistance in many human cancer types and its inhibition induces apoptosis in those cancer cells.Citation24,25,30 We therefore examined the effect of TrxR1 inhibition on the cell proliferation and apoptosis in bortezomib-resistant myeloma cells by using the TrxR1 inhibitor, auranofin, and TrxR1 specific siRNA. Treatment of RPMI8226-BR and U266-BR cells with auranofin significantly inhibited TrxR1 activity in a concentration dependent manner (). RPMI8226-BR and U266-BR cell proliferation was significantly reduced by approximately 60% and 80% following treatment with 4 μM and 8 μM auranofin, respectively (). Similarly, 4 μM auranofin significantly increased caspase-3 activity by 2-fold in both RPMI8226-BR and U266-BR cells indicating that TrxR1 inhibition leads to apoptosis in bortezomib-resistant myeloma cells ().
Figure 5. TrxR1 inhibition induces apoptosis in bortezomib-resistant myeloma cells. (A, B, C) RPMI8226-BR and U266-BR cells were treated with indicated concentrations of auranofin for 24 hours. The TrxR1 activity (A), cell proliferation (B), and apoptosis were assessed (C). Values indicate mean ± SEM of 3 independent experiments performed in triplicate. For caspase-3 activity assay (n = 3). One-way ANOVA followed by Tukey's post-test was employed. *, P < 0.0001 (compared to the 0 μM auranofin treatment). (D-G) RPMI8226-BR and U266-BR cells were transfected with 100 nmol/L of either control or TrxR1 specific siRNA. TrxR1 protein levels (48 hours) were analyzed by western blot (D, F). Cell viability was determined by using Trypan blue exclusion method 48 hours post-transfection (E, G). β-tubulin was used as a loading control. Western blots are the representative of 3 independent experiments. Values indicate mean ± SEM (n = 3). Unpaired student t test was employed. *, P < 0.05 (compared to the siControl).
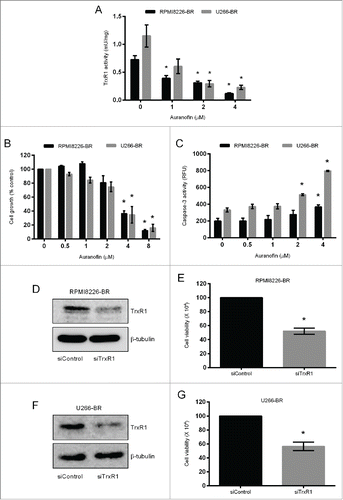
To ascertain if specific knockdown of TrxR1 could reproduce the effect of drug-induced TrxR1 inhibition on the growth of bortezomib-resistant myeloma cells, we used TrxR1 specific siRNA. Transfection of the siRNAs against TrxR1 suppressed TrxR1 protein expression compared to the control siRNA () and reduced RPMI8226-BR and U266-BR cell viability by approximately 40% ().
TrxR1 inhibition sensitizes bortezomib-resistant myeloma cells to bortezomib and reverses chemoresistance
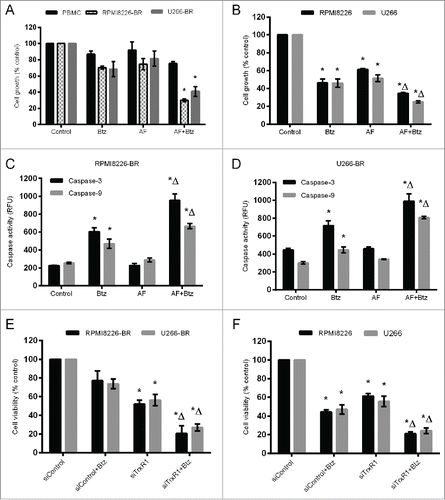
Inhibition of TrxR1 has been shown to reverse the chemoresistance in human cancers including chronic myeloid leukemia and ovarian cancer.Citation24,25 We investigated whether inhibition of increased TrxR1 restores the sensitivity to bortezomib in bortezomib-resistant myeloma cells and reverses the chemoresistance. Normal peripheral blood mononuclear cells (PBMCs), RPMI8226-BR, and U266-BR cells were treated with auranofin (2 μM) and bortezomib (10 nM) either alone or in combination for 48 hours. Treatment with auranofin or bortezomib alone had no statistically significant effect on cell proliferation. However, co-treatment with auranofin and bortezomib significantly reduced RPMI8226-BR and U266-BR cell proliferation by approximately 60%. Auranofin and bortezomib either alone or in combination exerted no significant cytotoxic effect on normal PBMCs (). While treatment of the parent RPMI8226 and U266 cells with auranofin (2 μM) and bortezomib (10 nM) alone significantly reduced their proliferation, co-treatment with auranofin (2 μM) and bortezomib (10 nM) for 48 hours further reduced the proliferation of these cells (). We then measured apoptosis in bortezomib-resistant myeloma cells treated with either auranofin or bortezomib alone or in combination. Higher caspase-3 and 9 activities were observed when cells were treated with auranofin and bortezomib together compared to the treatment with auranofin or bortezomib alone (). To confirm the results obtained by auranofin-induced TrxR1 inhibition, we used TrxR1 knockdown using TrxR1-specific siRNA in combination with bortezomib (10 nM) treatment, which significantly reduced RPMI8226-BR and U266-BR cell viability by 60% and 70%, respectively (). While TrxR1 knockdown using TrxR1-specific siRNA and bortezomib (10 nM) treatment alone significantly reduced parent RPMI8226 and U266 cell viability, co-treatment with TrxR1-specific siRNA and bortezomib for 48 hours further reduced the viability of these cells ().
Figure 6. TrxR1 inhibition sensitizes bortezomib-resistant myeloma cells to bortezomib and reverses chemoresistance. (A) PBMCs, RPMI8226-BR and U266-BR cells, and (B) parent RPMI8226 and U266 cells were treated with 2 μM auranofin (AF) and 10 nM bortezomib (Btz) either alone or in combination. Cell proliferation was analyzed by CellTiter Blue assay 48 hours post-treatment. (C, D) RPMI8226-BR and U266-BR cells were treated with 2 μM auranofin (AF) and 10 nM bortezomib (Btz) either alone or in combination. Apoptosis was assessed by measuring caspase-3 and caspase-9 activity 24 hours post-treatment in RPMI8226-BR (C) and U266-BR (D) cells. E, RPMI8226-BR and U266-BR cells, and F, parent RPMI8226 and U266 (F) cells were transfected with 100 nmol/L of either control or TrxR1 specific siRNA. Cells were treated with or without 10 nM bortezomib 4 hours post-transfection for 48 hours. Cell viability was measured by Trypan blue exclusion method. Values indicate mean ± SEM of 3 independent experiments performed in triplicate. For caspase-3 and 9 activity assay (n = 3). One-way ANOVA followed by Tukey's post-test was employed. * (compared to the untreated cells), Δ (compared to either AF alone or Btz alone panel B, C); (compared to either siTrxR1 alone or Btz alone panel D, E), P < 0.05.
TrxR1 inhibition suppresses NF-кβ signaling pathway in bortezomib-resistant myeloma cells
Increased NF-кβ expression was observed in bortezomib refractory primary myeloma patient samples and has been linked to drug resistance in those patients.Citation18 We initially examined the basal protein levels of NF-кβ subunit p65 in the parent and bortezomib-resistant U266 and RPMI8226 cells. Higher NF-кβ p65 protein levels were observed in the nucleus of bortezomib-resistant cells compared to the nucleus of parent cells whereas, its levels in the cytosol remained unchanged ().
Figure 7. TrxR1 inhibition suppresses NF-кβ signaling pathway in bortezomib-resistant myeloma cells. (A) NF-кβ subunit p65 protein levels were analyzed in the nuclear and cytosolic fractions of parent and bortezomib-resistant (BR) U266 and RPMI8226 cells by western blot analysis. (B) U266-BR cells were treated with the indicated concentrations of AF for 24 hours and with 4 μM AF for the indicated time periods. Expression of phospho-Iкβα was analyzed by western blot analysis. (C) NF-кβ p65 mRNA expression in U266-BR and RPMI8226-BR cells treated with or without 4 μM AF for 24 hours was analyzed by real time-qPCR (RT-qPCR). NF-кβ p65 mRNA expression was normalized against L32 housekeeping gene. (D) U266-BR cells were treated with AF (0–4 μM) for 24 hours. Nuclear, cytosolic, and total cell extracts were prepared and p65 protein levels were analyzed by western blot analysis. Lamin B1 and β-tubulin were used as loading controls for nuclear and cytosolic fractions, respectively. Western blots are the representative of 3 independent experiments. (E, F) U266-BR (E) and RPMI8226-BR (F) cells were treated with or without 4 μM AF for 24 hours. Expression of indicated NF-кβ-regulated genes was analyzed by RT-qPCR and normalized against L32. Values indicate mean ± SEM (n = 3). Unpaired student t test was employed. *, P < 0.05.
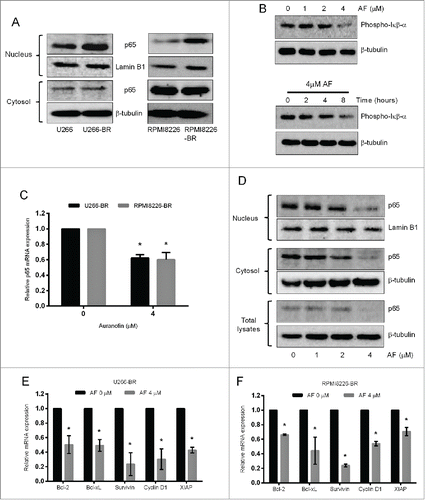
We next investigated whether TrxR1 inhibition overcomes bortezomib resistance in myeloma cells by inhibiting the NF-кβ pathway. The degradation of Iкβα and release of NF-кβ subunits is preceded by the phosphorylation of Iкβα at Ser32 and Ser36 residuesCitation31 Therefore, we first examined the effect of TrxR1 inhibition on Iкβα phosphorylation. Auranofin treatment markedly decreased the content of phosphorylated Iкβα in U266-BR cells in a concentration and time-dependent manner (). RT-qPCR data showed that 4 μM auranofin significantly reduced NF-кβ p65 mRNA expression in U266-BR and RPMI8226-BR (). In addition, western blot results showed that auranofin treatment significantly decreased NF-кβ p65 protein levels in the nucleus, cytosol, and total lysates in U266-BR cells in a concentration dependent manner (). We then analyzed the effect of TrxR1 inhibition on the downstream NF-кβ-regulated genes. RT-qPCR results showed a significant decrease in the mRNA expression levels of various NF-кβ target genes including Bcl−2, Bcl−xL, Survivin, Cyclin D1, and XIAP in auranofin-treated (4 μM) U266-BR () and RPMI8226-BR () cells compared to untreated cells.
Discussion
Although many anti-myeloma therapies have been developed in recent years, to date MM remains an incurable disease with drug resistance being the major impediment. Bortezomib is the first proteasome inhibitor to be approved by the FDA for the treatment of newly diagnosed and relapsed MM patients.Citation32 Bortezomib, in single agent therapy, has a response rate of approximately 30% and in combination with other conventional anti-myeloma therapeutic agents, the response rate increases to approximately 60–90% of MM patients.Citation33 Despite such a high response rate, MM patients acquire resistance to bortezomib when treated for a longer duration.Citation5,34 Hence, a thorough understanding of the molecular mechanisms for bortezomib resistance in MM and novel therapeutic approaches to overcome bortezomib resistance in MM are needed.
Here, for the first time we report the role of the cellular redox protein, TrxR1, in the bortezomib resistance of myeloma cells. It has been reported that MM patients having increased Copper-Zinc superoxide dismutase (CuZnSOD) expression have shorter overall and event free survival compared to patients with lower CuZnSOD levels.Citation35 Higher Trx1 levels in breast cancer patients also correlate with a low response rate to docetaxel compared to the patients with low Trx1 levels.Citation36 We here report that MM patients with higher TrxR1 levels have a shorter overall survival compared to patients with lower TrxR1 levels (). Thus, higher antioxidant levels may correlate with poor prognosis and clinical outcome in cancers including MM.
Several microenvironmental factors including hypoxia and interactions with the bone marrow stroma cells play a crucial role in acquisition of drug resistance in many human cancer types including hematological malignancies such as chronic myeloid leukemia.Citation37,38 Overcoming such drug resistance remains a great challenge to treat cancer. Our study highlights a novel strategy to overcome hypoxia-induced drug resistance in myeloma cells by targeting upregulated TrxR1. As discussed earlier, myeloma cells reside in the hypoxic BM region, which plays a crucial role in MM disease progression by conferring survival and chemoresistance.Citation8,26 Hypoxia promotes immature and stem-cell like phenotype in myeloma cells by decreasing CD138 and increasing CD34 expression.Citation9,39 Such hypoxic cells are resistant to proteasome inhibitors including bortezomib and carfilzomib.Citation9 In accordance to these studies, our results showed that hypoxic RPMI8226 and U266 cells became resistant to bortezomib compared to their normoxic counterparts. Moreover, our data showed increased TrxR1 protein levels in hypoxic myeloma cells suggesting a possible involvement of TrxR1 in the acquisition of hypoxia-induced bortezomib resistance in MM.
In recent years, targeting hypoxic myeloma cells has emerged as a novel therapeutic strategy to eliminate myeloma cells residing in the hypoxic environment. For example, the hypoxia-activated prodrug TH-302 induces apoptosis in hypoxic myeloma cells either alone or in combination with bortezomib.Citation10 In line with this study, our results showed that auranofin-induced TrxR1 inhibition exerted an anti-proliferative effect on hypoxic RPMI8226 and U266 cells (). Interestingly, unlike bortezomib, auranofin exerted significant anti-proliferative effects on myeloma cells growing under both normoxic and hypoxic conditions suggesting that TrxR1-inhibiting drugs may exert better anti-myeloma activity compared to other conventional drugs under different physiological conditions. Furthermore, we also showed that TrxR1 inhibition using a sub-lethal concentration of auranofin (2 μM) sensitized hypoxic myeloma cells to the cytotoxic effect of bortezomib (). Our results complement another study that showed that TrxR1 inhibition by a similar auranofin concentration (1.5 μM) has increased the bortezomib sensitivity of human colon carcinoma cell line HCT116 overexpressing Bcl−2 by 2-fold under normoxic conditions.Citation40 While many therapies targeting the hypoxic myeloma cells are being developed either as a single agent or combinatorial therapies,Citation8,10 our study provided evidence that targeting hypoxic myeloma cells using TrxR1 inhibitors, such as auranofin, provides a potentially useful and novel treatment strategy for MM.
This research, for the first time, shows the effect of hypoxia on the NF-кβ pathway and the role of TrxR1 in the regulation of hypoxia-induced NF-кβ p65 expression in myeloma cells. Hypoxia activates the NF-кβ signaling pathway and NF-кβ-mediated gene expression.Citation27,28 We show that exposure of U266 cells to hypoxia increased NF-кβ p65 protein levels in the nucleus. Increased nuclear p65 protein levels have been correlated with the acquisition of drug resistance in cancer cells.Citation13,29 Our results showed that auranofin-induced TrxR1 inhibition reduced the hypoxia-induced nuclear accumulation of NF-кβ p65 protein, reduced the mRNA levels of p65 and downstream NF-кβ-regulated genes, Survivin and Cyclin D1 (). Thus, TrxR1 inhibition induces myeloma cell death growing under a hypoxic microenvironment and overcomes hypoxia-induced bortezomib resistance by inhibiting the NF-кβ pathway.
Significant correlation between adaptation of cancer cells to oxidative stress by upregulating intracellular antioxidant systems and chemoresistance has been documented.Citation41 Increased CuZnSOD expression was shown to induce imexon-resistance in myeloma cells.Citation42 TrxR1 expression and activity is increased in adriamycin-resistant leukemia cells and cisplatin-resistant ovarian cancer cells, and its inhibition using auranofin induced apoptosis in these cells.Citation24,25 Here, for the first time we report that increased TrxR1 expression is associated with bortezomib resistance in myeloma cells. We showed that TrxR1 protein levels are upregulated in bortezomib-resistant myeloma cells (RPMI8226 & U266) compared to the parent cells () and its inhibition resulted in apoptosis in these cells (). Moreover, TrxR1 inhibition using either a sub-lethal concentration of auranofin or TrxR1-specific siRNAs significantly restored the sensitivity of bortezomib-resistant myeloma cells to bortezomib (). Our findings complement other studies that showed that modulation of antioxidant systems, glutathione and CuZnSOD, abrogates bortezomib resistance and resensitizes resistant myeloma cells to bortezomib.Citation15,35 Interestingly, despite higher Trx1 protein levels in bortezomib-resistant myeloma cells,Citation14 its inhibition using either a specific inhibitor PX-12 or Trx1 anti-sense plasmid did not sensitize bortezomib-resistant U266 cells to bortezomib (data not shown). However, the underlying mechanism for this effect remains to be elucidated. One possible explanation may be that TrxR1 have distinct downstream targets in addition to the common targets as Trx1, which may be involved in overcoming bortezomib resistance.Citation43 Thus, despite being part of the same antioxidant system TrxR1 inhibition may serve as a better and more effective therapeutic approach for treatment of bortezomib-resistant MM patients. Moreover, TrxR1-targeting drugs, like auranofin, can either be used as single agent therapy or in combination with bortezomib to treat relapsed MM patients.
Our study highlights a molecular mechanism by which TrxR1 inhibition overcomes bortezomib resistance in myeloma cells. Higher NF-кβ expression is seen in bortezomib refractory primary MM patientsCitation18 and bortezomib resistance in MM is associated with increased basal nuclear localization of NF-кβ p65 in bortezomib-resistant myeloma cells.Citation13 Here, we also show increased nuclear NF-кβ p65 levels in bortezomib-resistant U266 cells compared to the parent counterpart. We showed that TrxR1 inhibition using auranofin reduced NF-кβ p65 mRNA and protein expression, reduced the mRNA levels of downstream NF-кβ-regulated genes including anti-apoptotic and proliferation genes in bortezomib-resistant U266 cells (). These results indicated that TrxR1 inhibition suppresses the NF-кβ signaling pathway by inhibiting the phosphorylation and subsequent degradation of Iкβα and overcomes acquired bortezomib resistance in myeloma cells. In addition to the NF-кβ signaling pathway, TrxR1 inhibition, either by auranofin or TrxR1-specific si/shRNA, has also been shown to induce an expression of the oxidative stress responsive transcription factor, Nrf2.Citation44,45 Moreover, Nrf2 activation has been shown to inhibit the NF-кβ signaling pathway and attenuate NF-кβ-mediated inflammatory responses and induce apoptosis.Citation46 Nrf2 deficient mouse embryonic fibroblasts showed activation of NF-кβ,Citation47 suggesting that Nrf2 induction may inhibit the NF-кβ signaling pathway in the cells. Based on these findings, it may be hypothesized that Nrf2 may act as an intermediate between TrxR1 and NF-кβ, and TrxR1 inhibition may suppress the NF-кβ signaling pathway via Nrf2 induction. However, whether TrxR1 regulates the NF-кβ signaling pathway in MM via either Nrf2 or another mechanism remains undetermined and needs further investigation.
In conclusion, our research confirms that higher TrxR1 expression is associated with both the hypoxia-induced and acquired bortezomib resistance in myeloma cells, and its inhibition leads to apoptosis in these cells. Moreover, TrxR1 inhibition overcomes the bortezomib resistance in myeloma cells through inhibition of the NF-кβ signaling pathway. Therefore, we propose considering TrxR1-inhibiting drugs like auranofin, either for single agent or combination therapies to circumvent bortezomib resistance in MM and improve survival outcomes of MM patients.
Materials and methods
Cells and reagents
Two standard human multiple myeloma cell lines (RPMI8226 and U266) were obtained from Dr. Slavica Vuckovic (QIMR Berghofer Medical Research Institute). RPMI8226 cells were originally derived from the peripheral blood of a 61-year-old male with multiple myeloma (IgG lambda-type).Citation48 U266 cells were originally derived from the peripheral blood of a 53-year-old male with IgE-secreting myeloma (refractory).Citation49 Human peripheral blood mononuclear cells (PBMCs) were isolated from the whole blood of healthy volunteers and were collected under the ethical approval BPS/08/14/HREC. Cells were cultured in RPMI-1640 medium (Gibco) containing 10% (V/V) fetal bovine serum (FBS) (Bovagen), 200 mM L-glutamine, and 100 U/ml penicillin and 100 ug/ml streptomycin (Invitrogen). The monoclonal anti-TrxR1 antibody (https://www.rndsystems.com/products/mab7428) and polyclonal anti-phospho-Iкβα antibody (https://www.rndsystems.com/products/af4809) were purchased from R & D system. The polyclonal anti-NF-кβ p65 antibody (http://www.genetex.com/NFkB-p65-antibody-GTX102090.html) was purchased from GeneTex. The anti-β-tubulin (www.abcam.com/ab6046.html) and anti-Lamin B1 (www.abcam.com/ab133741.html) antibodies were purchased from Abcam. The TrxR1 inhibitor auranofin was purchased from Sigma (www.sigmaaldrich.com/catalog/product/sigma/a6733).
The TrxR1 validated small interfering RNA (siRNA), a pool of 4 siRNAs, and control siRNA were purchased from Santa Cruz Biotechnology (http://www.scbt.com/datasheet-36750.html).
Analysis of TrxR1 expression and clinical prognosis in primary MM patients
TrxR1 gene expression was analyzed using the gene expression profiling (GEP) data from MM patients receiving the total therapy (TT) 2 [thalidomide and stem-cell transplantation] and 3 [bortezomib, thalidomide, and stem-cell transplantation] deposited in the gene expression omnibus database GSE4581. The analysis was done as described previously.Citation35 Briefly, in this study MM patients were classified into high-risk and low-risk groups using the 70-gene model. The high-risk group comprised of the patients with shorter duration of complete remission and overall survival (OS). The Kaplan-Meier curve was created to estimate the OS in high-risk and low-risk patients. The TrxR1 expression was categorized into 2 groups, high and low, using upper (Q4) and lower quartiles (Q1, Q2, Q3) and OS was estimated with 95% confidence interval (CI) for TrxR1 as a continuous variable.
TrxR activity assays
The TrxR activity assays were performed as described previously.Citation50 Briefly, treated and untreated cells were lysed using 0.5% (v/v) Nonidet P-40 cell lysis buffer (150 mM NaCl, 50 mM Tris-Cl, pH 8; 0.5% (v/v) Nonidet P-50, 0.5 mM EDTA, 2 mM PMSF, 1 μl/ml protease inhibitor cocktail VI, 1X PBS). To omit non-TrxR1-specific DTNB reduction, cell lysates were treated with or without 8 μM auranofin for 30 minutes at room temperature. The TrxR activity was measured using a buffer containing 0.5 M potassium phosphate, 200 mM EDTA, 20 mM NADPH, and 125 mM DTNB. TNB production was measured by following an increase in absorbance at 412 nm for 10 minutes. The reaction rates obtained in the presence of auranofin were subtracted from the reaction rates obtained in the absence of auranofin to give the final corrected TrxR rates. Units of TrxR activity (µmoles of TNB produced/minute) were calculated using an extinction coefficient of 13.6 × 103 M−1 of TNB at 412 nm. The specific thioredoxin reductase activity was determined using the following equation: Specific activity (U/mg) = U/total protein.
Establishment of bortezomib-resistant cell lines
Bortezomib-resistant (BR) myeloma cell lines (RPMI8226-BR and U266-BR) were generated from the parent cell lines (RPMI8226 and U266) by multistep exposure of the cells to increasing doses of bortezomib (from 1 nM to 20 nM) for 10–12 weeks as described previously.Citation14 Bortezomib-resistance was confirmed by measuring cell proliferation and apoptosis in response to bortezomib exposure.
Western blot analysis
Whole cell extracts were prepared using 0.5% (v/v) Nonidet P-40 cell lysis buffer. Nuclear and cytosolic fractions were prepared using Nuclear Protein Extraction Kit (Cayman Chemicals) according to the manufacturer's guidelines. Western immunoblotting analysis was performed as described previously.Citation51 Blots were probed with various specific antibodies (TrxR1, phospho-Iкβα, p65, β-tubulin, Lamin B1) and ECL detection was done using GE ECL Western Blotting Substrate (GE Healthcare).
Cell proliferation assay
0.5 × 106 cells were treated with the appropriate drugs for specified period of time. Relative cell proliferation was assayed using the CellTiter-Blue Cell Viability Assay (Promega), as per the manufacturer's instructions.
Cell viability measurement
Cell viability was measured by the Trypan blue exclusion method as described previously.Citation14
Caspase-3 and 9 activity assays
Caspase-3 activity was determined in treated and untreated myeloma cells by using Ac-DEVD-AMC (Enzo Life Sciences, http://www.enzolifesciences.com/ALX-260-031), a caspase-3 substrate, as described previously.Citation14 Caspase-9 activity was determined following the cleavage of Ac-LEHD-AFC (Cayman Chemicals, https://www.caymanchem.com/catalog/17051), a caspase-9 substrate, in treated and untreated myeloma cells. Briefly, treated or untreated cells (0.5 × 106 cells) were pelleted, washed with PBS, re-suspended in 15 μl of PBS, and transferred to black-walled 96-wells plate. 85 μl of caspase assay buffer (100 mM HEPES, 0.1 M NaCl, 0.05 M EDTA, 10 mM DTT, 10% sucrose, 0.5% NP-40 at pH 7.5) containing 100 μM Ac-LEHD-AMC was added to the samples and the amount of AMC cleaved by caspase-9 was measured at 37°C by measuring the fluorescence at excitation wavelength of 405 nm and emission of 510 nm using the SpectraMax plate reader (Molecular Devices). Data were analyzed using SoftMax Pro software (Bio-strategy).
Transient transfections
Cells (2 × 106 per well) were transfected using the Amaxa nucleofector (T-001 program) using equivalent molar concentrations of all 4 TrxR1 specific siRNAs and control siRNA (final concentrations of 100 nmol/L) (Santa Cruz). Transfected cells were incubated for the specified time before the indicated treatments.
RNA extraction and quantitative real time-PCR
Total RNA was extracted from U266-BR and RPMI8226-BR cells (1 × 106 cells) using TRIsure™ Total RNA Lysis solution (Bioline) according to the manufacturer's instructions. Total RNA was reverse transcribed using the SensiFAST™ cDNA synthesis kit (Bioline). Resultant cDNA was analyzed by real-time quantitative PCR (RT-qPCR) using SensiFAST™ SYBR® No-Rox Kit (Bioline) and RT-qPCR primers for L32 [forward 5′-CAGGGTTCGTAGAAGATTCAAGGG-3′ and reverse 5′-CTTGGAGGAAAACATTGTGAGCGATC-3′],Citation52 NF-кβ p65 [forward 5′-ACCGCTGCATCCACAGTT-3′ and reverse 5′-GGATGCGCTGACTGATAGC-3′],Citation13 Bcl−2 [forward 5′-CACCTGCACACCTGGATCCAGGATAA-3′ and reverse 5′-CACCAG GGCCAAACTGAGCAG A-3′], Bcl−xL [forward 5′-CCTAGAGCCTTGGATCCAGGA GAA-3′ and reverse 5′-GGTTGAAGCGTTCCTGGCCCTTT-3′], Survivin [forward 5′-GAGCCAGACTTGGCCCAGTGTTT-3′ and reverse 5′-GATGGCACGGCGCACTTTCT-3′], Cyclin D1 [forward 5′-CGCCCTCGGTGTCCTACTTCAA-3′ and reverse 5′- CTG CAGGCGGCTCTTTTTCA-3], and XIAP [forward 5′-GAGGTTCTGGTTGCAGATCTA GTGA-3′ and reverse 5′-CTCCTCTTGCAGGCGCCTTA-3′] (Integrated DNA Technologies, IA, USA), on the Rotor-Gene 6000 Real-time PCR system (Corbett Life Sciences) according to the manufacturer's guidelines. Reaction conditions were 95°C for 2 minutes followed by 40 cycles of 95°C for 5 seconds, 60°C for 10 seconds, and 72°C for 20 seconds. The comparative cycle threshold algorithm (ΔΔCT) method was used to analyze gene expression. The mRNA was normalized against L32 expression.Citation52
Statistical analysis
Data were analyzed by using the software GraphPad Prism 6 (GraphPad Software). Values are presented as mean ± SEM. Statistical significance was determined by the specified statistical test. P < 0.05 was considered significant.
Abbreviations
| AF | = | auranofin |
| ANOVA | = | analysis of variance |
| BM | = | bone marrow |
| Btz | = | bortezomib |
| CuZnSOD | = | copper-zinc superoxide dismutase |
| GEP | = | gene expression profile |
| HIF-1α | = | hypoxia-inducible factor-1 α |
| HR | = | hazard ratio |
| Hx | = | hypoxia |
| IKKβ | = | inhibitor of nuclear factor kappa β |
| Iкβα | = | inhibitor of kappa β α |
| MM | = | multiple myeloma |
| NF-кβ | = | nuclear factor kappa β |
| Nrx | = | normoxia |
| OS | = | overall survival |
| PBMCs | = | peripheral blood mononuclear cells |
| RPMI8226-BR | = | bortezomib-resistant RMPI8226 |
| Trx1 | = | thioredoxin 1 |
| TrxR1 | = | thioredoxin reductase 1 |
| TT2 | = | total therapy 2 |
| TT3 | = | total therapy 3 |
| U266-BR | = | bortezomib-resistant U266 |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank A/Prof. Stephen Wood (Eskitis Institute, Brisbane, Australia) and his lab members for their help in setting up transfection experiments, Prof. George Mellick (Eskitis Institute, Brisbane, Australia) for collecting the blood samples from healthy individuals, and Tony Blick (Queensland University of Technology, Australia) for statistical advice.
Funding
This research was supported by a Griffith University Post-graduate Research Scholarship and a Griffith University International Post-graduate Scholarship (to P. R). We also thank Griffith University for providing financial support to carry out this research.
References
- Cavo M. Proteasome inhibitor bortezomib for the treatment of multiple myeloma. Leukemia 2006; 20:1341-52; PMID:16810203; http://dx.doi.org/10.1038/sj.leu.2404278
- Palumbo A, Facon T, Sonneveld P, Blade J, Offidani M, Gay F, Moreau P, Waage A, Spencer A, Ludwig H, et al. Thalidomide for treatment of multiple myeloma: 10 years later. Blood 2008; 111:3968-77; PMID:18245666; http://dx.doi.org/10.1182/blood-2007-10-117457
- Richardson PG, Blood E, Mitsiades CS, Jagannath S, Zeldenrust SR, Alsina M, Schlossman RL, Rajkumar SV, Desikan KR, Hideshima T, et al. A randomized phase 2 study of lenalidomide therapy for patients with relapsed or relapsed and refractory multiple myeloma. Blood 2006; 108:3458-64; PMID:16840727; http://dx.doi.org/10.1182/blood-2006-04-015909
- Palumbo A, Attal M, Roussel M. Shifts in the therapeutic paradigm for patients newly diagnosed with multiple myeloma: maintenance therapy and overall survival. Clin Cancer Res 2011; 17:1253-63; PMID:21411441; http://dx.doi.org/10.1158/1078-0432.CCR-10-1925
- Balsas P, Galan-Malo P, Marzo I, Naval J. Bortezomib resistance in a myeloma cell line is associated to PSMbeta5 overexpression and polyploidy. Leuk Res 2012; 36:212-8; PMID:21978467; http://dx.doi.org/10.1016/j.leukres.2011.09.011
- Kapoor P, Ramakrishnan V, Rajkumar SV. Bortezomib combination therapy in multiple myeloma. Semin Hematol 2012; 49:228-42; PMID:22726546; http://dx.doi.org/10.1053/j.seminhematol.2012.04.010
- Lonial S, Mitsiades CS, Richardson PG. Treatment options for relapsed and refractory multiple myeloma. Clin Cancer Res 2011; 17:1264-77; PMID:21411442; http://dx.doi.org/10.1158/1078-0432.CCR-10-1805
- Hu J, Handisides DR, Van Valckenborgh E, De Raeve H, Menu E, Vande Broek I, Liu Q, Sun JD, Van Camp B, Hart CP, et al. Targeting the multiple myeloma hypoxic niche with TH-302, a hypoxia-activated prodrug. Blood 2010; 116:1524-7; PMID:20530289; http://dx.doi.org/10.1182/blood-2010-02-269126
- Muz B, de la Puente P, Azab F, Luderer M, Azab AK. Hypoxia promotes stem cell-like phenotype in multiple myeloma cells. Blood Cancer J 2014; 4:e262; PMID:25479569; http://dx.doi.org/10.1038/bcj.2014.82
- Hu J, Van Valckenborgh E, Xu D, Menu E, De Raeve H, De Bryune E, Xu S, Van Camp B, Handisides D, Hart CP, et al. Synergistic induction of apoptosis in multiple myeloma cells by bortezomib and hypoxia-activated prodrug TH-302, in vivo and in vitro. Mol Cancer Ther 2013; 12:1763-73; PMID:23832122; http://dx.doi.org/10.1158/1535-7163.MCT-13-0123
- Kuhn DJ, Berkova Z, Jones RJ, Woessner R, Bjorklund CC, Ma W, Davis RE, Lin P, Wang H, Madden TL, et al. Targeting the insulin-like growth factor-1 receptor to overcome bortezomib resistance in preclinical models of multiple myeloma. Blood 2012; 120:3260-70; PMID:22932796; http://dx.doi.org/10.1182/blood-2011-10-386789
- Annunziata CM, Davis RE, Demchenko Y, Bellamy W, Gabrea A, Zhan F, Lenz G, Hanamura I, Wright G, Xiao W, et al. Frequent engagement of the classical and alternative NF-kappaB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell 2007; 12:115-30; PMID:17692804; http://dx.doi.org/10.1016/j.ccr.2007.07.004
- Murray MY, Zaitseva L, Auger MJ, Craig JI, MacEwan DJ, Rushworth SA, Bowles KM. Ibrutinib inhibits BTK-driven NF-kappaB p65 activity to overcome bortezomib-resistance in multiple myeloma. Cell Cycle 2015; 14:2367-75; PMID:25565020; http://dx.doi.org/10.1080/15384101.2014.998067
- Raninga PV, Di Trapani G, Vuckovic S, Bhatia M, Tonissen KF. Inhibition of thioredoxin 1 leads to apoptosis in drug-resistant multiple myeloma. Oncotarget 2015; 6:15410-24; PMID:25945832; http://dx.doi.org/10.18632/oncotarget.3795
- Nerini-Molteni S, Ferrarini M, Cozza S, Caligaris-Cappio F, Sitia R. Redox homeostasis modulates the sensitivity of myeloma cells to bortezomib. Br J Haematol 2008; 141:494-503; PMID:18341633; http://dx.doi.org/10.1111/j.1365-2141.2008.07066.x
- Chapman MA, Lawrence MS, Keats JJ, Cibulskis K, Sougnez C, Schinzel AC, Harview CL, Brunet JP, Ahmann GJ, Adli M, et al. Initial genome sequencing and analysis of multiple myeloma. Nature 2011; 471:467-72; PMID:21430775; http://dx.doi.org/10.1038/nature09837
- Lu S, Wang J. The resistance mechanisms of proteasome inhibitor bortezomib. Biomark Res 2013; 1:13; PMID:24252210; http://dx.doi.org/10.1186/2050-7771-1-13
- Markovina S, Callander NS, O'Connor SL, Kim J, Werndli JE, Raschko M, Leith CP, Kahl BS, Kim K, Miyamoto S. Bortezomib-resistant nuclear factor-kappaB activity in multiple myeloma cells. Mol Cancer Res 2008; 6:1356-64; PMID:18708367; http://dx.doi.org/10.1158/1541-7786.MCR-08-0108
- Tsubaki M, Satou T, Itoh T, Imano M, Komai M, Nishinobo M, Yamashita M, Yanae M, Yamazoe Y, Nishida S. Overexpression of MDR1 and survivin, and decreased Bim expression mediate multidrug-resistance in multiple myeloma cells. Leuk Res 2012; 36:1315-22; PMID:22819074; http://dx.doi.org/10.1016/j.leukres.2012.07.003
- Sewify EM, Afifi OA, Mosad E, Zaki AH, El Gammal SA. Cyclin D1 amplification in multiple myeloma is associated with multidrug resistance expression. Clin Lymphoma Myeloma Leuk 2014; 14:215-22; PMID:24468132; http://dx.doi.org/10.1016/j.clml.2013.07.008
- Lincoln DT, Ali Emadi EM, Tonissen KF, Clarke FM. The thioredoxin-thioredoxin reductase system: over-expression in human cancer. Anticancer Res 2003; 23:2425-33; PMID:12894524
- Li C, Thompson MA, Tamayo AT, Zuo Z, Lee J, Vega F, Ford RJ, Pham LV. Over-expression of Thioredoxin-1 mediates growth, survival, and chemoresistance and is a druggable target in diffuse large B-cell lymphoma. Oncotarget 2012; 3:314-26; PMID:22447839; http://dx.doi.org/10.18632/oncotarget.463
- Iwasawa S, Yamano Y, Takiguchi Y, Tanzawa H, Tatsumi K, Uzawa K. Upregulation of thioredoxin reductase 1 in human oral squamous cell carcinoma. Oncol Rep 2011; 25:637-44; PMID:21206984
- Liu JJ, Liu Q, Wei HL, Yi J, Zhao HS, Gao LP. Inhibition of thioredoxin reductase by auranofin induces apoptosis in adriamycin-resistant human K562 chronic myeloid leukemia cells. Pharmazie 2011; 66:440-4; PMID:21699084
- Marzano C, Gandin V, Folda A, Scutari G, Bindoli A, Rigobello MP. Inhibition of thioredoxin reductase by auranofin induces apoptosis in cisplatin-resistant human ovarian cancer cells. Free Radic Biol Med 2007; 42:872-81; PMID:17320769; http://dx.doi.org/10.1016/j.freeradbiomed.2006.12.021
- Azab AK, Hu J, Quang P, Azab F, Pitsillides C, Awwad R, Thompson B, Maiso P, Sun JD, Hart CP, et al. Hypoxia promotes dissemination of multiple myeloma through acquisition of epithelial to mesenchymal transition-like features. Blood 2012; 119:5782-94; PMID:22394600; http://dx.doi.org/10.1182/blood-2011-09-380410
- Koong AC, Chen EY, Giaccia AJ. Hypoxia causes the activation of nuclear factor kappa B through the phosphorylation of I kappa B alpha on tyrosine residues. Cancer Res 1994; 54:1425-30; PMID:8137243
- Taylor CT, Cummins EP. The role of NF-kappaB in hypoxia-induced gene expression. Ann N Y Acad Sci 2009; 1177:178-84; PMID:19845620; http://dx.doi.org/10.1111/j.1749-6632.2009.05024.x
- Liang Y, Zheng T, Song R, Wang J, Yin D, Wang L, Liu H, Tian L, Fang X, Meng X, et al. Hypoxia-mediated sorafenib resistance can be overcome by EF24 through Von Hippel-Lindau tumor suppressor-dependent HIF-1alpha inhibition in hepatocellular carcinoma. Hepatology 2013; 57:1847-57; PMID:23299930; http://dx.doi.org/10.1002/hep.26224
- Chen X, Shi X, Wang X, Liu J. Novel use of old drug: Anti-rheumatic agent auranofin overcomes imatinib-resistance of chronic myeloid leukemia cells. Cancer Cell Microenviron 2014; e415; PMID:25995993; http://dx.doi.org/10.14800/ccm.415
- Chen ZJ, Parent L, Maniatis T. Site-specific phosphorylation of IkappaBalpha by a novel ubiquitination-dependent protein kinase activity. Cell 1996; 84:853-62; PMID:8601309; http://dx.doi.org/10.1016/S0092-8674(00)81064-8
- Twombly R. First proteasome inhibitor approved for multiple myeloma. J Natl Cancer Inst 2003; 95:845; PMID:12813164; http://dx.doi.org/10.1093/jnci/95.12.845
- Murray MY, Auger MJ, Bowles KM. Overcoming bortezomib resistance in multiple myeloma. Biochem Soc Trans 2014; 42:804-8; PMID:25109961; http://dx.doi.org/10.1042/BST20140126
- Petrucci MT, Giraldo P, Corradini P, Teixeira A, Dimopoulos MA, Blau IW, Drach J, Angermund R, Allietta N, Broer E, et al. A prospective, international phase 2 study of bortezomib retreatment in patients with relapsed multiple myeloma. Br J Haematol 2013; 160:649-59; PMID:23293914; http://dx.doi.org/10.1111/bjh.12198
- Salem K, McCormick ML, Wendlandt E, Zhan F, Goel A. Copper-zinc superoxide dismutase-mediated redox regulation of bortezomib resistance in multiple myeloma. Redox Biol 2015; 4:23-33; PMID:25485927; http://dx.doi.org/10.1016/j.redox.2014.11.002
- Kim SJ, Miyoshi Y, Taguchi T, Tamaki Y, Nakamura H, Yodoi J, Kato K, Noguchi S. High thioredoxin expression is associated with resistance to docetaxel in primary breast cancer. Clin Cancer Res 2005; 11:8425-30; PMID:16322305; http://dx.doi.org/10.1158/1078-0432.CCR-05-0449
- Giuntoli S, Rovida E, Barbetti V, Cipolleschi MG, Olivotto M, Dello Sbarba P. Hypoxia suppresses BCR/Abl and selects imatinib-insensitive progenitors within clonal CML populations. Leukemia 2006; 20:1291-3; PMID:16710305; http://dx.doi.org/10.1038/sj.leu.2404224
- Damiano JS, Hazlehurst LA, Dalton WS. Cell adhesion-mediated drug resistance (CAM-DR) protects the K562 chronic myelogenous leukemia cell line from apoptosis induced by BCR/ABL inhibition, cytotoxic drugs, and gamma-irradiation. Leukemia 2001; 15:1232-9; PMID:11480565; http://dx.doi.org/10.1038/sj.leu.2402179
- Kawano Y, Kikukawa Y, Fujiwara S, Wada N, Okuno Y, Mitsuya H, Hata H. Hypoxia reduces CD138 expression and induces an immature and stem cell-like transcriptional program in myeloma cells. Int J Oncol 2013; 43:1809-16; PMID:24126540
- Wang X, Stafford W, Mazurkiewicz M, Fryknas M, Brjnic S, Zhang X, Gullbo J, Larsson R, Arner ES, D'Arcy P, et al. The 19S Deubiquitinase inhibitor b-AP15 is enriched in cells and elicits rapid commitment to cell death. Mol Pharmacol 2014; 85:932-45; PMID:24714215; http://dx.doi.org/10.1124/mol.113.091322
- Landriscina M, Maddalena F, Laudiero G, Esposito F. Adaptation to oxidative stress, chemoresistance, and cell survival. Antioxid Redox Signal 2009; 11:2701-16; PMID:19778285; http://dx.doi.org/10.1089/ars.2009.2692
- Samulitis BK, Landowski TH, Dorr RT. Correlates of imexon sensitivity in human multiple myeloma cell lines. Leuk Lymphoma 2006; 47:97-109; PMID:16321833; http://dx.doi.org/10.1080/10428190500266210
- Nordberg J, Arner ES. Reactive oxygen species, antioxidants, and the mammalian thioredoxin system. Free Radic Biol Med 2001; 31:1287-312; PMID:11728801; http://dx.doi.org/10.1016/S0891-5849(01)00724-9
- Kim NH, Oh MK, Park HJ, Kim IS. Auranofin, a gold(I)-containing antirheumatic compound, activates Keap1/Nrf2 signaling via Rac1/iNOS signal and mitogen-activated protein kinase activation. J Pharmacol Sci 2010; 113:246-54; PMID:20647686; http://dx.doi.org/10.1254/jphs.09330FP
- Cebula M, Schmidt EE, Arner ES. TrxR1 as a Potent Regulator of the Nrf2-Keap1 Response System. Antioxid Redox Signal 2015; 23:823-53; PMID:26058897; http://dx.doi.org/10.1089/ars.2015.6378
- Li W, Khor TO, Xu C, Shen G, Jeong WS, Yu S, Kong AN. Activation of Nrf2-antioxidant signaling attenuates NFkappaB-inflammatory response and elicits apoptosis. Biochem Pharmacol 2008; 76:1485-9; PMID:18694732; http://dx.doi.org/10.1016/j.bcp.2008.07.017
- Thimmulappa RK, Lee H, Rangasamy T, Reddy SP, Yamamoto M, Kensler TW, Biswal S. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J Clin Invest 2006; 116:984-95; PMID:16585964; http://dx.doi.org/10.1172/JCI25790
- Matsuoka Y, Moore GE, Yagi Y, Pressman D. Production of free light chains of immunoglobulin by a hematopoietic cell line derived from a patient with multiple myeloma. Proc Soc Exp Biol Med 1967; 125:1246-50; PMID:6042436; http://dx.doi.org/10.3181/00379727-125-32327
- Nilsson K, Bennich H, Johansson SG, Ponten J. Established immunoglobulin producing myeloma (IgE) and lymphoblastoid (IgG) cell lines from an IgE myeloma patient. Clin Exp Immunol 1970; 7:477-89; PMID:4097745
- Smith AD, Levander OA. High-throughput 96-well microplate assays for determining specific activities of glutathione peroxidase and thioredoxin reductase. Methods Enzymol 2002; 347:113-21; PMID:11898400; http://dx.doi.org/10.1016/S0076-6879(02)47012-7
- Karlenius TC, Shah F, Di Trapani G, Clarke FM, Tonissen KF. Cycling hypoxia up-regulates thioredoxin levels in human MDA-MB-231 breast cancer cells. Biochem Biophys Res Commun 2012; 419:350-5; PMID:22342720; http://dx.doi.org/10.1016/j.bbrc.2012.02.027
- Lafleur MA, Drew AF, de Sousa EL, Blick T, Bills M, Walker EC, Williams ED, Waltham M, Thompson EW. Upregulation of matrix metalloproteinases (MMPs) in breast cancer xenografts: a major induction of stromal MMP-13. Int J Cancer 2005; 114:544-54; PMID:15551360; http://dx.doi.org/10.1002/ijc.20763