
ABSTRACT
Cell division is strictly regulated by a diversity of proteins and lipids to ensure proper duplication and segregation of genetic material and organelles. Here we report a novel role of the putative lipid transporter ACAT-related protein required for viability 1 (Arv1) during telophase. We observed that the subcellular localization of Arv1 changes according to cell cycle progression and that Arv1 is recruited to the cleavage furrow in early telophase by epithelial protein lost in neoplasm (EPLIN). At the cleavage furrow Arv1 recruits myosin heavy chain 9 (MYH9) and myosin light chain 9 (MYL9) by interacting with IQ-motif-containing GTPase-activating protein (IQGAP1). Consequently the lack of Arv1 delayed telophase-progression, and a strongly increased incidence of furrow regression and formation of multinuclear cells was observed both in human cells in culture and in follicle epithelial cells of egg chambers of Drosophila melanogaster in vivo. Interestingly, the cholesterol-status at the cleavage furrow did not affect the recruitment of either IQGAP1, MYH9 or MYL. These results identify a novel function for Arv1 in regulation of cell division through promotion of the contractile actomyosin ring, which is independent of its lipid transporter activity.
Introduction
Successful cell division requires strict regulation in time and space to coordinate the sequential steps of DNA replication and segregation, mitotic spindle formation, morphological changes and finally physical cleavage via cytokinesis.Citation1 Prior to fulfilling the physical separation of daughter cells during cytokinesis animal cells form a contractile ring composed of F-actin and myosin (i.e. the actomyosin ring), at the cleavage furrow, which constricts and generates a thin intercellular bridge connecting the two daughter cells. Myosin is recruited to the cleavage furrow by alternative mechanisms, mainly via RhoA or Anillin.Citation2,3 Nucleation of F-actin is mediated by formins accumulating at the cleavage furrow by a RhoA-dependent mechanism.Citation2 Extensive studies throughout the years have identified numerous proteins operating upstream of RhoA and Anillin to modulate the assembly and constriction of the actomyosin ring.Citation4 Importantly, different types of lipids have also been assigned a crucial role for optimal furrow ingression.Citation5 For instance local accumulation of cholesterol, sphingolipids and phospholipids have been detected at the cleavage furrow and been demonstrated to drive cell division.Citation6-10 In particular phosphatidylinositol 4, 5-bisphosphate (PI(4,5)P2) is important for formation of the actomyosin ring by recruiting essential ring components such as Anillin and RhoA, which facilitate actin-polymerization.Citation11-13 Moreover, PI(4,5)P2 and sterol-enriched membrane domains interact with components of the actomyosin ring, thus anchoring it to the plasma membrane ensuring efficient ingression.Citation14-16
The membrane protein ACAT-related protein required for viability 1 (Arv1) has been attributed a role as a putative lipid transporter. It was first characterized in yeast arv1-mutants which displayed altered intracellular cholesterol distribution as well as abnormal phospholipid, sphingolipid and glycosylphosphatidylinositol-metabolism.Citation17-19 Similarly, in mice, antisense oligonucleotide targeting Arv1 transcripts caused accumulation of cholesterol in the endoplasmic reticulum at the expense of cholesterol levels in the plasma membrane.Citation20 Further it was recently reported that yeast arv1-mutants exhibit abnormal endoplasmic reticulum-morphology which potentially could also explain the observed impaired cholesterol transport.Citation21 Nonetheless yeast arv1 mutants have been shown to be mating defective due to disrupted formation of lipid-enriched domains and reduced MAP-kinase signaling, which is required for efficient mating.Citation22 In the current study we show that Arv1 facilitates telophase progression in human cells and drives cell division in its capacity of promoting efficient formation of the actomyosin ring.
Results
Cell cycle-dependent localization of Arv1
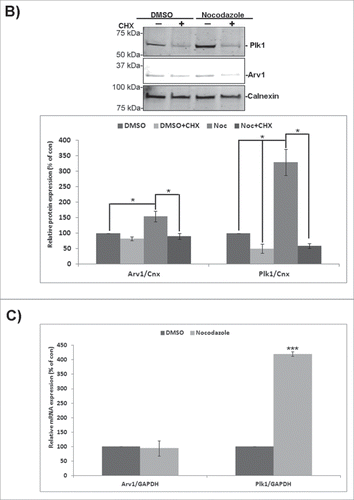
Upon assessing the intracellular localization of endogenous Arv1 in HeLa cells by confocal microscopy, we detected Arv1 on intercellular bridges connecting daughter cells late in cell division, and this motivated us to characterize the localization of Arv1 throughout mitosis in more detail (). During the interphase Arv1 colocalized with Plk1 on the centrosomes, and was also detected in the cytosol and in the nucleus. In contrast to Plk1, Arv1 was not found to be associated to the kinetochores early in prometaphase. The centrosomal localization of Arv1 persisted through progression of prometaphase and metaphase. During the anaphase some of the centrosomal Arv1 and Plk1 redistributed to decorate the equatorial ring which defines the localization of the contractile ring formed by actin and myosin.Citation1 As the cells proceeded into telophase, both Arv1 and Plk1 were found to translocate from the cleavage furrow to the intercellular bridge. However whereas Plk1 localized adjacent to the midbody dark zone as previously reported by Golsteyn and coworkers, Arv1 was predominantly found in the midbody dark zone.Citation23 The specificity of the Arv1 antibody was verified by the substantially decreased staining in Arv1 depleted cells (Fig. S1A). Furthermore, localization of Arv1 to the cleavage furrow was evident in several cell lines tested (HEpG2, BJ and RPE-1) (Fig. S1B). The cell cycle-regulated localization of Arv1 begged the question whether expression of Arv1 was accordingly regulated. To this end, the expression of Arv1 in exponentially growing and Nocodazole-synchronized HeLa cells was assessed. Arv1 protein levels were increased (two-fold (p< 0.05), but not as strongly as Plk1 (four-fold, p< 0.05) in the Nocodazole-synchronized cells (). Interestingly, the Arv1 mRNA levels were, however, unaffected upon mitotic entry in contrast to the induced Plk1 mRNA expression ().Citation23 This indicated that Arv1 was post-transcriptionally regulated during mitosis. Consistently, inhibition of protein synthesis by cycloheximide did abolish the Nocodazole-induced expression of Arv1 and Plk1 ().
Figure 1. Arv1 localization and expression are regulated according to mitotic progression. HeLa cells (A) grown on coverslips were fixed, permeabilized and stained with antibodies against Arv1 (red), Plk1 (green), Tubulin (white) and Hoechst (blue). Scale bar is 10 μm. (B) HeLa cells were treated with DMSO (vehicle) or 400 ng/ml Nocodazole (Noc) plus/minus 250 μg/ml cycloheximide (CHX) for 18 hours. The expression of Arv1 and Plk1 were determined by Western blotting and the intensities relative to Calnexin (loading control) were calculated using the Odyssey software. The graph represents the average of 3 separate experiments (± S.E), and significant differences are indicated as *p <0.05. (C) Relative expression of Arv1 and Plk1 mRNA of HeLa cells treated with DMSO (vehicle) or 400 ng/ml Nocodazole (18 hours) were determined by quantitative real time PCR. The graphs represent the average of at least 3 independent experiments (± S.E). Significantly different results are indicated (***p < 0.01).
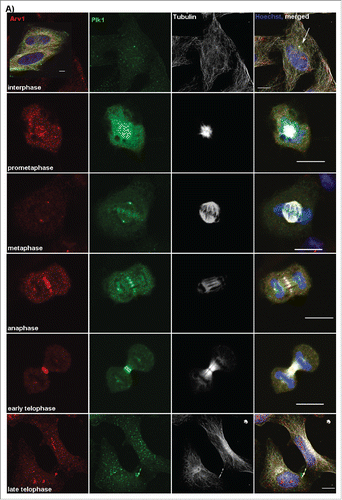
Figure 1. (Continued)
EPLIN recruits Arv1 to the cleavage furrow
To identify the proteins recruiting Arv1 to the cleavage furrow, a liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis of immunoprecipitated Arv1 and associated proteins was performed, which identified epithelial protein lost in neoplasm (EPLIN, also known as LIMA1) as a putative candidate (). Previously, EPLIN was shown to sustain proper cell division by regulating formation of the actomyosin ring driving furrow ingression.Citation24 The interaction between Arv1 and EPLIN was confirmed by co-immunoprecipitation and suggested that Arv1 associated stronger with EPLINβ compared to EPLINα (). Accordingly, we observed extensive co-localization of EPLIN and Arv1 at the cleavage furrow in dividing cells (, upper panel). Furthermore, when examining localization of Arv1 at the cleavage furrow in control and EPLIN knockdown cells (), 39% (p<0.01) less Arv1 was recruited in cells deprived of EPLIN (), thus showing that EPLIN is required for the accumulation of Arv1 at the cleavage furrow. That the levels of EPLIN in the cleavage furrow were not affected by depletion of Arv1 (Fig. S2), further supported our hypothesis that EPLIN works upstream of Arv1 at the cleavage furrow.
Figure 2. EPLIN recruits Arv1 to the cleavage furrow. (A) HeLa cells transiently transfected with pEGFP, pCA-EPLINα-pEGFP or pCA-EPLINβ-pEGFP were subjected to GFP-trap analysis. Lysate (5% of input) of cells expressing pEGFP (lane 1), EPLINα-pEGFP (lane 2) or EPLINβ-pEGFP (lane 3) was applied to verify equal amounts of Arv1 in the lysates used for immunoprecipitation. The amount of Arv1 co- immunoprecipitated with GFP (lane 4), EPLINα-GFP (lane 5) or EPLINβ-GFP (lane 6) was detected by Western blot. The Western blot was reprobed with anti-GFP to validate expression levels of the different constructs. A representative western blot is presented. (B) HeLa cells were transfected with 50 nM control or EPLIN siRNA for 48 hours. Efficient knockdown was verified by Western blotting analysis where Tubulin was used as loading control. Densitometry analysis of EPLIN/Tubulin-expression (±S.E) is indicated below the western blot. *** indicates significant differences, p<0.01. (C) Control or EPLIN depleted HeLa cells were fixed, permeabilized and stained with anti-EPLIN (green), anti-Arv1 (red), anti-Tubulin (white) and Hoechst (blue). Scale bars, 5 μm. The fluorescence intensities of EPLIN and Arv1 were determined by the LSM software. The graph represents the average of 3 independent experiments (± S.E). Significantly different results are indicated as ***p<0.01.
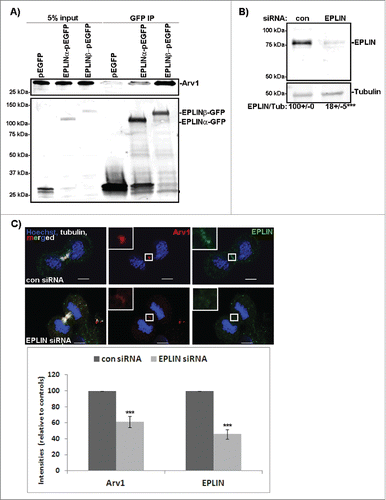
Table 1. Proteins interacting with Arv1 identified by LC-MS/MS analysis.
Arv1 depletion inhibits proliferation and causes a multinuclear phenotype
Studies in yeast have shown that normal cell growth is compromised in arv1 mutants.Citation22 To further explore whether Arv1 sustains growth in mammalian cells, proliferation was assessed in HeLa and HepG2 cells depleted of Arv1. Depletion of Arv1 protein (30% and 39% of control levels, p< 0.05) and mRNA (27% and 18% of control levels, p < 0.05) was confirmed by Western blotting and real time-PCR in HepG2 and HeLa cells, respectively (). Interestingly, Arv1 depletion reduced proliferation of HepG2 cells and HeLa cells by 25% and 13% (p < 0.05) compared to control cells, indicating that Arv1 is important for cell cycle progression in human cells ().
Figure 3. Arv1 is required for proper cell division. (A) HepG2 and HeLa cells were transfected with siRNA targeting an universal control or Arv1 and its depletion relative to Tubulin was confirmed by Western blot analysis. Average of 3 independent experiment (± S.E) is indicated below the Western blot, where significant differences is indicated as *p<0.01. (B) Depletion of Arv1 mRNA relative to GAPDH (loading control) was also assessed by quantitative real time PCR in both HepG2 and HeLa cells. The graph shown is based on the average of three independent experiments (± S.E). Significance are indicated as *p<0.05 and ***p<0.01. (C) Proliferation in Arv1 depleted cells and control cells were determined using the Click-iT EdU microplate assay. The average of three separate experiments, with six technical replicates per experiments, is presented (± S.E). Significant different results are indicated as ***p<0.01. (D) HeLa cells on cover slips were transfected with Arv1-pEGFP or Arv1(T806C,A809C)-pEGFP for 18 hours. Next the cells were transfected with control or Arv1 siRNA #1. 1) The cells were harvested after 48 hours, fixed, permeabilized and stained with anti-Tubulin (white) and Hoechst (blue). Representative confocal micrographs images of multinuclear cells (indicated by arrow heads) are shown (upper panel). Scale bar is 10 μm. At least 200 cells were imaged per experiment and used to quantify the percentage multinuclear cells induced by the different treatments. The graph shows the average of at least 4 separate experiments (± S.E). Significant differences are indicated as ***p<0.01. 2) The expression of Arv1-GFP under each condition was determined by Western blot analysis, where Calnexin represents the loading control. Densitometry analysis of GFP/Calnexin expression (±S.E) is indicated below. Significant difference is given as *p<0.05. (E) The percentage of multinuclear cells in control and Arv1 siRNA transfected HepG2 cells was determined using the 405-nm laser on an ImageStreamX imaging flow cytometer. The graph represents the average of two separate experiments (± S.E). (F) The number of egg chambers containing multinuclear follicle epithelial cells (FEs) (asterisk) was quantified in ovaries from wild type (w1118) and arv1-mutant Drosophila melanogasters. The average of 3 independent experiments is shown in the graph (± S.E) and significant differences are indicated (*** p<0.01). Scale bar, 5 μm.
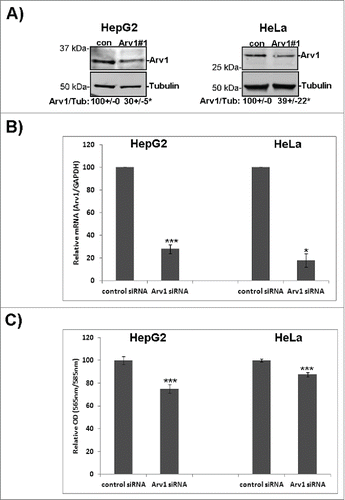
Figure 3. (Continued)
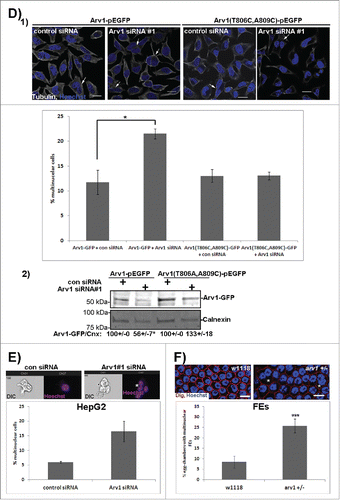
Our observation that Arv1 depletion increased the frequency of multinuclear cells by a two-fold (), further indicated that correct cell cycle progression depends on Arv1. Importantly, this multinuclear phenotype was only observed upon Arv1 depletion in cells expressing Arv1-GFP and not in cells expressing a siRNA-resistant version of Arv1-GFP, Arv1-(T806C,A809C)-GFP (). Compared to control siRNA-transfected cells, Arv1-GFP expression was reduced by 44% (p<0.05) in Arv1 siRNA-transfected cells (, lane 1 versus lane 2), whereas Arv1-(T806C,A809C)-GFP was not efficiently targeted by the Arv1 siRNA (, compare lane 3 and 4).
In addition, we used an imaging flow cytometer (ImageStreamX) to calculate the number of multinuclear HepG2 cells in response to the knockdown of Arv1. This system combines the advantage of analyzing large numbers of cells by flow cytometry while simultaneously acquiring image data from individual cells. Consistent with the microscopy data, a higher incidence of multinuclear cells was observed upon Arv1 depletion in the HepG2 cells (). Finally, the role of Arv1 during cell division was also verified in vivo using Drosophila melanogaster as model organism. Here the incidence of egg chambers displaying multinuclear follicle epithelial cells was 2.5 fold higher (p <0.01) in arv1 heterozygous mutant compared to wild type flies ().
Proper telophase progression is dependent on Arv1
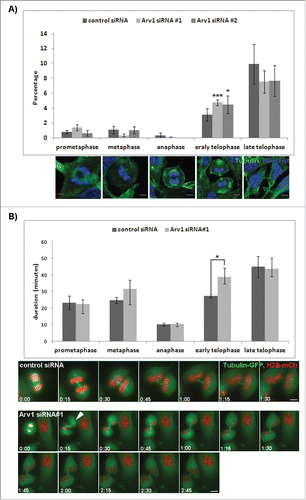
To further investigate the role of Arv1 during cell division, the relative distribution of cells in the different mitotic phases was compared in control and Arv1 depleted HeLa cells. The percentage of cells in telophase increased from 3% to 4.6% (p<0.05) upon Arv1 depletion, whereas the relative number of cells in prometaphase, metaphase, anaphase or cytokinesis remained unaffected (). Next we performed time-lapse microscopy to monitor cell division in control cells and Arv1 depleted cells (Supplementary Movie 1 and 2, respectively). Typically, the Arv1-deficient cells were temporarily halted in early telophase, where the two daughter cells appeared round and exhibited condensed chromosomes (still images, ). Consequently control cells used on average 27 minutes to complete early telophase, which we measured from furrow ingression to appearance of chromosome-decondensation and morphology-changes (i.e flattening of cells) (). In contrast Arv1 depleted cells used 39 minutes (p<0.05), which accompanied the increased frequency of telophase cells we observed in the fixed specimens (). The duration of the other mitotic phases remained unaffected in Arv1 depleted cells, suggesting that Arv1 specifically facilitates progression through telophase. Interestingly, in Arv1 depleted cells we often observed that at onset of telophase one of the emerging daughter cells became slightly out of focus as if it lost its adhesion to the substrate and was elevated (indicated with arrow at 0:15 in and shown in Supplementary Movie 2). We observed occasionally that Arv1 depleted cells aborted telophase, and the emerging daughter cells fused instead of completing cytokinesis, thus ending up as a multinuclear cell (Supplementary Movie 3). Detachment and lifting during telophase was previously correlated to inhibited actomyosin function.Citation25 Correspondingly, LC-MS/MS analysis identified myosin heavy chain 9 (MYH9), myosin light chain 6 and 12 A (MYL6 and MYL12A) as potential proteins interacting with Arv1 (). Interestingly, the IQ-motif-containing GTPase-activating protein (IQGAP1), a myosin-interacting protein associated with the cleavage furrow, was also identified in the LC-MS/MS analysis.Citation26
Figure 4. Normal telophase-progression is dependent on Arv1. (A) HeLa cells grown on coverslips were transfected with control or Arv1 siRNA, fixed, permeabilized and stained with anti-Tubulin (green) and Hoechst (blue). Confocal microscope images were acquired and used to calculate the number of cells in different mitotic phases defined by the structure of the mitotic spindle and chromosomes. Representative images of HeLa cells in the indicated mitotic phases are shown. Scale bar is 5 μm. At least 100 cells were randomly imaged per experiment and the average of 4 separate experiments (± S.E) is presented. Significant different results are depicted as *p<0.05 and ***p< 0.01. (B) HeLa Kyoto cells stably expressing GFP-tubulin (green) and mCherry Histone-2B (red) were transfected with control or Arv1 siRNA and synchronized by double Thymidin-treatment. Cell division was recorded by live cell imaging using the DeltaVision microscope the last 18 hours. The SoftWorx software was used to calculate the duration of each phase during cell division. The average of 4 separate experiments (± S.E) is shown, where at least 10 cells per experiment were analyzed. Significant differences are indicated as p<0.05. Still-images of control cell and Arv1 depleted cells are presented (lower panel) and time points from anaphase is indicated. Scale bar is 5 μm.
Arv1 facilitates recruitment of IQGAP1, MYH9 and MYL, but not F-actin, to the cleavage furrow
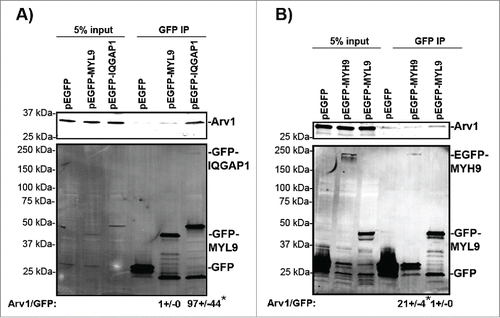
We performed a GFP-trap immunoprecipitation analysis to further assess the interaction between Arv1 and MYH9 or IQGAP1. Functional myosin II-complexes are hexamers composed of two MYH, two MYL and two essential light chains, where the expression of the different proteins are dependent of each other. Citation27,28 We wanted to assess the role of Arv1 for recruiting both MYH and MYL-proteins at the cleavage furrow, and since we were unable to obtain expression plasmids for either MYL12a or MYL6, we assessed whether Arv1 interacted with MYL9-GFP, since MYH9 forms complex with MYL9 in addition to MYL12a,b.Citation29 Indeed we observed that Arv1 associated MYL9-GFP, as well as IQGAP1-GFP and MYH9-GFP in Nocodazole-synchronized HeLa cells (). Calculating the amount of Arv1 co-immunoprecipiated with MYH9-GFP or IQGAP1-GFP compared to MYL9-GFP, the ratio of Arv1/MYH9-GFP and Arv1/IQGAP1-GFP was 21 and 97 fold higher (p<0.05), respectively, than the Arv1/MYL9-GFP. The immunoprecipitation analysis thus indicated that IQGAP1, and to less extent MYH9, interacted much more efficiently than MYL9 with Arv1 (). To further test the functional relationship between these proteins, the accumulation of MYH9, MYL9 and IQGAP1 at the cleavage furrow was determined in control and Arv1 depleted cells.
Figure 5. Arv1 interacts with IQGAP1, MYL and MYH9. HeLa cells transiently expressing pEGFP (A, B), pEGFP-MYL9 (A, B), pEGFP-IQGAP1 (A) or pEGFP-MYH9 (B) were synchronized in mitosis with 400 ng/ml Nocodazole (18 hours), lysed and used for GFP-trap immunoprecipiation analysis. To confirm equal expression levels of Arv1 in the cells expressing GFP (lane 1 in A-B), MYL9-GFP (lane 2 in A-B), IQGAP1-GFP (lane 3 in A) or MYH9-GFP (lane 3 in B), 5% of the lysate subjected to GFP-trap was analyzed by Western blotting analysis. Co-precipitated Arv1 and the expression of GFP-tagged proteins were determined by Western blotting (lanes 4–6). A representative experiment is shown. The Arv1/GFP-intensity ratios was quantified by the Odyssey Software and given below (± S.E). Significant different ratios relative to Arv1/GFP-MYL9 are indicated (*p< 0.05).
The depletion of Arv1 was confirmed by reduced accumulation at the cleavage furrow in telophase cells shown by confocal images consistent with the reduced total cellular expression shown by Western blot analysis (). Interestingly, the Arv1 depleted cells accumulated less IQGAP1 at the cleavage furrow, whereas the cellular expression of IQGAP1 was unaffected (). On the other hand, depleting IQGAP1 did not affect localization of Arv1 at the cleavage furrow, indicating that IQGAP1 works downstream of Arv1 (Fig. S3). Approximately 50% less (p<0.05) MYH9 accumulated at the cleavage furrow of Arv1-depleted cells, although the total cellular expression of MYH9 was not affected by Arv1 knockdown (). Further, the recruitment of MYL was clearly reduced in Arv1 knockdown cells (50% of control levels, p<0.05), even though the total cellular expression was slightly increased (). Furrow ingression is mediated by constriction of the actomyosin ring, which is composed of both myosin and F-actin.Citation1 As a result of this, we further assessed whether Arv1 was implicated in recruitment of F-actin to the cleavage furrow as well. Interestingly, identical, or slightly increased, levels of phalloidin-stained F-actin were detected at the cleavage furrow of control cells as compared to Arv1 deprived cells (), whereas the total expression levels of actin was not affected upon Arv1 depletion. Together, these results indicate that Arv1 specifically recruits only one component of the actomyosin ring, namely myosin.
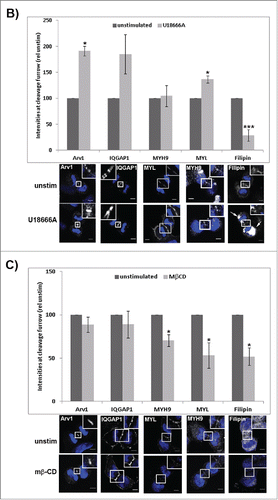
Figure 6. Arv1 promotes recruitment of IQGAP1, MYH9 and MYL, but not F-actin, to the cleavage furrow. (A) HeLa cells grown on coverslips were depleted or not of Arv1 for 48 hours. The cells were fixed, permeabilized and stained with Hoechst (blue) and anti-Arv1 (white, 1), anti-IQGAP1 (white, 2), anti-MYH9 (white, 3), anti-MYL (white, 4)), phalloidin (white, 5) or 50 ng/ml Filipin (white, 6). DRAQ5 (blue) was used to stain the nucleus of Filipin-stained cells Scale bar is 5 μm. The intensities of 10 randomly selected cells per experiment were determined by the LSM software (version 3.2) and the average of at least 3 separate experiments are shown (± S.E). Western blot analysis was performed to determine the total cellular expression of the different proteins investigated (lower panel). The intensities of the immunoreactive bands relative to the loading control (Tubulin) were quantified using the Odyssey software and the average of 3 separate (± S.E) are given below typical Western blot results. Significant different results are indicated (*p<0.05 and ***p<0.01). (B, C) HeLa cells seeded on coverslips were treated or not with 1 μg/ml U18666A for 24 hours (B) or 5 mM metyl-β -cyclodextrin (MβCD) for 60 minutes (C), fixed, permeabilized and stained with Hoechst (blue) and anti-Arv1 (white), anti-IQGAP1 (white), anti-MYH9 (white), anti-MYL (white) or 50 ng/ml Filipin (white). DRAQ5 (blue) was used to stain the nucleus of Filipin-stained cells. Scale bars, 5 μm. The fluorescence intensities were determined by the LSM Software (version 3.2) and the graph shows the average of 3 independent experiments (± S.E), where significant different results are indicated as *p<0.05 and ***p<0.01.
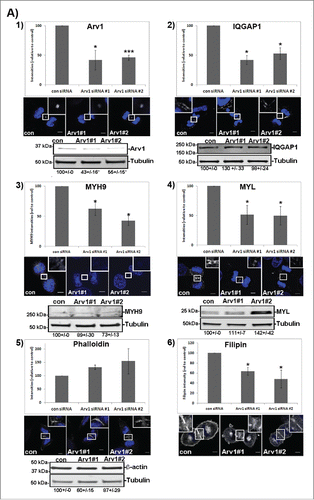
Figure 6. (Continued)
As myosin is recruited to the cleavage furrow by alternative pathways besides IQGAP1 (ref 26), we wanted to explore whether Arv1 influenced any of these. Therefore, the accumulation of RhoA and Anillin at the cleavage furrow of control and Arv1 depleted cells were determined. The apparently increased accumulation of RhoA at the cleavage furrow reflected most likely the slightly increased expression levels of RhoA in Arv1 knockdown cells (Fig. S4A). Compared to control cells, Anillin was found accumulated at the cleavage furrow of Arv1 depleted cells (Fig. S4B). Together our observations indicate that Arv1 facilitates recruitment of MYL and IQGAP1 to the cleavage furrow by a molecular pathway independent of RhoA and Anillin.
Cholesterol-levels at the cleavage furrow are dispensable for localization of IQGAP1 and myosin
Given the importance of lipids in regulating formation of the actomyosin ring, we further explored whether Arv1-modulated recruitment of MYH9, MYL and IQGAP1 was dependent on its modulation of intracellular cholesterol transport. Previous studies identified Arv1 to be a putative cholesterol transporter as its depletion increased the intracellular cholesterol level at the expense of the plasma membrane cholesterol level.Citation17-19 Accordingly, HeLa cells depleted of Arv1 accumulated 45% less (p<0.05) cholesterol at the cleavage furrow compared to control cells, consistent with its role during intracellular cholesterol transport (). To determine whether Arv1 recruits IQGAP1, MYH9 and MYL in a cholesterol-dependent manner, we assessed the accumulation of these proteins in cells treated with the oxidosqualene cyclase inhibitor U18666A, which efficiently impairs intracellular cholesterol transport from late endosomes.Citation30 Accordingly, we observed reduced cholesterol-levels at the cleavage furrow (61%, p<0.01) accompanied with accumulation of cholesterol in endocytotic compartments in the perinuclear region (indicated with arrow heads) upon U18666A treatment. In contrast to Arv1 depletion, U18666A-treatment modestly stimulated accumulation of both IQGAP1 and MYL at the cleavage furrow (). Moreover, Arv1 also accumulated at the cleavage furrow probably due to the sterol-induced expression of Arv1 (data not shown). The impaired cholesterol-transport to the cleavage furrow did not affect the accumulation of neither IQGAP1 nor MYH9 at the cleavage furrow. On the other hand, slightly more MYL (36%, p<0.05) was detected at the cleavage furrow upon U18666A-treatment ().
Alternatively, the plasma membrane-cholesterol content was reduced by treating the cells with methyl-β-cyclodextrin (MβCD). Treating cells with 5mM MβCD for 60 minutes reduced the total cellular cholesterol levels by 49% (p<0.05) (). In contrast to U18666A, Arv1 localization was not affected in cells treated with MβCD, due to short treatment (only 60 minutes). Interestingly, accumulation of IQGAP1 at the cleavage furrow remained unaffected. However, localization of MYL and MYH9 were reduced by 47% and 30% (p<0.05), respectively.
Arv1 and IQGAP1 drive telophase progression
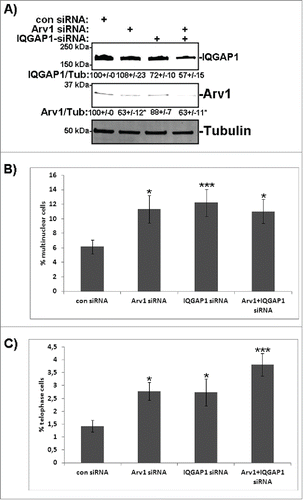
We have shown that Arv1 specifically recruits IQGAP1, MYH9 and MYL to the cleavage furrow without affecting Anillin, RhoA (Fig. S4) or F-actin (), suggesting that Arv1 represents yet another mechanism to drive actomyosin ring-formation. Even though Arv1 was found to co-precipitate with MYH9 and MYL9, the association to IQGAP1 was much more efficient (), indicating that Arv1 indirectly recruits myosin to the cleavage furrow via IQGAP1. To test this further, HeLa cells were depleted of Arv1, IQGAP1 or both () and the percentage multinuclear cells and cells in telophase were determined. Compared to control cells, knockdown of Arv1 or IQGAP1 doubled (p<0.05) the number of multinuclear cells, and, interestingly, to the same extent separately as in combination (). The same tendency was observed when quantifying cells in telophase, where 1.4% of the control cell-population was in telophase in contrast to 2.8%, 2.7% or 3.8% of Arv1-, IQGAP1- or Arv1+IQGAP1-depleted cells (p<0.05), respectively (). Together these observations indicate that Arv1 and IQGAP1 function by a common molecular pathway to promote telophase progression.
Figure 7. Depletion of Arv1 and IQGAP1 give rise to multinuclear cells. (A) HeLa cells were transfected with siRNA targeting control, Arv1, IQGAP1 or the latter two in combination for 48 hours. Western blot analysis were performed to validate knockdown of IQGAP1 (upper panel) and Arv1 (middle panel), where Tubulin (lower panel) was used as loading control. Densitometric calculation of Arv1 or IQGAP1 expression relative to Tubulin (±S.E) is indicated below the Western blots. The number of multinuclear cells (B) and cells in telophase (C) were quantified in control and Arv1+/− IQGAP1-depleted cells using confocal fluorescence microscopy. The average of at least 3 separate experiments (± S.E) is presented. Statistical significance is reported as ***p<0.01 and *p<0.05.
Discussion
In this study we have shown that Arv1 localizes at the centrosomes, the cleavage furrow and the intercellular bridge during cell division. The LC-MS/MS analysis identified EPLIN, IQGAP1, MYH9, MYL12a and 6, as Arv1-interacting proteins. We demonstrated that EPLIN facilitates recruitment of Arv1 to the cleavage furrow in early telophase. At the cleavage furrow, Arv1 seems to provide a platform recruiting IQGAP1 and subsequently MYH9 and MYL to sustain an efficient actomyosin ring formation and furrow ingression. In support of this, live cell imaging of Arv1 depleted cells, showed a high incidence of transient detachment and swirling of cells, a phenomenon associated with defective actomyosin ring formation.Citation25 Consequently we found that cells deprived of Arv1 display prolonged telophase, occasionally resulting in cytokinesis failure as evident by the presence of multinuclear cells both in vitro and in vivo. This indicates a novel role for Arv1 in telophase progression.
The localization of Arv1 to the cleavage furrow assigns a novel role for Arv1, in addition to its putative cholesterol transporter activity previously described. We show that EPLIN recruits Arv1 to the cleavage furrow at the onset of telophase. Interestingly, EPLIN was previously shown to facilitate accumulation of myosin, in addition to actin, at the cleavage furrow.Citation24,31 In accordance with our findings, it is intriguing that both EPLIN and Arv1 are required for efficient myosin accumulation at the cleavage furrow, and it is tempting to speculate that Arv1 may be a downstream target of EPLIN in this pathway. The GFP-immunoprecipitation analysis indicated that Arv1 interacts more efficiently with EPLINβ compared to EPLINα. Curiously, the two EPLIN-isoforms seems to be differently expressed in different cell-types, but the functional interpretation of this remains elusive. Both isoforms were previously reported to facilitate actomyosin-ring formation by associating both myosin and actin, but this does not exclude the possibility of unique molecular mechanisms where EPLINα and β collaborate with different proteins, such as Arv1. Citation24
Myosin together with actin assembles into the actomyosin ring which contracts at early telophase.Citation1 Interestingly, the depletion of Arv1 compromised the recruitment of MYH9 and MYL to the cleavage furrow, whereas F-actin, on the other hand, rather accumulated at the cleavage furrow of Arv1-deprived cells. The accumulated F-actin could be expected to override or neutralize the reduced myosin-recruitment during Arv1 depletion. However, an in vitro system for actomyosin ring-formation and constriction clearly indicated that myosin, not actin-dynamics, was essential for the efficient constriction.Citation32 Thus it seems that myosin and actin are independently regulated at the cleavage furrow, also in line with our findings. Formation of the actomyosin ring is mediated by alternative pathways through RhoA and Anillin thus providing a robust regulation of the process.Citation2,3,33 Importantly, Arv1 modulated accumulation of MYH9 and MYL oppositely from RhoA and Anillin. The induced accumulation of RhoA and Anillin at the cleavage furrow may indeed facilitate nucleation of F-actin, and explain the common accumulation of RhoA, Anillin and F-actin observed upon Arv1 depletion. Furthermore, myosin has been reported to regulate the actin dynamics in the contractile ring by facilitating F-actin disassembly.Citation34,35 Therefore the impaired recruitment of myosin to the cleavage furrow may further contribute to the accumulation of F-actin observed in Arv1 depleted cells. Nevertheless the current study implies that Arv1 mediates myosin recruitment independently of RhoA and Anillin, thus representing an additional pathway which adds greater robustness to the formation of the actomyosin ring during cell division.
Interestingly, the scaffold protein IQGAP1 was identified in LC-MS/MS analysis and GFP-trap analysis further validated its interaction with Arv1. Moreover we found IQGAP1 to facilitate cell division and to localize to the cleavage furrow in accordance to previous studies.Citation36,37 Interestingly, this localization of IQGAP1 was observed to be Arv1-dependent, but not vice versa, suggesting that IQGAP1 functions downstream of Arv1. Based on our immunoprecipiation analysis indicating that IQGAP1 associates with Arv1 much more efficiently than MYH9 and MYL9, and previous studies in yeast indicating that IQGAP promotes myosin recruitment (see refs Citation38,39), it seems plausible that Arv1 recruits MYH9 and MYL indirectly by first recruiting IQGAP1 to the cleavage furrow. Consistently, compared to the localization of MYH9 and MYL at the cleavage furrow, IQGAP1 correlated more directly with the accumulation of Arv1 when depleting cholesterol from the plasma membrane. Alternatively, myosin is recruited to the cleavage furrow by several pathways, in addition to the IQGAP1-Arv1.
Intriguingly, in yeast the IQGAP-like protein was reported to participate in the F-actin-independent mechanism of myosin recruitment.Citation40 Likewise, the current study indicates that human cells utilize IQGAP1 in a similar pathway, which is modulated by Arv1. The role of IQGAP in cell division has been extensively studied in yeast. In contrast to yeast, human cells express three IQGAP proteins, where IQGAP1 and 3 have been assigned roles during cell division.Citation41 Recently, IQGAP3, but not IQGAP1, was shown to regulate cytokinesis by recruiting RhoA and Anillin.Citation42 That IQGAP1 and IQGAP3 regulate cell division by separate pathways was further illustrated by the additive effect on multinucleation when cells were depleted of both IQGAP1 and IQGAP3. The current study hypothesizes that IQGAP1 regulates furrow ingression by recruiting MYH9 and MYL to the cleavage furrow, which is regulated by Arv1 rather than Anillin. Moreover, we provide epistatic evidence that Arv1 and IQGAP1 are members of the same molecular pathway. In context of IQGAP1 function as a scaffold interacting with numerous proteins, we cannot exclude the possibility that IQGAP1 modulates cell division by additional means than recruiting myosin to the cleavage furrow. Consistently, IQGAP1 was recently shown to stimulate nuclear pore reformation.Citation37
Interestingly, that manipulation of plasma membrane-cholesterol and Arv1 depletions affected the recruitment of IQGAP1, MYH9 and MYL differently indicates a scaffolding role of Arv1 during the assembly of the actomyosin ring independently of its cholesterol-transporting capacity. The compromised intracellular transport due to the U18666A-treatment yielded an accumulation of Arv1 protein, which further indicates that lipids modulate its expression (data not shown, will be addressed further). Consequently, the U18886A-stimulated accumulation of Arv1 at the cleavage furrow may therefore account for the moderate accumulation of both IQGAP1 and MYL we observed upon U18666A-treatment. In contrast, we did observe that U18886A increased accumulation of F-actin at the cleavage furrow (data not shown), which could further stabilize MYL as previously reported.Citation43 Depleting the plasma membrane-cholesterol content acutely using 5mM MβCD for 60 minutes, assessed directly the potential lipid-dependent recruitment to the cleavage furrow without the influence of possible lipid-regulated gene expression of the different proteins. Equal amounts of Arv1 were detected at the cleavage furrow in unstimulated and MβCD-treated cells, implying that the U18666A-induced accumulation reflected induced Arv1 expression. Interestingly, MβCD-treatment reduced the localization of MYH9 and MYL, most likely due to the reduced RhoA-accumulation, which is reported to associate the cleavage furrow in by lipid-dependent mechanism. Citation44 Interestingly, the correlative accumulation of Arv1 and IQGAP1 observed further imply that Arv1 directly recruits IQGAP1 to the cleavage furrow.
In summary, reducing the cholesterol-content at the cleavage furrow instantly did not mimick the reduced localization of IQGAP1 and myosin as observed by Arv1 depletion. Therefore our data are consistent with the view that efficient formation of the actomyosin ring is regulated by modulating both the protein and lipid composition at the cleavage furrow. In conclusion, we provide evidence that Arv1 operates in one of the several parallel pathways that recruit myosin to the cleavage furrow. Arv1 thus represents a novel molecular component that together with EPLIN, IQGAP1 and myosin ensures optimal furrow ingression.
Materials and methods
Antibodies and reagents
Rabbit anti-Arv1 (ab107080), mouse anti-Plk1 (ab17056) and mouse anti-IQGAP1 (ab56529) were from Abcam. Mouse-anti-β actin (A5316) and mouse-anti-α−Tubulin (T5168) were obtained from Sigma Aldrich. Mouse anti-EPLIN (clone 1B7, LS-C134069) was from LifeSpan BioSciences, Inc. Goat anti-Calnexin (sc-6465) was provided by Santa Cruz. Mouse anti-MYL (3672S) was from Cell Signaling. Rabbit anti-MYH9 (GTX113236) was bought from GeneTex. Mouse anti-GFP (11814460001) was from Roche. Mouse anti-Dlg was from Developmental Studies Hybridoma Bank. All secondary antibodies used for confocal microscopy were purchased from Jackson ImmunoResearch Laboratories, whereas the secondary antibodies used for Western blot analysis were from LI-COR® Biosciences GmbH. Hoechst 33342 and Rhodamine-phalloidin were obtained from Life Technologies. DRAQ5 was from Biostatus. Protease Inhibitor Cocktail was provided by Roche. Laemmli loading buffer was from Bio-Rad. N-ethylmaleimide, Filipin III (F4767), Phosphatase Inhibitor Cocktail 2, Thymidine, MβCD and Nocodazole were from Sigma Aldrich. Leupeptin was provided by Peptide Institute Inc., All other chemicals were of analytical grade.
Plasmid construction
Wild type, human Arv1 cDNA was amplified by RT-PCR using the primer pair (forward: 5′-AGCTGAAGCTTGCCACCATGGGCAACG-3′ and reverse: 5′-ACCGGTGGATCCGC- GAAGTCCTGAGATTTAAAG-3′) and cloned into the BamHI/HindIII-site upstream of GFP in pEGFP-N1 (Clonetech). This Arv1-GFP plasmid was further used as a template to generate T806C- and A809C-Arv1-GFP targeting the siRNA-Arv1 sequence without changing the corresponding amino acids. Mutageneses were carried out using QuickChange II XL Mutagenesis Kit (Agilent Technologies) according to the manufacturer's instructions. The oligonucleotide sequences used for mutagenesis are 5′-CCAGAGTATGGAATGGGATGTCGGCAGTGATTATGCC-3′ and its complementary 5′-GGCATAATCACTGCCGACATCCCATTCCATACTCTGG-3′. The siRNA target sequence (in bold) covers nucleotides 788–818 of human Arv1 (NM_022786) with the in vitro mutated nucleotide underlined. Both constructs were sequenced to validate correct sequence and mutagenesis.
Cell cultures, transfections and siRNA transfections
HeLa cells were grown in DMEM (Gibco) supplemented with 10% fetal calf serum, 2 mM L-glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin (Medium A). HeLa Kyoto cells stably expressing GFP-tubulin and mCherry Histone-2B was grown in Medium A. HepG2 cells were cultured in MEM (Gibco) supplemented with 10% fetal calf serum, 2 mM L-glutamine, 100 U/ml penicillin, 100μg/ml streptomycin and 1% Non Essential Amino Acids (Medium B). All the cell lines were cultured in an atmosphere containing 5% CO2 at 37°C according to ATCC.
For knockdown analysis 4 × 105 cells were reversely transfected with 40 nM control siRNA (SI04380467, Qiagen), Arv1#1 siRNA (SI04159113, Qiagen), Arv1#2 siRNA (L-014217-01-0005, Dharmacon), 25 nM IQGAP1 siRNA (cat no, SI2173416, Qiagen), 25 nM EPLIN siRNA (cat no, SI04159743, Qiagen) using RNAiMax transfection reagent according to the manufacturer (Life Technologies). For double knockdown experiments the cells were transfected with 65 nM control siRNA, 40 nM Arv1 siRNA plus 25 nM control siRNA, 25 nM IQGAP1 siRNA plus 40 nM Arv1 or 40 nM control siRNA. Cells were used and harvested 48 hours after transfection. For rescue experiments HeLa cells were first transfected with 0.25 μg pEGFP, 1 μg Arv1-pEGFP or 1 μg Arv1(T806C,A809C)-pEGFP using FuGene ® 6 Transfection reagents (Promega). After 18 hours the cells were re-transfected with 40 nM control or Arv1 siRNA using RNAiMax. The cells were harvested 48 hours after siRNA transfection.
For GFP trap precipitation experiments 4 × 105 HeLa cells (per well in a 6-well plate) were seeded and transfected with either 0.25 μg pEGFP or 1 μg pCA-EPLINα-EGFP, 1 μg pCA-EPLINβ-EGFP, 1 μg pEGFP-MYH9 (11347, Addgene), 1 μg pEGFP-MYL9 (35680, Addgene) or 1 μg pEGFP-IQGAP1 (30112, Addgene) using the FuGENE ® 6 Transfection reagent as described by the manufacturer (Promega). The cells were grown for 30 hours, synchronized in mitosis using 400 ng/ml Nocodazole for another 18 hours and finally harvested for immunoprecipiation analysis.
Cell proliferation and viability assay
HepG2 and HeLa cells (4 × 105) were transfected with 40 nM control or Arv1 siRNA as described above. The cells were splitted further into 96 well plates after 48 hours in order to determine proliferation using the Click-iT EdU microplate assay (Life Technologies). The cells were labeled with 10 μM EdU for 12 hours and the further assay was performed as described by the manufacturer (Life Technologies). The remaining of the cells were harvested and used to determine the knockdown efficiency.
LC-MS/MS and protein identification
Nocodazole-synchronized HeLa cells were subjected for immunoprecipitation with anti-Arv1 or rabbit IgG using Dynabeads® M-280 coated with sheep anti-rabbit IgG (Life Technologies). Precipitated proteins were eluted from the beads using 1M Glycine (pH=2.0). A sample volume of 100 µl corresponding to a protein concentration of approximately 55 µg was withdrawn from each sample, the proteins were denaturated and reduced by addition of DTT in a 1DTT+25protein (w+w) ratio and incubated at 65°C for 30 minutes. After cooling the sample to room temperature, the proteins were alkylated using IAM in a 1IAM+10protein (w+w) ratio in the dark at room temperature for 30 minutes. The pH was adjusted with triethylammonium bicarbonate and subsequently trypsin was added in a 1trypsin+20protein (w+w) ratio for protein digestion. The digestion was done overnight in room temperature. The reduction, alkylation and digestion were carried out with gentle mixing. Protein digestion was attenuated by deactivating the trypsin using 3 μl formic acid (50 %). The LC-MS/MS analysis was carried out on the Easy nLC1000 system and the Q-Exactive mass spectrometer with a nanospray interface (Thermo Fisher Scientific (Waltham, MA, USA) as previously described.Citation45 Briefly, an aliquot of 2 μl of each sample was loaded on a C18 monolithic column (0.1 × 60 mm) using 0.1 % formic acid as loading mobile phase at a flow rate set to 4 µl/min. The sample was subsequently transferred to the C18 monolithic column (0.1 mm × 100 cm).Citation46 The analytical column was placed in a column oven (Mistral LC oven, Spark-Holland, Emmen, Netherlands) for a controlled column temperature. The temperature was set to 80°C. A 25 minute gradient was used to separate the peptides. Mobile phase A consisted of 0.1 % formic acid and mobile phase B was ACN. The gradient increased linearly from 5 to 40 % B within 20 minutes, before it increased to 95 % B in 5 minutes. The flow rate was set to 2 µl/min. The Q-Exactive MS was operated in data dependent mode. The ESI spray voltage was +2.5 kV and the capillary temperature was 250°C. All mass spectra were acquired in positive mode in the range between m/z 350 and 1850 at a resolution of 35,000 (MS) and 17,500 (MS/MS). Higher-energy collisional dissociation (HCD) with 25% normalized collision energy was used for fragmentation. Charge state screening and exclusion were enabled, excluding all unassigned, single charged and peptides with a charge state higher than 7. The default charge state was set to 2. The ten most intense peptide ions were isolated with 1 repeated count. XCalibur 2.2 (Thermo Fisher Scientific) was used to control both the Easy nLC-1000 and Q-Exactive mass spectrometer.
Proteome Discoverer (v1.3.0, Thermo Scientific), using the Sequest search engine, was applied for protein identification. All spectra were searched along the Uniprot, human 9606 database using a decoy database search, using trypsin and 2 missed cleavages as parameters. Precursor mass and fragment mass tolerance were set to 10 ppm and 0.3 Da, respectively. Cysteine carbamidomethylation was set as static (fixed) modification, while methionine oxidation was used as dynamic modification. Peptides with medium confidens (> 95 %) were used for protein identification.
Real time PCR
Total RNA was isolated using RNA blood isolation kit (Qiagen) according to the manufacturers' protocol, including the DNase1 treatment steps. During cDNA synthesis 200 ng total-RNA was reversely transcribed using AffinityScript QPCR cDNA synthesis kit according to the manufacturer (Stratagene). Quantiative PCR (Agilent Technologies Stratagene MX3005P) was performed using cDNA QuantiTect SYBR Green PCR kit (Qiagen) with initial denaturation for 10 minutes at 95°C, followed by 40 cycles, each consisting of 30 seconds, at 95°C, 1 minute at 60°C, 1 minute at 72°C. Specificity of the PCR-products was verified by agarose-electrophoresis and amplicon sequencing. Relative mRNA expression was calculated by the 2-ΔΔCt method and adjusted for PCR efficiencies. Citation47 GAPDH was used as the reference gene. Two technical replicates of each biological sample were PCR amplified. The different primer pairs used are listed below.
Oligonucleotides
The Primer 3 software was used to design the oligonucleotides which were synthesized by Integrated DNA Technologies. The sequences of the forward (f) and reverse (r) primers used for quantitative real time PCR analysis is given below.
ARV1-f1: 5′-TCCTGTGGGTAGAACGGCCCA-3′
ARV1-r1: 5′-TGCTTTCCAGCAGTAAGCCACTC-3′
GAPDH-f1: 5′-TGCACCACCACCTGCTTAGC-3′
GAPDH-r1: 5′-GGCATGGACTGTGGTCATGAG-3′
PLK1-f1: 5′-CGAGTTCTTTACTTCTGGCTAT-3′
PLK1-r1: 5′-GGAGACTCAGGCGGTATGT-3′
Western blotting
Cells were rinsed with PBS and lysed [in 25mM HEPES (pH=7.2), 125 mM potassium acetate, 2.5 mM magnesium acetate, 5 mM EGTA, 0.5% NP-40, 1 mM DTT and protease inhibitor cocktail] on ice for 30 minutes. Nuclei and cell debris were cleared by spinning the samples at 20,800g for 10 minutes (4°C). Equal amounts of proteins were separated by SDS-PAGE and blotted onto PVDF membranes (Millipore Corporation). The membranes were incubated with primary antibodies (4°C overnight), washed with PBS-0.01% Tween, incubated with fluorescently labeled secondary antibodies (LI-COR® Biosciences GmbH) for 1 hour at room temperature and rinsed with PBS-0.01% Tween and finally PBS. The membranes were scanned using the Odyssey developer and the intensities of the immunoreactive bands were quantified by the Odyssey software (LI-COR® Biosciences GmbH).
Confocal immunoflourescence microscopy
Cells grown on coverslips were fixed in 4% formaldehyde (30 minutes, room temperature) or methanol (−20°C, 10 minutes), before they were permeabilized with PEM buffer [80 mM PIPES, 5 mM MgCl2 × 6H2O, pH=6.8) containing 0.05% saponin] for 5 minutes (room temperature) and stained with primary antibodies (4°C, overnight). The coverslips were washed 3 times with PBS-0.05% saponin and stained with secondary antibodies for 1 hour at room temperature. After washing, the coverslips were mounted in Mowiol containing Hoechst 33342 or DRAQ5 (where indicated) for nuclear staining. Specifically, PFA-fixed cells were stained with 50 ng/ml Filipin (room temperature, 1 hour) prior to permeabilization and incubation with primary antibodies. The specimens were examined using a LSM780 Zeiss confocal microscope. Images were acquired using the same settings below saturation when they were used for quantifications. The intensities of the different immunostainings were calculated by the LSM Software (version 3.2).
Live cell imaging
HeLa Kyoto cells stably expressing GFP-tubulin and mCherry Histone-2B were used for live cell imaging. Cells (4 × 105) were transfected with 40 nM control or Arv1 siRNA using RNAiMax as described above. Approximately 2 hours after transfection, the cells were treated with 2.5 mM Thymidin to start cell cycle synchronization. After 17 hours the cells were washed with fresh medium and released for 8 hours, before they were stimulated with 2.5 mM Thymidin for another 17 hours. The cells were released from the second Thymidin-block, rinsed in fresh phenol-free Medium A and cultured for another 6 hours. Live cell-imaging was performed using a DeltaVision microscope equipped with Elite TruLight Illumination System (Applied Precision) and 40x oil objective. The images were captured in 10 minutes intervals. The Z sections were projected by the SoftWorx software. The progression through cell division was evaluated by the structure of mitotic spindle and chromosomes, visualized by GFP-tubulin and mCherry-Histone-2B, respectively. Typical images of cells in the different states of cell division are presented in . Specifically, we define telophase onset when furrow ingression starts and telophase persists as long the emerging daughter cells are circular, exhibit condensed chromosomes and short intercellular bridge. Cytokinesis starts when the emerging daughter cells spread, the chromosomes de-condense and the intercellular bridge elongates. Abscission at the midbody defines cytokinesis-termination.
Quantification of polynuclear cells with ImageStreamX
HepG2 cells were siRNA transfected as previously described, fixed in 2% PFA for 20 minutes and permeabilized with PEM buffer containing 0.05% saponin for 5 minutes (room temperature). Further, the nuclei were stained with 2 μg/ml Hoechst 33342 for 30 minutes, before the cells were pelleted, resuspended in 70 μl PBS, and post-fixed in 2% PFA. The cells were analyzed using an ImageStreamX imaging flow cytometer (Amnis, Seattle, USA), where the images were acquired using the 405 nm-laser and 40x objective. Acquired data were processed using the IDEAS 4.0 analysis software. Apoptotic cells have bright and punctuate nuclear staining and were excluded from the analysis by gating on cells with low nuclear area (Area_Treshold (M07, Ch07, 50%)) and high bright field contrast (Contrast_M01_Ch01). Multinuclear cells were distinguished from mono or bi-nuclear cells based on the higher DNA content (Intensity_MC_Ch07) and visually inspection of the nuclear images.
GFP-trap immunoprecipitation
HeLa cells were seeded in 6-well plates at the density 3 × 105 cells per well. The next day the cells were transfected with 0.25 μg GFP, 1 μg pCA-EPLINα-EGFP, 1 μg pCA-EPLINβ-EGFP, 1 μg pEGFP-MYH9, 1 μg pEGFP-MYL9 or 1 μg pEGFP-IQGAP1 using FuGene6® Transfection Reagent (Promega) and harvested after 48 hours. Enrichment of mitotic cells was obtained by treating the cells with 400 ng/ml Nocodazole the last 18 hours. GFP trap immunoprecipiation was performed according to the manufacturer (Chromotek). Specifically, transfected cells from a 6-well plate were pooled, lysed before cell debris and nuclei were removed by centrifugation (100000g, 5 min, 4°C). The post-nuclear supernatant was diluted with Washing buffer and incubated with 15 μl GFP beads for 1 hour (4°C). Beads with bound proteins were washed before the precipitated proteins were eluted by boiling (10 minutes) in Laemmli sample buffer (Bio-Rad) containing 100 mM DTT. The eluted proteins were subjected to SDS-PAGE and immunoblotted with antibodies against Arv1 and GFP.
Fly stocks and immunostaining of ovaries from Drosophila melanogaster
To determine the putative effects of Arv1 for cell division in vivo, ovaries were dissected from 2–3 d old wild type (w1118) and heterozygous arv1 mutant (BDSC27023, Bloomington Drosophila Stock Center) females fed yeast paste. Ovaries were fixed in 4% formaldehyde (EM grade, Polysciences) for 30 min (room temperature), permeabilized (3x 15 min) and blocked (30 min) in PBS + 0.3% bovine serum albumin (BSA) + 0.3% Triton X-100 (PBT) (room temperature). Specimens were incubated with the primary antibody (anti-Dlg) diluted in PBT at 4°C (overnight), washed 3x 15 minutes in PBT before being incubated with secondary antibody and 1μg/ml Hoechst for 2 hours and finally washed 3x 15 minutes in PBT. The ovaries were mounted in Vectashield (Vector laboratories) and examined using a Zeiss LSM780 confocal microscope.
Statistical analysis
Significance was calculated using a paired, 2-tailed Student's t-test and the p-values are indicated in each experiment.
Abbreviations
| Arv1 | = | ACAT-related protein required for viability |
| EPLIN | = | epithelial protein lost in neoplasm |
| IQGAP | = | IQ-motif-containing GTPase-activating protein |
| MβCD | = | methyl-β-cyclodextrin |
| MYH | = | myosin heavy chain |
| MYL | = | myosin light chain |
| LC-MS/MS | = | liquid chromatography-tandem mass spectrometry |
| PI(4,5)P2 | = | phosphatidylinositol 4,5-bisphosphate |
Disclosure of potential conflicts of interest
The authors declare that they have no conflicts of interest.
Authors' contributions
L.M has performed the experiments presented in , , Figs. S1–4, and Supplementary Movies. L.M initiated and supervised the study. Hi.S performed the experiments shown in . Hi.S and L.M wrote the manuscript. The experiment presented in and the corresponding Material and Method-legends were performed by V.S-G and Hi.S. The LC-MS/MS analysis () was performed by H.M-F. The experiments and analysis performed in Drosophila melanogaster were supervised by K.H. The manuscript was edited by Ha.S. All the authors reviewed and agreed the final version of the manuscript.
KCCY_S_1146834.zip
Download Zip (8.4 MB)Acknowledgments
We would like to thank Dr Camilla Raiborg and Dr Eva Wenzel (Centre for Cancer Biomedicine, the Norwegian Radium Hospital, Oslo University Hospital, Oslo, Norway) for providing helpful discussions regarding cloning, live cell imaging and analysis of mitotic cells. We are grateful to Dr Randi Sjylåsen (Department of Radiation biology, the Norwegian Radium Hospital, Oslo University Hospital, Oslo, Norway) for valuable input in the design and analysis of cell cycle profiles using imaging flow cytometry. The pCA-EPLINα-EGFP and pCA-EPLINβ-EGFP were generously provided by Dr. Masatoshi Takeichi (Laboratory for Cell Adhesion and Tissue Patterning, RIKEN Center for Developmental Biology, 2-2-3 Minatojima-Minamimachi, Chuo-ku, Kobe 650-0047, Japan).
Funding
This work was partly supported by the Research Council of Norway through its Centres of Excellence funding scheme, project number 179571.
Related Research Data
References
- Fededa JP, Gerlich DW. Molecular control of animal cell cytokinesis. Nat Cell Biol 2012; 14:440-7; PMID:22552143; http://dx.doi.org/10.1038/ncb2482
- Jordan SN, Canman JC. Rho GT. Pases in animal cell cytokinesis: an occupation by the one percent. Cytoskeleton (Hoboken, N.J) 2012; 69:919-30.
- Piekny AJ, Maddox AS. The myriad roles of Anillin during cytokinesis. Semin Cell Dev Biol 2010; 21:881-91; PMID:20732437; http://dx.doi.org/10.1016/j.semcdb.2010.08.002
- D'Avino PP, Giansanti MG, Petronczki M. Cytokinesis in animal cells. Cold Spring Harbor Perspect Biol 2015; 7:a015834; PMID:25680833; http://dx.doi.org/10.1101/cshperspect.a015834
- Cauvin C, Echard A. Phosphoinositides: Lipids with informative heads and mastermind functions in cell division. Biochimica et Biophysica Acta 2015; 1851:832-43; PMID:25449648; http://dx.doi.org/10.1016/j.bbalip.2014.10.013
- Atilla-Gokcumen GE, Castoreno AB, Sasse S, Eggert US. Making the cut: the chemical biology of cytokinesis. ACS Chem Biol 2010; 5:79-90; PMID:20014865; http://dx.doi.org/10.1021/cb900256m
- Ng MM, Chang F, Burgess DR. Movement of membrane domains and requirement of membrane signaling molecules for cytokinesis. Dev Cell 2005; 9:781-90; PMID:16326390; http://dx.doi.org/10.1016/j.devcel.2005.11.002
- Fernandez C, Lobo Md Mdel V, Gomez-Coronado D, Lasunción MA. Cholesterol is essential for mitosis progression and its deficiency induces polyploid cell formation. Exp Cell Res 2004; 300:109-20; PMID:15383319; http://dx.doi.org/10.1016/j.yexcr.2004.06.029
- Atilla-Gokcumen GE, Bedigian AV, Sasse S, Eggert US. Inhibition of glycosphingolipid biosynthesis induces cytokinesis failure. J Am Chem Soc 2011; 133:10010-3; PMID:21668028; http://dx.doi.org/10.1021/ja202804b
- Emoto K, Inadome H, Kanaho Y, Narumiya S, Umeda M. Local change in phospholipid composition at the cleavage furrow is essential for completion of cytokinesis. J Biol Chem 2005; 280:37901-7; PMID:16162509; http://dx.doi.org/10.1074/jbc.M504282200
- Liu J, Fairn GD, Ceccarelli DF, Sicheri F, Wilde A. Cleavage furrow organization requires PIP(2)-mediated recruitment of anillin. Curr Biol 2012; 22:64-9; PMID:22197245; http://dx.doi.org/10.1016/j.cub.2011.11.040
- Yoshida S, Bartolini S, Pellman D. Mechanisms for concentrating Rho1 during cytokinesis. Genes Dev 2009; 23:810-23; PMID:19339687; http://dx.doi.org/10.1101/gad.1785209
- Logan MR, Mandato CA. Regulation of the actin cytoskeleton by PIP2 in cytokinesis. Biol Cell / Under Auspices Euro Cell Biol Organization 2006; 98:377-88; PMID:16704377; http://dx.doi.org/10.1042/BC20050081
- Takeda T, Kawate T, Chang F. Organization of a sterol-rich membrane domain by cdc15p during cytokinesis in fission yeast. Nat Cell Biol 2004; 6:1142-4; PMID:15517003; http://dx.doi.org/10.1038/ncb1189
- Wachtler V, Rajagopalan S, Balasubramanian MK. Sterol-rich plasma membrane domains in the fission yeast Schizosaccharomyces pombe. J Cell Sci 2003; 116:867-74; PMID:12571284; http://dx.doi.org/10.1242/jcs.00299
- Field SJ, Madson N, Kerr ML, Galbraith KA, Kennedy CE, Tahiliani M, Wilkins A, Cantley LC. PtdIns(4,5)P2 functions at the cleavage furrow during cytokinesis. Curr Biol 2005; 15:1407-12; PMID:16085494; http://dx.doi.org/10.1016/j.cub.2005.06.059
- Tinkelenberg AH, Liu Y, Alcantara F, Khan S, Guo Z, Bard M, Sturley SL. Mutations in yeast ARV1 alter intracellular sterol distribution and are complemented by human ARV1. J Biol Chem 2000; 275:40667-70; PMID:11063737; http://dx.doi.org/10.1074/jbc.C000710200
- Swain E, Stukey J, McDonough V, Germann M, Liu Y, Sturley SL, Nickels JT Jr. Yeast cells lacking the ARV1 gene harbor defects in sphingolipid metabolism. Complementation by human ARV1. J Biol Chem 2002; 277:36152-60; PMID:12145310; http://dx.doi.org/10.1074/jbc.M206624200
- Kajiwara K, Watanabe R, Pichler H, Ihara K, Murakami S, Riezman H, Funato K. Yeast ARV1 is required for efficient delivery of an early GPI intermediate to the first mannosyltransferase during GPI assembly and controls lipid flow from the endoplasmic reticulum. Mol Biol Cell 2008; 19:2069-82; PMID:18287539; http://dx.doi.org/10.1091/mbc.E07-08-0740
- Tong F, Billheimer J, Shechtman CF, Liu Y, Crooke R, Graham M, Cohen DE, Sturley SL, Rader DJ. Decreased expression of ARV1 results in cholesterol retention in the endoplasmic reticulum and abnormal bile acid metabolism. J Biol Chem 2010; 285:33632-41; PMID:20663892; http://dx.doi.org/10.1074/jbc.M110.165761
- Georgiev AG, Johansen J, Ramanathan VD, Sere YY, Beh CT, Menon AK. Arv1 regulates PM and ER membrane structure and homeostasis but is dispensable for intracellular sterol transport. Traffic (Copenhagen, Denmark) 2013; 14:912-21.
- Villasmil ML, Ansbach A, Nickels JT, Jr. The putative lipid transporter, Arv1, is required for activating pheromone-induced MAP kinase signaling in Saccharomyces cerevisiae. Genetics 2011; 187:455-65; PMID:21098723; http://dx.doi.org/10.1534/genetics.110.120725
- Golsteyn RM, Schultz SJ, Bartek J, Ziemiecki A, Ried T, Nigg EA. Cell cycle analysis and chromosomal localization of human Plk1, a putative homologue of the mitotic kinases Drosophila polo and Saccharomyces cerevisiae Cdc5. J Cell Sci 1994; 107 (Pt 6):1509-17; PMID:7962193
- Chircop M, Oakes V, Graham ME, Ma MP, Smith CM, Robinson PJ, Khanna KK. The actin-binding and bundling protein, EPLIN, is required for cytokinesis. Cell Cycle 2009; 8:757-64; PMID:19221476; http://dx.doi.org/10.4161/cc.8.5.7878
- Eppinga RD, Li Y, Lin JL, Lin JJ. Tropomyosin and caldesmon regulate cytokinesis speed and membrane stability during cell division. Arch Biochem Biophys 2006; 456:161-74; PMID:16854366; http://dx.doi.org/10.1016/j.abb.2006.06.015
- Pathmanathan S, Hamilton E, Atcheson E, Timson DJ. The interaction of IQGAPs with calmodulin-like proteins. Biochem Soc Trans 2011; 39:694-9; PMID:21428964; http://dx.doi.org/10.1042/BST0390694
- Tan JL, Ravid S, Spudich JA. Control of nonmuscle myosins by phosphorylation. Annu Rev Biochem 1992; 61:721-59; PMID:1497323; http://dx.doi.org/10.1146/annurev.bi.61.070192.003445
- Trybus KM. Assembly of cytoplasmic and smooth muscle myosins. Curr Opin Cell Biol 1991; 3:105-11; PMID:1854473; http://dx.doi.org/10.1016/0955-0674(91)90172-U
- Park I, Han C, Jin S, Lee B, Choi H, Kwon JT, Kim D, Kim J, Lifirsu E, Park WJ, et al. Myosin regulatory light chains are required to maintain the stability of myosin II and cellular integrity. Biochem J 2011; 434:171-80; PMID:21126233; http://dx.doi.org/10.1042/BJ20101473
- Liscum L, Faust JR. The intracellular transport of low density lipoprotein-derived cholesterol is inhibited in Chinese hamster ovary cells cultured with 3-beta-[2-(diethylamino)ethoxy]androst-5-en-17-one. J Biol Chem 1989; 264:11796-806
- Smith TC, Fang Z, Luna EJ. Novel interactors and a role for supervillin in early cytokinesis. Cytoskeleton (Hoboken, N.J) 2010; 67:346-64; http://dx.doi.org/10.1002/cm.20449
- Mishra M, Kashiwazaki J, Takagi T, Srinivasan R, Huang Y, Balasubramanian MK, Mabuchi I. In vitro contraction of cytokinetic ring depends on myosin II but not on actin dynamics. Nat Cell Biol 2013; 15:853-9; PMID:23770677; http://dx.doi.org/10.1038/ncb2781
- Rincon SA, Paoletti A. Mid1/anillin and the spatial regulation of cytokinesis in fission yeast. Cytoskeleton (Hoboken, N.J) 2012; 69:764-77.
- Guha M, Zhou M, Wang YL. Cortical actin turnover during cytokinesis requires myosin II. Curr Biol 2005; 15:732-6; PMID:15854905; http://dx.doi.org/10.1016/j.cub.2005.03.042
- Murrell MP, Gardel ML. F-actin buckling coordinates contractility and severing in a biomimetic actomyosin cortex. Proc Natl Acad Sci U S A 2012; 109:20820-5; http://dx.doi.org/10.1073/pnas.1214753109
- Bielak-Zmijewska A, Kolano A, Szczepanska K, Maleszewski M, Borsuk E. Cdc42 protein acts upstream of IQGAP1 and regulates cytokinesis in mouse oocytes and embryos. Developmental biology 2008; 322:21-32; PMID:18662680; http://dx.doi.org/10.1016/j.ydbio.2008.06.039
- Lian AT, Hains PG, Sarcevic B, Robinson PJ, Chircop M. IQGAP1 is associated with nuclear envelope reformation and completion of abscission. Cell Cycle 2015; 14:2058-74; PMID:25928398; http://dx.doi.org/10.1080/15384101.2015.1044168
- Boyne JR, Yosuf HM, Bieganowski P, Brenner C, Price C. Yeast myosin light chain, Mlc1p, interacts with both IQGAP and class II myosin to effect cytokinesis. J Cell Sci 2000; 113 Pt 24:4533-43.
- Shannon KB, Li R. A myosin light chain mediates the localization of the budding yeast IQGAP-like protein during contractile ring formation. Curr Biol 2000; 10:727-30; PMID:10873803; http://dx.doi.org/10.1016/S0960-9822(00)00539-X
- Takaine M, Numata O, Nakano K. Fission yeast IQGAP maintains F-actin-independent localization of myosin-II in the contractile ring. Genes Cells: Devoted Mol Cell Mech 2013; 19:161-76; PMID:24330319
- Hedman AC, Smith JM, Sacks DB. The biology of IQGAP proteins: beyond the cytoskeleton. EMBO Rep 2015; 16:427-46; PMID:25722290; http://dx.doi.org/10.15252/embr.201439834
- Adachi M, Kawasaki A, Nojima H, Nishida E, Tsukita S. Involvement of IQGAP family proteins in the regulation of mammalian cell cytokinesis. Genes Cells: Devoted Mol Cell Mech 2014; 19:803-20; http://dx.doi.org/10.1111/gtc.12179
- Dean SO, Rogers SL, Stuurman N, Vale RD, Spudich JA. Distinct pathways control recruitment and maintenance of myosin II at the cleavage furrow during cytokinesis. Proc Natl Acad Sci U S A 2005; 102:13473-8; http://dx.doi.org/10.1073/pnas.0506810102
- Abe M, Makino A, Hullin-Matsuda F, Kamijo K, Ohno-Iwashita Y, Hanada K, Mizuno H, Miyawaki A, Kobayashi T. A role for sphingomyelin-rich lipid domains in the accumulation of phosphatidylinositol-4,5-bisphosphate to the cleavage furrow during cytokinesis. Mol Cell Biol 2012; 32:1396-407; PMID:22331463; http://dx.doi.org/10.1128/MCB.06113-11
- Rogeberg M, Wilson SR, Malerod H, Lundanes E, Tanaka N, Greibrokk T. High efficiency, high temperature separations on silica based monolithic columns. Journal of chromatography. A 2011; 1218:7281-8;
- Miyamoto K, Hara T, Kobayashi H, Morisaka H, Tokuda D, Horie K, Koduki K, Makino S, Núñez O, Yang C, et al. High-efficiency liquid chromatographic separation utilizing long monolithic silica capillary columns. Anal Chem 2008; 80:8741-50; PMID:18947204; http://dx.doi.org/10.1021/ac801042c
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods (San Diego, Calif) 2001; 25:402-8.