
Abstract
Both RNF4 and KAP1 play critical roles in the response to DNA double-strand breaks (DSBs), but the functional interplay of RNF4 and KAP1 in regulating DNA damage response remains unclear. We have previously demonstrated the recruitment and degradation of KAP1 by RNF4 require the phosphorylation of Ser824 (pS824) and SUMOylation of KAP1. In this report, we show the retention of DSB-induced pS824-KAP1 foci and RNF4 abundance are inversely correlated as cell cycle progresses. Following irradiation, pS824-KAP1 foci predominantly appear in the cyclin A (-) cells, whereas RNF4 level is suppressed in the G0-/G1-phases and then accumulates during S-/G2-phases. Notably, 53BP1 foci, but not BRCA1 foci, co-exist with pS824-KAP1 foci. Depletion of KAP1 yields opposite effect on the dynamics of 53BP1 and BRCA1 loading, favoring homologous recombination repair. In addition, we identify p97 is present in the RNF4-KAP1 interacting complex and the inhibition of p97 renders MCF7 breast cancer cells relatively more sensitive to DNA damage. Collectively, these findings suggest that combined effect of dynamic recruitment of RNF4 to KAP1 regulates the relative occupancy of 53BP1 and BRCA1 at DSB sites to direct DSB repair in a cell cycle-dependent manner.
Introduction
One essential aspect of the DNA damage response (DDR) is to choose the proper repair pathway. There are 2 prominent pathways: non-homologous end joining (NHEJ) or homologous recombination (HR) to repair DNA double-strand breaks (DSBs). The choice between these 2 pathways is, at least in part, affected by the presence (or not) of a homologous template at different stages of cell cycle and the complexity of chromatin. In brief, NHEJ is mediated by 53BP1 and occurs primarily in the G1-phase, whereas BRCA1 facilitates HR repair during the S-/G2-phases.Citation1,2 Emerging evidence indicates that post-translational modifications (PTMs) of many DSB repair proteins dynamically modulate their recruitment to DSB sites or interactions with other DSB repair proteins (reviewed inCitation3-5 and references therein). However, the cell cycle-dependent crosstalk between DDR signaling cascades and their corresponding underlying mechanisms that regulate the choice of DSB repair pathways during cell cycle transition remain unclear.
SUMO-targeted ubiquitin E3 ligases (STUbLs), for example, RING finger protein 4 (RNF4), contain SUMO-interacting motifs (SIMs) that interact with the SUMO or SUMO-like domains of its ubiquitylation targets.Citation6 Published reports have contributed extensively to our understanding of how RNF4 recruits substrates in response to DSBs and its role in connecting the SUMO and ubiquitin pathways.Citation7-13 However, it is still unclear whether RNF4, or other STUbLs, target distinct substrate(s) for timely cell cycle-dependent degradation during DDR and their impact on genome stability. Recently, RNF4 has been reported to prevent replication re-initiation at the collapsed replication fork and to remove Faconi anemia (FA) protein complexes during DNA interstrand crosslink repair,Citation14,15 suggesting that RNF4 plays critical roles in the cells undergoing DNA replication. Another study demonstrates that the E3 ligase activity of RNF4 is enhanced by cyclin-dependent kinase 2 (CDK2)-mediated phosphorylation during S-phase of cell cycle to favor HR repair by degrading MDC1,Citation16 further attesting to the potential role of RNF4 in mediating repair pathway choice.
KRAB domain-associated protein 1 (KAP1) is a multi-functional protein and is subjected to several types of PTM.Citation17 For example, that PIKK family, including ATM, phosphorylates Ser (S)-824 of KAP1 (pS824-KAP1) is a critical event for proper DSB repair and DDR.Citation18,19 KAP1 depletion rescues NHEJ repair defects caused by ATM inhibition during G1-phase.Citation20,21 On the other hand, HR repair is restored by knocking down KAP1 in ATM-inhibited G2 cells.Citation22,23 It is thereby suggested that 53BP1 enriches pS824-KAP1 signal within heterochromatic region to enable NHEJ repair.Citation21 In contrast, a recent report suggests that 53BP1 is also required for promoting pS824-KAP1 foci at heterochromatin for HR repair in G2-phase.Citation24 Altogether, results from these studies imply that KAP1 may engage in distinct DSB repair pathways depending on the stage of cell cycle. Intriguingly, the chromatin retention of KAP1 has been found to be largely reduced in G2-phase,Citation22,25 suggesting the association of KAP1 with chromatin is possibly regulated in a cell cycle-dependent manner and it may be a critical determinant affecting DSB repair pathway choice.
While much is learned about the recruitment and assembly of DDR proteins at DSB sites, less is known on the events affecting the dynamics or limiting DDR-protein complexes or foci in different phases of cell cycle. Given the short half-life of DSB-induced pS824-KAP1 species and its critical role in chromatin relaxation, we therefore investigated how pS824-KAP1 level is regulated during the course of cell cycle-dependent DSB repair and explored its underlying mechanism(s). Our previous studies have shown that RNF4 engages an active process to accelerate the turnover of S824-phosphoylated, SUMOylated KAP1 through an arginine-rich motif (ARM). Here, we report that the RNF4 expression is cell cycle-regulated at the post-translational level, and inversely correlates with the magnitude and/or duration of pS824-KAP1 foci. During the G1-phase, RNF4 is subjected to proteasome-mediated degradation and pS824-KAP1 foci are prominent to block HR repair and to promote NHEJ repair. When cells progress to S-/G2-phases, RNF4 accumulates, in cooperation with p97 segregase, to degrade pS824-KAP1 in order to relieve the brake on HR repair. Our results suggest that the RNF4 level sets a threshold for the preferential recruitment of RNF4 to KAP1 and contributes, at least in part, to the DSB repair pathway choice during cell cycle progression.
Results
The appearance of pS824-KAP1 foci and the abundance of RNF4 are inversely correlated during the course of cell cycle progression
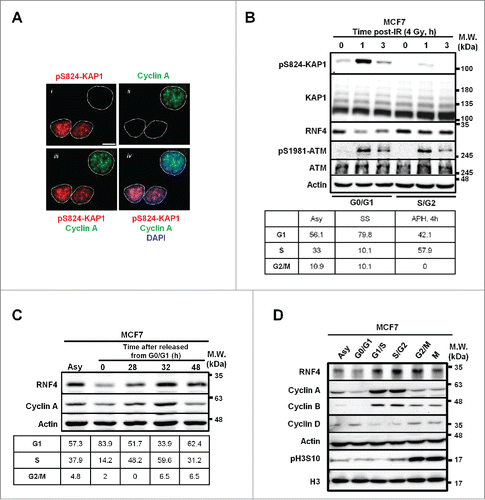
Our immunofluorescent (IF) imaging studies of pS824-KAP1 foci revealed that not all MCF7 breast cancer cells exhibit discrete pS824-KAP1 foci at 1-h post-irradiation (IR, 4 Gy) (, panel i). To examine whether the retention of pS824-KAP1 foci is cell cycle-regulated, co-staining of cyclin A, a known marker for S-/G2-phase,Citation26 was performed (, panel ii). unexpectedly shows that pS824-KAP1 foci mainly appeared in the cyclin A (-) cells (panels iii and iv). Next, to validate the results from the imaging studies, MCF7 cells, maintained in the phenol red-free culture media, were synchronized by serum starvation (24-h), resulting in 80% of cells were at the boundary of G0-/G1-phases. In a separate experiment, MCF7 cells were synchronized to the boundary of G1-/S-phases by the treatment of aphidicolin (APH) for 24-h and then released in the complete media for additional 4-h, resulting in about 57% of cells reached S/G2 boundary (, lower panel). These individually G0-/G1- and S-/G2-synchronous cells were irradiated at 4 Gy and pS824-KAP1 signal was found to be enriched predominantly at 1-h post-IR and decreased at 3-h post-IR in the G0-/G1-cells. We could still observe a minor induction of pS824-KAP1 at 1-h post-IR in the S-/G2-synchronous cells because there were still nearly 40% of cells remaining in G1-phase. Interestingly, a reduced RNF4 level was observed in the G0-/G1-cells, while ATM level and pS1981-ATM signal remained largely comparable. (, upper panel). To investigate whether the RNF4 abundance changed during cell cycle progression, we synchronized and released the cells from G0-/G1-phases to assess RNF4 level. As shown in , RNF4 expression was not static during cell cycle progression and was relatively in parallel with cyclin A expression. RNF4 was reduced in G0-/G1-phases, compared with asynchronous cells, and then increased as the cells progressed to S-phase, peaked at G2-phase and decreased again when the cells re-entered to G0-/G1-phases. This notion was validated by observations that when cells were individually synchronized to different stages of cell cycle, RNF4 expression also changed accordingly (). In addition, we observed this phenomenon not only in MCF7 cells, but also in MDA-MB-231 cells, another breast cancer cell line (Fig. S1A). However, the change of RNF4 abundance during cell cycle progression was less prominent in the immortalized human fibroblasts (Fig. S1B). Taken together, these results revealed an inverse correlation between the appearance of pS824-KAP1 signal and RNF4 abundance at different stages of cell cycle.
Figure 1. Inverse correlation between pS824-KAP1 foci and RNF4 abundance. (A) pS824-KAP1 foci are mainly present in cyclin A (-) cells. Co-staining of pS824-KAP1 and cyclin A in cycling MCF7 cells at 1-h post-IR (4 Gy). The foci were visualized with immunofluorescence (IF) microscopy using indicated antibodies and DAPI. Scale bar: 10 μm. (B) pS824-KAP1 is abundantly present in G0-/G1-phases, but not in S-/G2-phases. MCF7 cells were synchronized and then irradiated as indicated. Upper panel: Western blot analyses were performed to assess the abundance of indicated proteins. Lower panel: cell cycle analyses of synchronous cells. Numbers represent the percentage of cells in each cell cycle phase. Asy: asynchronous; SS: serum starvation; APH, 4h: release from aphidicolin treatment for 4-h. (C) RNF4 abundance is regulated during cell cycle progression. MCF7 cells were synchronized by serum starvation and then released as indicated. Upper panel: RNF4 abundance was assessed by Western blot analysis. Lower panel: cell cycle analyses of synchronous cells. Numbers represent the percentage of cells in each cell cycle phase. (D) Dynamic level of RNF4 at different stages of cell cycle. MCF7 cells were synchronized at different stages of cell cycle as indicated. Western blot analyses were used to assess the abundance of the indicated proteins. (B-D) N = 3, representative blots were shown.
RNF4 is down-regulated in G0-/G1-phases of cell cycle by proteasomal degradation
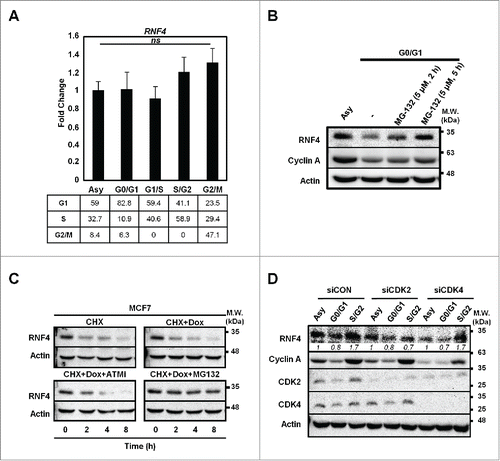
To understand whether the change of RNF4 abundance during cell cycle progression is operated at transcriptional or translational level, quantitative RT-PCR (qRT-PCR) analyses were performed to assess RNF4 mRNA levels in the respective synchronous cells. shows that the cell cycle progression did not affect RNF4 message levels. Next, pre-treatment of G0-/G1-cells with a proteasome inhibitor, MG132, was able to raise the RNF4 level (), implicating the involvement of proteasomal degradation in suppressing RNF4 expression in G0-/G1-cells. Moreover, we examined whether DNA damage signal affected the stability of RNF4. By treating the cells with cycloheximide (CHX) to inhibit de novo protein biosynthesis, a rapid turnover of RNF4 (half-life <2-h) was noted (). However, the turnover rate of RNF4 was not affected by a DSB-inducing agent, doxorubicin (Dox) and the decrease of RNF4 was restored by MG132, but not by ATM inhibitor (). Lastly, because CDK2 has been reported to regulate RNF4 function in S-phase,Citation16 we knocked down CDK2 or CDK4 to evaluate whether RNF4 abundance is controlled by the key kinases driving cell cycle progression. As shown in , knockdown of CDK2 prevented RNF4 from accumulation in S-/G2-phases, and knockdown CDK4 did not affect the dynamics of RNF4 abundance in different cell cycle phases. Taken together, these data suggest that the RNF4 expression during cell cycle progression was regulated, at least in part, by proteasome-mediated protein degradation and CDK2.
Figure 2. Cell cycle-dependent regulation of RNF4. (A) Static RNF4 mRNA level during cell cycle progression. Upper panel: RNF4 message levels were assessed by quantitative RT-PCR (qRT-PCR) in MCF7 cells synchronized at different stages of cell cycle. Bars: mean ± SD, N = 3; ns: not significant (one-way ANOVA). Lower panel: cell cycle distribution in synchronous MCF7 cells. Numbers represent the percentage of cells in each cell cycle phase. (B) RNF4 is stabilized by MG132 upon serum starvation. Serum-starved MCF7 cells were pre-treated with MG132 for the indicated time periods prior to harvesting. RNF4 levels were detected by Western blot analyses. (C) The stability of RNF4 is not affected by DNA damage. MCF7 cells treated with cycloheximide (CHX) in the presence of doxorubicin (Dox), ATM inhibitor (ATMi) Ku55933, or proteasome inhibitor MG132 were assessed for RNF4 protein abundance by Western blot analyses. (D) Knocking down CDK2 prevents the accumulation of RNF4 in S-/G2-phases. MCF7 cells were transfected with control siRNA (siCON), siCDK2, siCDK4, followed by synchronization at different cell cycle stages. RNF4 protein abundance was assessed by Western blot analyses. Italic numbers represent the quantification of RNF4 protein levels. (B, C) N = 3, representative blots were shown.
RNF4 co-opts with p97 to suppress pS824-KAP1 foci formation
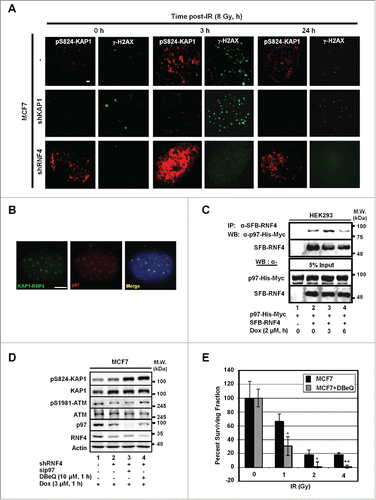
Previously, we have shown that RNF4 targets phospho-SUMO-KAP1 for degradation, and the depletion of RNF4 results in the accumulation of pS824-KAP1 signal.Citation13 To understand whether RNF4 regulates DNA repair by targeting KAP1 for degradation in response to DNA damage, IF image analyses were performed. As shown in , the steady-state level of pS824-KAP1 foci was higher in the MCF7 cells harboring a short hairpin (sh)RNA targeting RNF4 (MCF7/shRNF4) than that in MCF7 cells. To further characterize the co-localization of pS824-KAP1 and γ-H2AX foci, we engineered HEK293 cells to overexpress an RNF4 mutant that lacked the E3 ligase domain (N'-SIM-ARM). This mutation caused the enlargement of both pS824-KAP1 and γ-H2AX foci, and it was more evident that the pS824-KAP1 and γ-H2AX foci co-localized in these cells (Fig. S2A). Together with our previous report,Citation13 we postulated that RNF4 is responsible for removing pS824-KAP1 signal from the damage sites. Next, bimolecular fluorescence complementation (BiFC) assay, a cell-based protein-protein interaction assay that we have previously used to examine DNA damage-induced RNF4-KAP1 interaction was performed.Citation13 We found that p97, an AAA-type ATPase, was identified to be co-localized with the RNF4-KAP1 foci which were resulted from the interaction between RNF4 and KAP1 due to the complementation of 2 fragmented green fluorescent molecules fused to RNF4 and KAP1 (). This finding was in consistence with a recent report showing that RNF4 works with DVC1-p97 complex to extract FA complex from chromatin.Citation14 Next, co-immunoprecipitation assays were performed to confirm the interaction between RNF4 and p97. As shown in , RNF4 interacted with p97 and the interaction increased at 3-h, and then decreased at 6-h post Dox-treatment. Notably, the abundance of p97 was stable throughout cell cycle progression (Fig. S2B), indicating that the change of RNF4 abundance is the main determinant for their dynamic interaction. In addition, pS824-KAP1 signal further accumulated in MCF7 cells depleted with both RNF4 and p97, compared with that in MCF7/shRNF4 cells (). Similarly, treatment with DBeQ, a small molecule p97 inhibitor,Citation27 in MCF7/shRNF4 cells caused an accumulation of pS824-KAP1 signal (). Lastly, pharmacological or genetic inhibition of p97 by DBeQ or sip97, respectively, was able to sensitize MCF7 cells to IR ( and Fig. S2C). Altogether, we concluded that RNF4 cooperates with p97 to target pS824-KAP1 for degradation to facilitate DDR.
Figure 3. p97 partakes in the regulation of pS824-KAP1. (A) Time-dependent formation and degradation of pS824-KAP1 foci. The pS824-KAP1 foci were examined in irradiated MCF7, MCF7/shKAP1 and MCF7/shRNF4 cells (8 Gy). Cells were fixed at the indicated time points post-IR and pS824-KAP1 and γ-H2AX foci visualized by IF staining. (B) p97 is co-localized to where KAP1 and RNF4 interact. KAP1 (625-835)-VN173 and RNF4 (WT)-CC155 were co-transfected into U2OS cells. After 48-h, cells were pre-treated with MG132 (5 μM) for 4-h prior to IR exposure. Cells were fixed at 1-h following irradiation (4 Gy). Green: KAP1-RNF4 interaction; Red: p97. Scale bar: 5 µm. (C) Interaction of RNF4 with p97. HEK293 cells were co-transfected with SFB-RNF4 and p97-His-Myc followed by treating with Dox (3 μM) for the indicated time periods. Cell lysates were incubated with S-beads to pull down SFB-RNF4-associated complex followed by Western blot analysis. p97-His-Myc was detected by anti-Myc antibody. 5% input control was blotted with the indicated antibodies. (D) pS824-KAP1 signal was further enhanced in MCF7/shRNF4 cells treated with DBeQ or sip97. Cells were transfected with siRNA against p97 or treated with DBeQ (10 μM) in the presence of vehicle or Dox (3 μM). pS824-KAP1, total KAP1, pS1981-ATM, total ATM were assessed by blotting with the corresponding antibodies. β-actin served as loading control. (E) p97 inhibitor sensitizes MCF7 cells to IR. MCF7 cells were treated with DBeQ (200 nM) and increasing dose of IR. Colonies were fixed and stained with crystal violet at 12-days after treatment. Surviving fraction was calculated with a correction for the plating efficiency. Bars: mean ± SD; *: p < 0.02.
DNA damage-induced pS824-KAP1 and BRCA1 foci are mutually exclusive
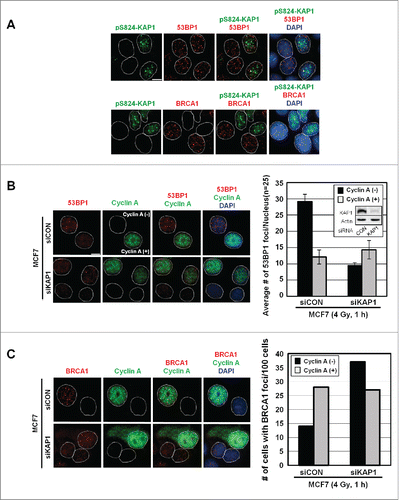
Given that the appearance of pS824-KAP1 foci was cell cycle-dependent, we determined whether pS824-KAP1 foci commensurate with the foci of other known DSB repair markers, such as 53BP1 and BRCA1. At 1-h post-IR, pS824-KAP1 foci were visualized in cells exhibiting 53BP1 foci (, upper panel), but not present in the cells that had BRCA1 foci (, lower panel). Based on mutual exclusion of pS824-KAP1 and BRCA1 foci, we hypothesized that pS824-KAP1 regulated the occupancy of 53BP1 or BRCA1 at DSB sites. To test this possibility, a small interfering RNA (siKAP1) was utilized to knock down KAP1 in MCF7 cells (, inset). Clearly, KAP1 knockdown resulted in a marked decrease in the number of 53BP1 foci in cyclin A (-) cells, whereas the number 53BP1 foci remained largely unaffected in cyclin A (+) cells (). In contrast, more BRCA1 foci were noted in with KAP1 depleted, cyclin A (-) cells (). Conceivably, pS824-KAP1 likely prevents BRCA1 accumulation, but promotes 53BP1 loading at DSB sites during G1-phase.
Figure 4. Irradiation induces the co-appearance of pS824-KAP1 foci with 53BP1, but not BRCA1 foci. (A) Lack of co-appearance of DSB-induced pS824-KAP1 and BRCA1 foci in MCF7 cells. Cycling MCF7 cells were irradiated (4 Gy). At 1-h post-IR, cells were fixed and immunostained with anti-pS824-KAP1, anti-53BP1 or anti-BRCA1 antibodies to detect foci. Scale bar: 10 μm. (B) Knockdown of KAP1 impairs the recruitment of 53BP1 to DSB sites in cyclin A (-) cells. Co-staining of 53BP1 and cyclin A in MCF7 cells transfected with a control siRNA or an siRNA that targeted KAP1. Knockdown efficiency of siRNA targeting KAP1 is validated by Western blot (inset). Following IR (4 Gy), cells were allowed to recover for 1-h. At least 30 cells were analyzed for 53BP1 foci and the corresponding level of cyclin A (right panel, bars: mean ± SD). (C) Depletion of KAP1 enhances BRCA1 recruitment to DSB sites in cyclin A (-) cells. Co-staining of BRCA1 and cyclin A in MCF7 cells transfected with control siRNA or an siRNA that targeted KAP1. Following IR (4 Gy), cells were allowed to recover for 1-h, and then 100 cells were analyzed for BRCA1 foci and the corresponding level of cyclin A.
KAP1 and RNF4 have opposite effect on homologous recombination DSB repair
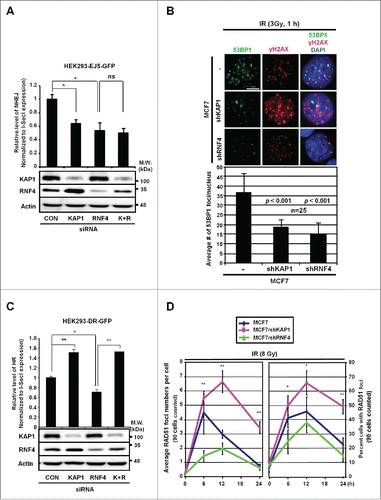
Based on the co-existence of pS824-KAP1 foci with 53BP1 but not BRCA1 foci upon DSB induction (), we speculated that RNF4 or KAP1 might affect how DSBs are repaired. To examine this possibility, we measured the repair of DSBs in HEK293 cells that were specifically generated by an endonuclease, I-SceI.Citation28 Using an integrated EJ5-GFP reporter, we showed that NHEJ repair was impaired in KAP1 and RNF4-knockdown cells (). This finding is supported by our observation that knocking down either KAP1 or RNF4 in MCF7 cells caused a 50% decrease in the number of 53BP1 foci (). To determine their respective effect on HR repair, we used an integrated DR-GFP reporter. While knocking down RNF4 clearly impaired HR, knocking down KAP1 boosted HR (). Moreover, the HR frequency was rescued when RNF4 and KAP1 were both depleted, compared with RNF4-knockdown cells (), suggesting that RNF4 and KAP1 are in the same axis to regulate HR. To further clarify the role of KAP1 and RNF4 in regulating HR repair process, we measured DNA end-resection, the first step of recombination in HR repair,Citation29 by quantifying camptothecin (CPT)-induced RPA2 signal as RPA2 and the other 2 RPA subunits form complex to cover the resected, single-stranded DNA (ssDNA).Citation30 As shown in Fig. S3A, CPT has been shown to induce RPA2 signal in S-phase cells.Citation30 Knockdown of CtIP, a key molecule promoting DNA-end resection,Citation31 significantly reduced the percentage of RPA-positive cells in S-phase upon CPT treatment. However, knockdown of KAP1 or RNF4 did not affect CPT-induced DNA-end resection. Next, we quantified the signal of RAD51, which is essential for strand exchange during HR,Citation32 in MCF7 cells after IR exposure. It has been shown that more RAD51 foci appear after exposure to higher dosage of IRCitation33 and more RAD51 foci co-localized to γ-H2AX at a later timepoint post-IR.Citation34 Therefore, we used a higher dosage of IR (8 Gy) to trace RAD51 foci for 6-, 12-, and 24-h. Consistent with the results using the DR-GFP reporter, in MCF7/shKAP1 cells, there was a time-dependent increase in the formation of RAD51 foci, compared to MCF7 cells. In contrast, MCF7/shRNF4 cells had a significant decrease in the number of RAD51 foci, compared to MCF7/shKAP1 cells ( and Fig. S3B). In summary, we conclude that RNF4 is required for both HR and NHEJ, consistent with the report that RNF4-deficiency causes persistent IR-induced DNA damage and signals.Citation7,12
Figure 5. RNF4 and KAP1 have opposite effects on HR repair. (A) Both KAP1 and RNF4 contribute to NHEJ repair. HEK293 cells were transfected with control siRNA, siKAP1 or siRNF4 or both siKAP1 and siRNF4. The knockdown efficiency of KAP1 and RNF4, and the transfection efficiency of pCBASce were confirmed by Western blot. *: p < 0.005; ns: not significant. The relative frequencies of NHEJ repair of EJ5-GFP, which results in GFP-positive cells, are shown for each transfection. (B) KAP1 or RNF4-depletion reduces 53BP1 foci in MCF7 cells. MCF7, MCF7/shKAP1 and MCF7/shRNF4 cells were fixed and stained for 53BP1 after exposure to IR (3 Gy) and allowed to recover for 1-h. Enumeration of 53BP1 foci was done in 25 nuclei for each cell type. The average number of 53BP1 foci per nucleus was 36.72 ± 9.49 in MCF7 cells, 18.40 ± 3.85 in MCF7/shKAP1 cells (p < 0.001), and 15.28 ± 5.31 in MCF7/shRNF4 cells (p < 0.001). (C) Knockdown of KAP1 enhances HR repair. The relative frequencies of the repair of DR-GFP by HR, which results in GFP-positive cells, in HEK293 cells transfected with control siRNA, siKAP1, siRNF4 or both are shown. *: p < 0.005; **: p < 0.001. (D) KAP1 or RNF4-depletion has opposite effects on RAD51 recruitment. Cells were fixed at the indicated times post-IR. The number of RAD51 foci per cell and percent cells with RAD51 foci out of 90 cells in each cell line is shown for each time point. *: p < 0.05; **: p < 0.01. (A-D) Bars: mean ± SD.
Discussion
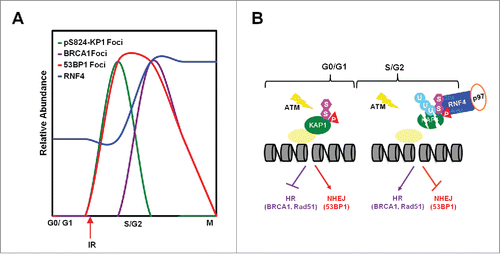
The findings presented here indicate that in response to genotoxic stress, the appearance of pS824-KAP1 foci and the abundance of RNF4 are cell cycle-regulated. pS824-KAP1 foci specifically appear in G0-/G1-phases, in which RNF4 abundance is lower. BRCA1 foci are exclusively present in S-/G2-phases while RNF4 abundance is higher and no pS824-KAP1 foci exist (). Mechanistically, cell cycle-regulated RNF4 works with p97 to target pS824-SUMO-KAP1 for degradation during S-/G2-phases to promote the recruitment of BRCA1/RAD51 to DSB sites for HR repair (). Our findings thereby suggesting that pS824-KAP1 plays a role in G0-/G1-phases to promote NHEJ repair and to block HR repair, presumably by inhibiting BRCA1 recruitment to DSB sites.
Figure 6. RNF4 regulates the choice of DNA repair pathways. (A) The relative abundance of pS8234-KAP1 foci, BRCA1 foci, 53BP1 foci and RNF4 level in different phases of cell cycle. (B) Diagram depicting cell cycle-dependent recruitment of RNF4 to pS824-SUMO-KAP1 regulates the accumulation of 53BP1 and BRCA1 foci at DSB sites, hence the choice of DNA repair pathways.
NHEJ and HR are 2 main pathways to repair DSBs in human cells. It is believed that while BRCA1 favors HR repair, 53BP1 inhibits end resection and engages NHEJ pathway for DSB repair. Given that RNF4 targets pS824-SUMO-KAP1 for degradation,Citation13 indeed, it is conceivable that the degradation of KAP1 by RNF4 represents one of the means by which to balance DNA repair pathway selection. Notably, we observed that knockdown of KAP1 did not affect RPA2 loading to the resected ssDNA, but increased BRCA1 and RAD51 recruitment, implying that KAP1 plays a role in inhibiting the loading of BRCA1 and RAD51 to DSB sites. However, the exact mechanism(s) utilized by pS824-KAP1 to inhibit BRCA1 recruitment is still unclear. One attractive possibility is that KAP1 directly interacts with 53BP1,Citation35 thus favoring 53BP1 foci formation. In turn, 53BP1 retards BRCA1 recruitment during G1-phase.Citation36-38 The second possibility is that KAP1 and BRCA1 compete for HP1. HP1 is required for KAP1 to localize to DSB sites.Citation39-41 We and others have shown that HP1 is important for the recruitment of critical HR factors, including BRCA1 and RAD51.Citation40,42,43 We surmise that KAP1 is anchored by HP1 at DSB sites during G1-phase and its degradation by RNF4 allows the recruitment of HR factors to HP1 in S-/G2-phases.
RNF4 has been reported to regulate both NHEJ and HR repair by targeting several DDR proteins.Citation7,9,10,12 It also plays a role in the DSB repair exclusively occurring during S-/G2-phases.Citation14,15,44 However, how RNF4 is timely activated to target DDR proteins in S-/G2-phases remains unknown. Recently, a study demonstrates that RNF4 is phosphorylated by an S-phase-specific kinase CDK2 and the phosphorylation enhances the ligase activity of RNF4, thereby promoting the degradation of MDC1 to facilitate HR repair.Citation16 We also observed that RNF4 accumulation in S-/G2-phases was dependent on CDK2. Therefore, we propose that RNF4 abundance is dynamic during cell cycle progression to accommodate the choice of DSB repair pathways and speculate that the cell cycle-dependent phosphorylation/de-phosphorylation might promote RNF4 accumulation during S-/G2-phases.
In this study, we also identify another player, p97, involving in the RNF4-KAP1 axis to regulate DDR. p97 participates in DSB signaling by assembling and disassembling of protein complexes near the DSB sites, leading to proper DDR to maintain genome stability.Citation45-47 Nie et al. have shown that Cdc48 (p97)-Ufd1-Npl4 interacts with SUMO proteins through the C-terminal SIM in Ufd1 in yeasts, and demonstrated that Ufd1 and Slx8, a STUbL, work together to regulate SUMO chain homeostasis.Citation48 In mammalian cells, RNF4 removes FANCI/FANCD2 complex through DVC1-p97 complex in response to replication stress.Citation14 We found that p97 was co-localized to where RNF4 and KAP1 interact and it was in the same complex with RNF4. Altogether, we propose that p97 plays a critical role in promoting the extraction of pS824-KAP1 from chromatin by RNF4 and speculate that inhibition of p97 or RNF4 may result in the retention of pS824-KAP1 on chromatin to impair proper DDR by inhibiting HR repair. One recent study shows that KAP1 chromatin retention caused by SET overexpression leads to defective DDR and the blockage of HR repair during G2-phase.Citation49 Also, the association of KAP1 with chromatin is significantly reduced during S-/G2-phases,Citation22,25 implicating that the removal pS824-KAP1 signal during S-/G2-phases is critical for allowing HR repair to process. Based on our data, we believe that cell cycle-dependent regulation of RNF4 abundance sets a threshold to determine the retention of pS824-KAP1 foci (or not), thereby regulating DSB repair choices.
In summary, our studies revealed a novel pathway, S-/G2-dependent accumulation of RNF4, to suppress pS824-KAP1 foci retention. Consistent with the published reports,Citation35,50 pS824-KAP1 promotes DSB repair using the NHEJ pathway in the G0/G1-phase in which the RNF4 expression is lower. Furthermore, pS824-KAP1 accumulation prevents the recruitment of BRCA1 to DSB repair sites, thereby inhibiting HR repair. Together, we and others indicate that RNF4 participates in both NHEJ and HR repair pathways.Citation7,9,10,12 Especially during S-/G2-phases, accumulated RNF4 targets pS824-KAP1 for degradation, which regulates, at least in part, DSB repair pathway choice. Given the prominent role of KAP1 in maintaining epigenetic stability during the transition of mouse oocytes into embryosCitation51 and in regulating lymphoid development,Citation52,53 our findings indicate that perturbing the cell cycle-dependent balance of RNF4 with pS824-KAP1 could affect a wide spectrum of biological processes beyond the DDR.
Materials and methods
Cell cultures and reagents
MCF7, U2OS and HEK293 cells were maintained (37°C, 5% CO2) in Dulbecco's modified Eagle's medium (DMEM) supplemented with fetal bovine serum (10%), penicillin (50 units/ml) and streptomycin (50 μg/ml). MCF7/shKAP1 and MCF7/shRNF4 cells were cultured in MCF7 medium with puromycin (2 μg/ml, Sigma-Aldrich P8833). Proteasome inhibitor MG132 (Calbiochem 474790) was used at 5 μM in this study. Cycloheximide (CHX, Sigma-Aldrich C4859) was used at 100 μg/ml to inhibit protein biosynthesis. p97 inhibitor, N2,N4-dibenzylquinazoline-2,4-diamine (DBeQ) was kindly provided by Dr. Frank Schoenen (University of Kansas).Citation27
Cell cycle synchronization and cell cycle analysis
Phenol red, serum-free DMEM was used to synchronize MCF7 cells to G0-/G1-boundary. Aphidicolin (3 µM, Sigma-Aldrich, A0781) was used to synchronize MCF7 cells to S-/G2-boundary and then washed off to release the cells into S-/G2-phases. Nocodazole (1 µM, Sigma-Aldrich, M1404) was used to synchronize MCF7 cells to G2-/M-boundary and then washed off to release the cells into M-phase. Cells were trypsinized, washed with cold PBS, fixed in 70% ethanol, treated with ribonuclease A (RNase A, 10 ng/ml, Sigma-Aldrich R4875) at 37°C and stained with propidium iodide (PI, 10 ng/ml, Sigma-Aldrich P4170). DNA contents were analyzed by Accuri C6 Flow Cytometer (BD Biosciences) and data were processed by Accuri C6 software (BD Biosciences). Cell cycle distribution was modeled by ModFit LT 4.0 (Verity Software House).
Cloning and construction of plasmids
SFB-RNF4, pBiFC-RNF4-CC155 and pBiFC-KAP1-VN173 were generated as previously described.Citation13 pCDNA3-p97-His-Myc was a kind gift from Dr. Raymond Deshaies (Caltech).
Western blot analysis and antibodies
SDS-PAGE and Western blot were performed as previously described.Citation13 Antibodies used on Western blots were KAP1 (Bethyl A300-274A), phospho-S824-KAP1 (Bethyl A300-767A), RNF4 (a gift of Drs. Ronald Hay and Jorma PalvimoCitation54), phospho-S1981-ATM and p97 (Abcam, ab81292 and ab109240, respectively), ATM, CDK2, CDK4 and CtIP (GeneTex GTX70103, GTX101226, GTX112842 and GTX108857, respectively), Myc, cyclin A and cyclin D1 (Santa Cruz Biotechnology sc-40, sc-751 and sc-753, respectively), cyclin B (GeneTex GTX100911), phospho-S10-H3 and β-actin (Millipore 06-570 and MAB1501, respectively), H3 (Active Motif 39763). Blots were visualized by enhanced chemiluminescence (ECL-Plus, GE Biosciences) using a Versadoc 3000 Imaging System (Bio-Rad). Densitometric tracing data were obtained and analyzed with Quantity One Software (Bio-Rad). Western analyses shown are representative of 2 to 4 independent experiments.
Bimolecular fluorescence complementation (BiFC) assay
U2OS cells transfected with pBiFC-RNF4-CC155 and pBiFC-KAP1-VN173 were treated with MG132 (5 μM) for 4-h prior to IR exposure. Cells were fixed at 1-h following irradiation (4 Gy). p97 was immunostained as described below in “Immunofluorescence staining and imaging.”
Ionizing radiation (IR)
Shepherd Mark I Cesium-137 γ irradiator was used to irradiate cells at a fixed dose rate of 1 to 2 Gy/min.
Immunofluorescence staining and imaging
The staining was done as previously described.Citation13 Antibodies used were phopsho-S824-KAP1 (Bethyl A300-767A, 1:500), γ-H2AX (Millipore 05-636, 1:500), 53BP1 (Mouse: Millipore MAB3802, 1:500; Rabbit: Santa Cruz Biotechnology sc-22760, 1:500), BRCA1 (Santa Cruz Biotechnology, Mouse: sc6954, Rabbit: sc-652, 1:200), cyclin A (Santa Cruz Biotechnology sc-271682, 1: 100), RAD51 (Santa Cruz Biotechnology sc-8349, 1:200), p97 (Abcam ab109240, 1:1000)_in incubation buffer (PBS containing 2% BSA). Cells were then washed with PBS-Tween 20 and incubated with a fluorescent secondary antibody (Alexa Fluor 488 goat anti-mouse or Alexa Fluor 568 goat anti-rabbit, Invitrogen). Slides were mounted with ProLong Gold antifade reagent with 4′,6-diamidino-2-phenylindole (DAPI) (Molecular Probe P36935), viewed using a 60X objective on an inverted IX81 microscope or a 100X objective on an AX70 fluorescence microscope. Images were collected and processed using Image-Pro 6.3 software.
Total RNA isolation, reverse transcription and quantitative PCR
Total RNA was extracted using RNeasy kit (QIAGEN) following manufacturer's instructions. 1 μg of total RNA was used for synthesizing cDNA by reverse transcription (iScript, BioRad). Quantitative PCR analysis of RNF4 mRNA was performed with iQ SYBR Green Supermix (BioRad), a fraction of each cDNA sample, and primers targeting RNF4 (forward: 5′-ATGAGTACAAGAAAGCGTCG-3′; reverse: 5′- AGTGAGGTCCACAATTTCATC-3′). PCR amplification and fluorescence detection were performed by using MyiQ real-time PCR detection system, and the threshold cycles were determined by iQ5 program (default setting). Fold induction was determined using the ΔΔCt method against 18S rRNA.
Co-immunoprecipitation (Co-IP)
Co-IP was performed as previously described.Citation13 In short, cells were lysed with NETN buffer (0.5% Nonidet P-40, 20 mM Tris-HCL (pH 8.0), 100 mM NaCl, and 1 mM EDTA) and mixed with S protein-agarose (Novagen 69704) to pull down SFB-RNF4. Eluates were analyzed on Western blots probed with anti-Myc (Santa Cruz Biotechnology sc-40) and anti-FLAG (Sigma-Aldrich F3165) antibodies.
Clonogenic survival assay
Cells were seeded in triplicate in 6-well plates at 100 cells/well in complete growth medium 1-day before treatments. Medium was changed every 3-days. After 12-day incubation, cells were fixed in ice-cold methanol for 10-mins and stained with crystal violet solution (0.2%, 10-mins). Colonies were counted and the surviving fraction was calculated using the plating efficiency.
Production of lentivirus and lentiviral transduction
Lentiviral vectors pLKO.1-shRNF4, pΔ8.7, and pVSV-G were constructed and used to produce lentivirus in HEK293FT cells, as previously described.Citation55 Viral supernatant was collected, pooled, concentrated and stored as previously described.Citation55 For lentiviral infection, MCF7 cells were plated 1-day prior to infection and cultured overnight to ˜70% confluency. The culture medium was then aspirated and fresh medium was added that contained concentrated lentiviruses with an empty vector, a short hairpin RNA (shRNA) against human KAP1, RNF4 or a random sequence (as a control). Cells and virus were incubated for 24-h in the presence of polybrene (8 µg/ml). All studies used mixed populations of transduced cells.
Plasmid and siRNA transfection
Lipofectamine 2000 (Invitrogen 11668019) was used for the plasmid DNA transfections. Control siRNA and siRNA against KAP1, RNF4, CDK2, CDK4 and CtIP were obtained from Santa Cruz Biotechnology (sc-38550, sc-38236, sc-29259, sc-29261 and sc-37765 respectively). Lipofectamine RNAiMAX (Invitrogen 13778150) was used for the siRNA transfections according to the manufacturer's protocol. The transfected cells were analyzed 48-h after transfection.
Repair assay
Repair assays were performed as previously described.Citation28 Approximately 2 × 105 HEK293/EJ5-GFP or HEK293/DR5-GFP cells were first transfected with the desired siRNA (10 nM) alone, and then co-transfected with pCBSce (1 μg) and the desired siRNA (10 nM) the next day. GFP-positive cells were quantified by flow cytometry using a Gallios Flow cytometer (Beckman) 72-h after the second transfection. The remaining cells were collected and lysed with Laemmli's sample buffer and analyzed on Western blots using antibodies against HA-I-Sce-I, KAP1 and RNF4. The percentage of GFP-positive cells was normalized to the pCBASce transfection efficiency to determine the activity of NHEJ or HR repair.
Statistical analysis
Error bars represent the standard deviation of the mean. Unpaired Student's t-tests, assuming equal variances, were performed using Microsoft Excel to determine significant differences between groups; one-way ANOVA was performed using Prism (GraphPad) when applicable; p < 0.05 is considered significant.
Abbreviations
| 53BP1 | = | p53-binding protein 1 |
| ARM | = | arginine-rich motif |
| ATM | = | ataxia telangiectasia mutated |
| BiFC | = | bi-molecular fluorescence complementation |
| BRCA1 | = | breast cancer 1 |
| CDK | = | cyclin-dependent kinase |
| CHX | = | cycloheximide |
| CtIP | = | C-terminal binding protein-interacting protein |
| CPT | = | camptothecin |
| DBeQ | = | N2,N4-dibenzylquinazoline-2,4-diamine |
| DDR | = | DNA damage response |
| Dox | = | doxorubicin |
| DSBs | = | DNA double-strand breaks |
| FA | = | Faconi anemia |
| γ- H2AX | = | phosphorylated H2A on serine 139 |
| HP1 | = | Heterochromatin protein 1 |
| HR | = | homologous recombination |
| IF | = | immunofluorescence |
| IR | = | irradiation |
| KAP1 | = | KRAB-associated protein 1 |
| KRAB | = | Krüppel-associated box |
| NHEJ | = | non-homologous end joining |
| PIKK | = | phosphatidylinositol 3-kinase-related kinase |
| pS1981-ATM | = | phosphorylated ATM on serine 1981 |
| pH3S10 | = | phosphorlated H3 on serine 10 |
| pS824-KAP1 | = | phosphorylated KAP1 on serine 824 |
| PTM | = | post-translational modification |
| RNF4 | = | RING finger protein 4 |
| SFB | = | S-tag/FLAG/Streptavidin-binding peptide |
| shRNA | = | short hairpin RNA |
| SIM | = | SUMO-interacting motifs |
| siRNA | = | small interfering RNA |
| ssDNA | = | single-strand DNA |
| STUbL | = | SUMO-targeted ubiquitin ligase |
| SUMOylation | = | small ubiquitin-like modification |
| qRT-PCR | = | quantitative reverse transcription PCR |
| RPA | = | replication protein A |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplemental Files
Download MS Word (2.3 MB)Acknowledgments
We thank Drs. Ronald Hay, Jorma Palvimo, Frank Schoenen, Raymond Deshaies and the RNAi consortium at Academia Sinica for valuable reagents and helpful advice; and members of the Ann laboratory for helpful discussions.
Funding
This work was supported in part by National Institute of Health Research Grants R01DE10742 and R01DE14183 (to DKA), R01CA120954 (to JMS), National Science Council Grants 99-2321-B-001-010, 98-2321-B-001-012 (to HMS), and P30CA33572
References
- Rothkamm K, Kruger I, Thompson LH, Lobrich M. Pathways of DNA double-strand break repair during the mammalian cell cycle. Mol Cell Biol 2003; 23:5706-15; PMID:12897142; http://dx.doi.org/10.1128/MCB.23.16.5706-5715.2003
- Chapman JR, Taylor MR, Boulton SJ. Playing the end game: DNA double-strand break repair pathway choice. Mol Cell 2012; 47:497-510; PMID:22920291; http://dx.doi.org/10.1016/j.molcel.2012.07.029
- Bekker-Jensen S, Mailand N. The ubiquitin- and SUMO-dependent signaling response to DNA double-strand breaks. FEBS Lett 2011; 585:2914-9; PMID:21664912; http://dx.doi.org/10.1016/j.febslet.2011.05.056
- Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell 2010; 40:179-204; PMID:20965415; http://dx.doi.org/10.1016/j.molcel.2010.09.019
- Polo SE, Jackson SP. Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev 2011; 25:409-33; PMID:21363960; http://dx.doi.org/10.1101/gad.2021311
- Perry JJ, Tainer JA, Boddy MN. A SIM-ultaneous role for SUMO and ubiquitin. Trends Biochem Sci 2008; 33:201-8; PMID:18403209; http://dx.doi.org/10.1016/j.tibs.2008.02.001
- Vyas R, Kumar R, Clermont F, Helfricht A, Kalev P, Sotiropoulou P, Hendriks IA, Radaelli E, Hochepied T, Blanpain C, et al. RNF4 is required for DNA double-strand break repair in vivo. Cell Death and Differ 2013; 20:490-502; PMID:23197296; http://dx.doi.org/10.1038/cdd.2012.145
- Poulsen SL, Hansen RK, Wagner SA, van Cuijk L, van Belle GJ, Streicher W, Wikstrom M, Choudhary C, Houtsmuller AB, Marteijn JA, et al. RNF111/Arkadia is a SUMO-targeted ubiquitin ligase that facilitates the DNA damage response. J Cell Biol 2013; 201:797-807; PMID:23751493; http://dx.doi.org/10.1083/jcb.201212075
- Yin Y, Seifert A, Chua JS, Maure JF, Golebiowski F, Hay RT. SUMO-targeted ubiquitin E3 ligase RNF4 is required for the response of human cells to DNA damage. Genes Dev 2012; 26:1196-208; PMID:3371408; http://dx.doi.org/10.1101/gad.189274.112
- Luo KT, Zhang HX, Wang LW, Yuan J, Lou ZK. Sumoylation of MDC1 is important for proper DNA damage response. EMBO J 2012; 31:3008-19; PMID:22635276; http://dx.doi.org/10.1038/emboj.2012.158
- Guzzo CM, Berndsen CE, Zhu J, Gupta V, Datta A, Greenberg RA, Wolberger C, Matunis MJ. RNF4-dependent hybrid SUMO-ubiquitin chains are signals for RAP80 and thereby mediate the recruitment of BRCA1 to sites of DNA damage. Sci Signal 2012; 5:ra88; PMID:23211528; http://dx.doi.org/10.1126/scisignal.2003485
- Galanty Y, Belotserkovskaya R, Coates J, Jackson SP. RNF4, a SUMO-targeted ubiquitin E3 ligase, promotes DNA double-strand break repair. Genes Dev 2012; 26:1179-95; PMID:3371407; http://dx.doi.org/10.1101/gad.188284.112
- Kuo C-Y, Li X, Kong X-Q, Luo C, Chang C-C, Chung Y, Shih H-M, Li KK, Ann DK. An arginine-rich motif of ring finger protein 4 (RNF4) oversees the recruitment and degradation of the phosphorylated and SUMOylated KAP1/TRIM28 during genotoxic stress. J Biol Chem 2014; 289(30):20757-72; PMID:24907272; http://dx.doi.org/10.1074/jbc.M114.555672
- Gibbs-Seymour I, Oka Y, Rajendra E, Weinert Brian T, Passmore Lori A, Patel Ketan J, Olsen Jesper V, Choudhary C, Bekker-Jensen S, Mailand N. Ubiquitin-SUMO circuitry controls activated fanconi anemia ID complex dosage in response to DNA damage. Mol Cell 2015; 57:150-64; PMID:25557546; http://dx.doi.org/10.1016/j.molcel.2014.12.001
- Xie J, Kim H, Moreau LA, Puhalla S, Garber J, Al Abo M, Takeda S, D'Andrea AD. RNF4-mediated polyubiquitination regulates the Fanconi anemia/BRCA pathway. J Clin Invest 2015; 125:1523-32; PMID:25751062; http://dx.doi.org/10.1172/JCI79325
- Luo K, Deng M, Li Y, Wu C, Xu Z, Yuan J, Lou Z. CDK-mediated RNF4 phosphorylation regulates homologous recombination in S-phase. Nucleic Acids Res 2015; 43:5465-75; PMID:25948581; http://dx.doi.org/10.1093/nar/gkv434
- Cheng C-T, Kuo C-Y, Ann DK. KAPtain in charge of multiple missions: Emerging roles of KAP1. World J Biol Chem 2014; 5:308-20; PMID:25225599; http://dx.doi.org/10.4331/wjbc.v5.i3.308
- White DE, Negorev D, Peng H, Ivanov AV, Maul GG, Rauscher FJ, 3rd. KAP1, a novel substrate for PIKK family members, colocalizes with numerous damage response factors at DNA lesions. Cancer Res 2006; 66:11594-9; PMID:17178852; http://dx.doi.org/10.1158/0008-5472.CAN-06-4138
- Ziv Y, Bielopolski D, Galanty Y, Lukas C, Taya Y, Schultz DC, Lukas J, Bekker-Jensen S, Bartek J, Shiloh Y. Chromatin relaxation in response to DNA double-strand breaks is modulated by a novel ATM and KAP-1 dependent pathway. Nat Cell Biol 2006; 8:870-U142; PMID:16862143; http://dx.doi.org/10.1038/ncb1446
- Goodarzi AA, Noon AT, Deckbar D, Ziv Y, Shiloh Y, Lobrich M, Jeggo PA. ATM signaling facilitates repair of DNA double-strand breaks associated with heterochromatin. Mol Cell 2008; 31:167-77; PMID:18657500; http://dx.doi.org/10.1016/j.molcel.2008.05.017
- Noon AT, Shibata A, Rief N, Lobrich M, Stewart GS, Jeggo PA, Goodarzi AA. 53BP1-dependent robust localized KAP-1 phosphorylation is essential for heterochromatic DNA double-strand break repair. Nat Cell Biol 2010; 12:177-84; PMID:20081839; http://dx.doi.org/10.1038/ncb2017
- Beucher A, Birraux J, Tchouandong L, Barton O, Shibata A, Conrad S, Goodarzi AA, Krempler A, Jeggo PA, Lobrich M. ATM and Artemis promote homologous recombination of radiation-induced DNA double-strand breaks in G2. EMBO J 2009; 28:3413-27; PMID:2752027; http://dx.doi.org/10.1038/emboj.2009.276
- Shibata A, Conrad S, Birraux J, Geuting V, Barton O, Ismail A, Kakarougkas A, Meek K, Taucher-Scholz G, Lobrich M, et al. Factors determining DNA double-strand break repair pathway choice in G2 phase. EMBO J 2011; 30:1079-92; PMID:3061033; http://dx.doi.org/10.1038/emboj.2011.27
- Kakarougkas A, Ismail A, Klement K, Goodarzi AA, Conrad S, Freire R, Shibata A, Lobrich M, Jeggo PA. Opposing roles for 53BP1 during homologous recombination. Nucleic Acids Res 2013; 41:9719-31; PMID:23969417; http://dx.doi.org/10.1093/nar/gkt729
- Goodarzi AA, Noon AT, Jeggo P. The impact of heterochromatin on DSB repair. Biochem Soc Trans 2009; 37:569-76; PMID:19442252; http://dx.doi.org/10.1042/BST0370569
- Pagano M, Pepperkok R, Verde F, Ansorge W, Draetta G. Cyclin A is required at two points in the human cell cycle. EMBO J 1992; 11:961-71; PMID:1312467
- Chou TF, Brown SJ, Minond D, Nordin BE, Li K, Jones AC, Chase P, Porubsky PR, Stoltz BM, Schoenen FJ, et al. Reversible inhibitor of p97, DBeQ, impairs both ubiquitin-dependent and autophagic protein clearance pathways. Proc Natl Acad Sci U S A 2011; 108:4834-9; PMID:21383145; http://dx.doi.org/10.1073/pnas.1015312108
- Bennardo N, Cheng A, Huang N, Stark JM. Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet 2008; 4:e1000110; PMID:18584027; http://dx.doi.org/10.1371/journal.pgen.1000110
- Huertas P. DNA resection in eukaryotes: deciding how to fix the break. Nat Struct Mol Biol 2010; 17:11-6; PMID:20051983; http://dx.doi.org/10.1038/nsmb.1710
- Forment JV, Walker RV, Jackson SP. A high-throughput, flow cytometry-based method to quantify DNA-end resection in mammalian cells. Cytometry Part A 2012; 81A:922-8; PMID:22893507; http://dx.doi.org/10.1002/cyto.a.22155
- Sartori AA, Lukas C, Coates J, Mistrik M, Fu S, Bartek J, Baer R, Lukas J, Jackson SP. Human CtIP promotes DNA end resection. Nature 2007; 450:509-14; PMID:17965729; http://dx.doi.org/10.1038/nature06337
- Forget AL, Kowalczykowski SC. Single-molecule imaging brings Rad51 nucleoprotein filaments into focus. Trends Cell Biol 2010; 20:269-76; PMID:2862779; http://dx.doi.org/10.1016/j.tcb.2010.02.004
- Haaf T, Golub EI, Reddy G, Radding CM, Ward DC. Nuclear foci of mammalian Rad51 recombination protein in somatic cells after DNA damage and its localization in synaptonemal complexes. Proc Natl Acad Sci U S A 1995; 92:2298-302; PMID:7892263
- Paull TT, Rogakou EP, Yamazaki V, Kirchgessner CU, Gellert M, Bonner WM. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr Biol 2000; 10:886-95; PMID:10959836; http://dx.doi.org/10.1016/S0960-9822(00)00610-2
- Lin Y-H, Yuan J, Pei H, Liu T, Ann DK, Lou Z. KAP1 Deacetylation by SIRT1 Promotes Non-Homologous End-Joining Repair. PLoS ONE 2015; 10:e0123935; PMID:25905708; http://dx.doi.org/10.1371/journal.pone.0123935
- Bunting SF, Callen E, Wong N, Chen HT, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez-Capetillo O, Cao L, et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 2010; 141:243-54; PMID:20362325; http://dx.doi.org/10.1016/j.cell.2010.03.012
- Chapman JR, Barral P, Vannier JB, Borel V, Steger M, Tomas-Loba A, Sartori AA, Adams IR, Batista FD, Boulton SJ. RIF1 is essential for 53BP1-dependent nonhomologous end joining and suppression of DNA double-strand break resection. Mol Cell 2013; 49:858-71; PMID:23333305; http://dx.doi.org/10.1016/j.molcel.2013.01.002
- Escribano-Diaz C, Orthwein A, Fradet-Turcotte A, Xing M, Young JT, Tkac J, Cook MA, Rosebrock AP, Munro M, Canny MD, et al. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol Cell 2013; 49:872-83; PMID:23333306; http://dx.doi.org/10.1016/j.molcel.2013.01.001
- Ryan RF, Schultz DC, Ayyanathan K, Singh PB, Friedman JR, Fredericks WJ, Rauscher FJ, 3rd. KAP-1 corepressor protein interacts and colocalizes with heterochromatic and euchromatic HP1 proteins: a potential role for Kruppel-associated box-zinc finger proteins in heterochromatin-mediated gene silencing. Mol Cell Biol 1999; 19:4366-78; PMID:10330177; http://dx.doi.org/10.1128/MCB.19.6.4366
- Baldeyron C, Soria G, Roche D, Cook AJ, Almouzni G. HP1alpha recruitment to DNA damage by p150CAF-1 promotes homologous recombination repair. J Cell Biol 2011; 193:81-95; PMID:3082177; http://dx.doi.org/10.1083/jcb.201101030
- White D, Rafalska-Metcalf IU, Ivanov AV, Corsinotti A, Peng H, Lee SC, Trono D, Janicki SM, Rauscher FJ, 3rd. The ATM substrate KAP1 controls DNA repair in heterochromatin: regulation by HP1 proteins and serine 473/824 phosphorylation. Mol Cancer Res 2012; 10:401-14; PMID:22205726; http://dx.doi.org/10.1158/1541-7786.MCR-11-0134
- Soria G, Almouzni G. Differential contribution of HP1 proteins to DNA end resection and homology-directed repair. Cell Cycle 2013; 12:422-9; PMID:23287531; http://dx.doi.org/10.4161/cc.23215
- Lee YH, Kuo CY, Stark JM, Shih HM, Ann DK. HP1 promotes tumor suppressor BRCA1 functions during the DNA damage response. Nucleic Acids Res 2013; 41:5784-98; PMID:23589625; http://dx.doi.org/10.1093/nar/gkt231
- Ragland RL, Patel S, Rivard RS, Smith K, Peters AA, Bielinsky AK, Brown EJ. RNF4 and PLK1 are required for replication fork collapse in ATR-deficient cells. Genes Dev 2013; 27:2259-73; PMID:24142876; http://dx.doi.org/10.1101/gad.223180.113
- Davis EJ, Lachaud C, Appleton P, Macartney TJ, Näthke I, Rouse J. DVC1 (C1orf124) recruits the p97 protein segregase to sites of DNA damage. Nat Struct Mol Biol 2012; 19:1093-100; PMID:23042607; http://dx.doi.org/10.1038/nsmb.2394
- Meerang M, Ritz D, Paliwal S, Garajova Z, Bosshard M, Mailand N, Janscak P, Hubscher U, Meyer H, Ramadan K. The ubiquitin-selective segregase VCP/p97 orchestrates the response to DNA double-strand breaks. Nat Cell Biol 2011; 13:1376-82; PMID:22020440; http://dx.doi.org/10.1038/ncb2367
- Mosbech A, Gibbs-Seymour I, Kagias K, Thorslund T, Beli P, Povlsen L, Nielsen SV, Smedegaard S, Sedgwick G, Lukas C, et al. DVC1 (C1orf124) is a DNA damage–targeting p97 adaptor that promotes ubiquitin-dependent responses to replication blocks. Nat Struct Mol Biol 2012; 19:1084-92; PMID:23042605; http://dx.doi.org/10.1038/nsmb.2395
- Nie M, Aslanian A, Prudden J, Heideker J, Vashisht AA, Wohlschlegel JA, Yates JR, 3rd, Boddy MN. Dual recruitment of Cdc48 (p97)-Ufd1-Npl4 ubiquitin-selective segregase by small ubiquitin-like modifier protein (SUMO) and ubiquitin in SUMO-targeted ubiquitin ligase-mediated genome stability functions. J Biol Chem 2012; 287:29610-9; PMID:22730331; http://dx.doi.org/10.1074/jbc.M112.379768
- Kalousi A, Hoffbeck A-S, Selemenakis Platonas N, Pinder J, Savage Kienan I, Khanna Kum K, Brino L, Dellaire G, Gorgoulis Vassilis G, Soutoglou E. The Nuclear Oncogene SET Controls DNA Repair by KAP1 and HP1 Retention to Chromatin. Cell Rep 2015; 11:149-63; PMID:25818296; http://dx.doi.org/10.1016/j.celrep.2015.03.005
- Liu JP, Xu LL, Zhong JN, Liao J, Li J, Xu XZ. Protein phosphatase PP4 is involved in NHEJ-mediated repair of DNA double-strand breaks. Cell Cycle 2012; 11:2643-9; PMID:22732494; http://dx.doi.org/10.4161/cc.20957
- Messerschmidt DM, de Vries W, Ito M, Solter D, Ferguson-Smith A, Knowles BB. Trim28 is required for epigenetic stability during mouse oocyte to embryo transition. Science 2012; 335:1499-502; PMID:22442485; http://dx.doi.org/10.1126/science.1216154
- Santoni de Sio FR, Massacand J, Barde I, Offner S, Corsinotti A, Kapopoulou A, Bojkowska K, Dagklis A, Fernandez M, Ghia P, et al. KAP1 regulates gene networks controlling mouse B-lymphoid cell differentiation and function. Blood 2012; 119:4675-85; PMID:22452978; http://dx.doi.org/10.1182/blood-2011-12-401117
- Santoni de Sio FR, Barde I, Offner S, Kapopoulou A, Corsinotti A, Bojkowska K, Genolet R, Thomas JH, Luescher IF, Pinschewer D, et al. KAP1 regulates gene networks controlling T-cell development and responsiveness. FASEB J 2012; 26:4561-75; PMID:22872677; http://dx.doi.org/10.1096/fj.12-206177
- Hakli M, Karvonen U, Janne OA, Palvimo JJ. SUMO-1 promotes association of SNURF (RNF4) with PML nuclear bodies. Exp Cell Res 2005; 304:224-33; PMID:15707587; http://dx.doi.org/10.1016/j.yexcr.2004.10.029
- Nguyen HV, Chen JL, Zhong J, Kim KJ, Crandall ED, Borok Z, Chen Y, Ann DK. SUMOylation attenuates sensitivity toward hypoxia- or desferroxamine-induced injury by modulating adaptive responses in salivary epithelial cells. Am J Pathol 2006; 168:1452-63; PMID:16651613; http://dx.doi.org/10.2353/ajpath.2006.050782