
ABSTRACT
MicroRNAs (miRNAs) in the AGO-containing RISC complex control messenger RNA (mRNA) translation by binding to mRNA 3′ untranslated region (3′UTR). The relationship between miRNAs and other regulatory factors that also bind to mRNA 3′UTR, such as CPEB1 (cytoplasmic polyadenylation element-binding protein), remains elusive. We found that both CPEB1 and miR-15b control the expression of WEE1, a key mammalian cell cycle regulator. Together, they repress WEE1 protein expression during G1 and S-phase. Interestingly, the 2 factors lose their inhibitory activity at the G2/M transition, at the time of the cell cycle when WEE1 expression is maximal, and, moreover, rather activate WEE1 translation in a synergistic manner. Our data show that translational regulation by RISC and CPEB1 is essential in cell cycle control and, most importantly, is coordinated, and can be switched from inhibition to activation during the cell cycle.
Introduction
The importance of 3′UTR (3′ untranslated region) in the control of translation and mRNA stability has been known for many years. One of the best-studied RNA regulatory sequences located in 3′UTRs is the CPE (cytoplasmic polyadenylation element). CPE is bound by CPEB1 (cytoplasmic polyadenylation element binding protein), a 4-member protein family.
CPEB1 was initially found to be important for Xenopus oocyte maturation.Citation1 It was later shown to be involved in learning and memory Citation2,3 and in the regulation of the mammalian cell cycle.Citation4-7 During Xenopus oocyte maturation, CPEB1 controls meiosis progression from prophase I to metaphase II, triggering tightly controlled waves of polyadenylation at various phases of meiosis,Citation8 as well as during the embryonic cell-cycle.Citation5 In mammals, CPEB1 is also implicated in senescence Citation4,6 and in controlling the translation of proteins involved in cell-cycle checkpoints.Citation7 CPEB1 is a conserved, sequence-specific RNA-binding protein containing a zinc finger and 2 RNA recognition motifs (RRMs).Citation1,8-10 Xenopus studies show that CPEB1 can both promote and inhibit RNA translation by respectively elongating or shortening mRNA poly(A) tails since CPEB1 recruits adenylating and/or de-adenylating protein complexes. This dual action of CPEB1 changes over the course of the cell cycle, depending on CPEB1 post-transcriptional modifications and on the number and location of the CPEs to which CPEB1 binds. The CPEB1-containing complex in Xenopus includes: symplekin, which may be a platform protein upon which multi-component complexes are assembled; poly(A) ribonuclease (PARN), which is a deadenylating enzyme, and germ-line-development factor 2 (Gld2), which is a poly(A) polymerase.Citation11,12 Induction of cytoplasmic polyadenylation is mediated by activation of the serine/threonine kinase Aurora A/Eg2, possibly via repression of glycogen synthase kinase 3.Citation10,13 When phosphorylated on either S174 or T171 (which is species-dependent), CPEB1 promotes polyadenylation by stimulating the activity of Gld-2,Citation11 an atypical poly(A) polymerase.Citation14 The newly elongated tail is then bound by the poly(A)-binding protein (PABP), which promotes translation by facilitating assembly of the eIF4F initiation complex.Citation15
The miRNA (microRNA) system is another well-known regulator of mRNA translation. MicroRNAs are single-stranded RNA molecules of about 21–23 nucleotides in length, which are transcribed as 70–90 nt precursors and further processed to short double-stranded sequences by the endonuclease DICER. MiRNAs regulate gene expression by forming miRNA-induced silencing complexes (miRISCs). MiRISCs inhibit translation by binding through the microRNA strand to imperfectly matched sequences in the 3′UTR of target mRNAs. The MiRNA mode of action is a much-debated issue. However, there are exist experimental proofs supporting collaboration between the RISC complex, which contains the proteins argonaute 1 and 2 (AGO1 and AGO2), and the deadenylation complex.Citation16,17 The mRNA targets of miRNAs are frequently subject to deadenylation,Citation18,19 further supporting the idea that the length of the poly(A) tail is a key element in the control of translation by miRNAs. Thus, both miRNAs and CPEB1 control the length of mRNA poly(A) tails, raising the possibility that they may cooperate to regulate common targets.
CPEB1 and RISC complexes have been found in processing bodies (P-bodies), which are sites of mRNA degradation and storage, as well as in stress granules, where translation initiation complexes are stored under various stress conditions. It is worth mentioning that DDX6 (rck/p54), a DEAD-box helicase that interacts with AGO1 and AGO2 in cells and is essential in P-bodies Citation20 and stress granules, associates with CPEB1 in both Spisula (clam p47) and Xenopus Citation21 and could therefore constitute a link between CPEB1 and the RISC complex.
All these observations suggest that CPEB1 and RISC cooperate. Here, we address this issue by investigating the functional interaction between CPEB1 and a nearby miRNA-binding site on the 3′UTR of WEE1 mRNA. WEE1 is a kinase component of the G2/M cell-cycle checkpoint. WEE1 determines the time of entry into mitosis, thereby influencing the size of daughter cells. Loss of WEE1 results in smaller than normal daughter cells, due to premature cell division. Although WEE1 kinase has long been characterized as a key inhibitor of cyclin-dependent kinase 1 (Cdk1) and of mitotic entry in eukaryotes, the regulation of WEE1 expression and activity is still not fully understood. WEE1 is regulated at the post-translational level by phosphorylation.Citation22 During Xenopus oocyte maturation, WEE1 mRNA translation is regulated by a CPE sequence located in its 3′UTR.Citation23 WEE1 mRNA CPE is conserved in the human. In addition, the 3′UTR of human WEE1 mRNA contains a miR-15b binding site, and WEE1 is one of the high-score predicted targets for miR-15b (using 2 algorithms, Targetscan and Microcosm). And it was indeed proved that WEE1 could be a target of miR-15b under a certain physiological condition.Citation24 We therefore explored how each of these 2 elements of WEE1 3′UTR, the CPE and miR-15b binding sites, function during the mammalian cell cycle.
We found that the CPE and miR-15b sequences in WEE1 3′UTR are both important for WEE1 expression, being independent inhibitors of WEE1 3′UTR-dependent translation in G1 and S cells. CPEB1 and AGO2 (RISC) complexes interacted in an RNA- and DDX6 (rck/p54)-dependent manner. Importantly, we observed that the inhibitory effects of miR-15/RISC and CPEB1 were abolished during G2/M transition. Moreover, the 2 sequences together had an activating effect at this stage of the cell cycle when WEE1 protein is maximal, as documented by luciferase assays or polysomal profiles of reporter mRNAs, as well by analyzing WEE1 endogenous protein in CPEB1- and/or miR-15b-depleted cells.
Our data demonstrate that WEE1 expression is regulated at the translational level in a cell-cycle-dependent manner. Moreover, they show that the regulatory activities of 2 RNA binding complexes are modified in a coordinated manner, switching from being activators to inhibitors during the cell cycle.
Results
WEE1is regulated at the prost-transcriptional level in a 3′UTR-dependent manner
WEE1 expression is regulated at the level of translation during Xenopus oocyte maturation.Citation23 In HeLa cells synchronized by a double thymidine block, WEE1 protein expression was regulated in a cell-cycle dependent manner (), increasing at the G2/M transition. This regulation occurred mostly at the post-transcriptional level, since WEE1 mRNA did not vary significantly during cell-cycle progression ().
Figure 1. Regulation of WEE1 in HeLa cells depends on CPE and miR-15b binding sites. (A) Cell cycle distribution (by FACS analysis) of HeLa cells at indicated times after release from a double thymidine block. (B) Western blot analyses of WEE1 expression in HeLa cells after release from a double thymidine block; actin is used as a loading control. (C) QPCR analyses of WEE1 mRNA, in the same cells; Cyclo A was used to standardize the data. (D) Schematic representation of R-luc WEE1 3′UTR fusions (WEE1 3′UTR was introduced into the 3′UTR of Renilla luciferase reporters). (E) Luciferase expression of the reporter constructs described in D following transfection into HeLa cells; Renilla enzymatic activity (LucR) was normalized to Firefly (control, LucF). Statistics: Student's t test; *: t = 0.05.
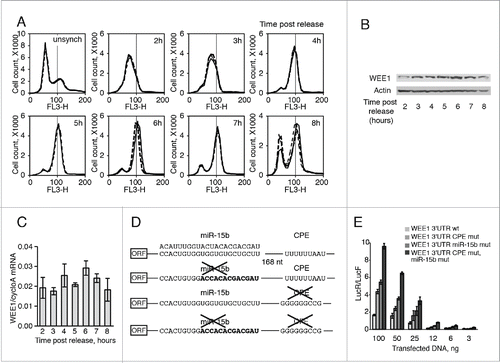
In Xenopus oocyte, Wee1 3′UTR includes 3 CPEs, and Wee1 mRNA translation is controlled by CPEB1 (Charlesworth et al., 2000). Human WEE1 mRNA contains few potential CPEs sites as well as few potential hexonucleotide sites (Fig. S1). Interestingly, an miR-15b binding site is located 168 nucleotides upstream of the first CPE-site (). In order to explore the role of these elements in WEE1 regulation, we generated Renilla-luciferase reporter constructs containing, either wild type or mutated WEE1 3′UTR in the first CPE or the miR-15b sites, alone or in combination (). We focused our attention on the first CPE, because of its close location to the miR-15b site. In HeLa cells, reporter activity was stimulated by mutations in the CPE or miR-15b site (). Mutating the 2 sites led to higher expression than mutating each one alone, and the effects were additive. These data suggest that both CPEB1 and miR-15b control WEE1 expression in an independent manner. To further confirm this hypothesis, we used CPEB1Citation25 and/or miR-15b Citation26 loss-of-function assays. First, we confirmed the activity of the inhibitors by Western blot analysis and QRT-PCR, respectively (). Treating the cells with either one of the inhibitors increased the activity of the wild-type WEE1 3′UTR reporter, while simultaneous depletion of CPEB1 and inhibition of miR-15b was even more effective (), well correlated with the results obtained by mutating CPEB1 and miR-15b binding sites (). It is important to note that depletion of CPEB1 did not affect luciferase activity in the absence of the CPE sequence, nor did inhibition of miR-15b affect luciferase in the absence of the miR-15b binding site (), underscoring the specificity of the inhibition. Thus both CPEB1 and the RISC complexes appear to regulate WEE1 mRNA through their respective binding sites. This hypothesis was further supported by the subcellular localization of WEE1 reporter mRNA, CPEB1, and miR-15b (). The three components were observed in dots that are likely to be P-bodies or stress granules, since they contain DDX6/p54 or TIA1 (Fig. S2 A, B, C). Some granules containing the reporter mRNA also contained both miR-15b and CPEB1. Mutation of each binding site alone did not reduce the presence of the reporter mRNA in dots containing the other partner, but mutation of the 2 sites did abolish the recruitment of the reporter mRNA into granules ( and S3A, B).
Figure 2. MiR-15b and CPEB1 control WEE1 expression. (A) Depletion of CPEB1 by siRNAs (Western blot). (B) Depletion of miR-15b by an antisense LNA (QPCR); (C) Influence of CPEB1 and/or miR-15b depletion on WEE1 3′UTR reporter luciferase activity transfected into HeLa cells along with inhibitors as indicated. Controls were miRNA (Applied Biosystems) and AllStars (Qiagen). Numbers indicate the ratio LucR/LucF for each sample. (D) Localization of WEE1 mRNA, CPEB1, and miR-15b in HeLa cells. HeLa cells were transfected with Cy5-labeled miR-15b, CPEB1-Cherry, FITC-labeled WEE1 3′UTR (wt or mutant as indicated). Arrows point to granules with co-localized WEE1 mRNA, CPEB1, and/or miR-15b. (E) CPEB1 and miR-15b control WEE1 protein expression. For gain-of-function assays, HeLa cells were transfected with a construct expressing CPEB1-Cherry (or Cherry as a control), or with miR-15b precursor (or a control miRNA); for loss-of-function assays, cells were transfected with the inhibitors described in A and B; WEE1 expression was monitored 48h later by Western blot. (F) Overexpression of miR-15b monitored by QPCR. (G) Overexpression of CPEB1 monitored by Western blot.
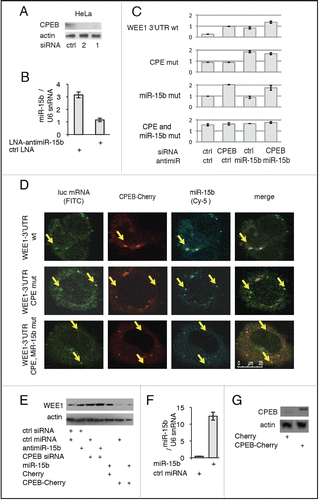
We next tested whether endogenous WEE1 protein was affected by modulating CPEB1 and/or miR-15b levels. When CPEB1 and/or miR-15b were down-regulated or inhibited (), endogenous WEE1 protein was increased (, S4B). Importantly, depletion of CPEB1 and/or miR-15b had little effect on WEE1 mRNA levels (Fig. S4C). Conversely, overexpressing CPEB1 () reduced WEE1 protein expression (). Overexpressing miR-15b () did not impact on WEE1 protein, possibly due to the fact that this miRNA is already highly expressed in HeLa cells.
Interaction between CPEB1 and miR-15b/RISC
Since the CPE and miR-15b binding sites are in close proximity, next we used co-immunoprecipitation experiments to investigate whether CPEB1 and RISC could physically interact. AGO2 co-immunoprecipitated with overexpressed FLAG-Myc-CPEB1, but this interaction was RNA-dependent (). Conversely, CPEB1 co-immunoprecipitated with exogenous FLAG-HA-AGO2, and this was also dependent on the presence of RNA (). Thus, the 2 complexes may bind to common mRNA targets.
Figure 3. Interaction between CPEB1 and AGO2 is RNase and DDX6 dependent. (A) Co-immunoprecipitation of endogenous AGO2 with CPEB1-Myc-FLAG; cells were transfected with a tagged CPEB1 (CPEB1-Myc-FLAG) expression vector or an empty vehicle vector as a control (vector); cell extracts were immunoprecipitated using anti-FLAG antibodies, following RNase treatment when indicated, and monitored by Western blot; inp: input; IP: immunoprecipitates. (B) Cells were transfected with a tagged AGO2 (AGO2-FLAG-HA) expression vector or an empty vehicle vector as a control (vector); cell extracts were immunoprecipitated using anti-FLAG antibodies, following RNase treatment when indicated. (C) DDX6 is required for AGO2/CPEB1 interaction. Cells treated for 48h with an siRNA to DDX6 (or a control siRNA) were transfected and treated as in A. (D) Cells were transfected with CPEB1-Myc-FLAG and AGO2-FLAG-HA expression vectors, along with wild type (wt) or transdominant DDX6 helicase dead mutant (mut) expression vectors; extracts were immunoprecipitated with anti-HA antibodies and analyzed as in A. (E) Cells were transfected with wild-type DDX6 (wt) or transdominant DDX6 mutant (mut) together with CPEB1-Myc-FLAG or AGO2-FLAG-HA expression vectors; extracts were immunoprecipitated with anti-FLAG antibodies and analyzed as in A and B.
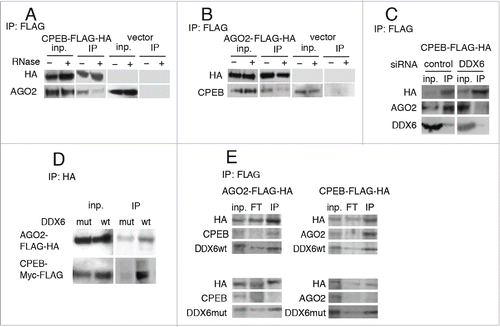
As previously published,Citation20,21 we detected the helicase DDX6 in both CPEB1 and AGO2 FLAG-tag immunoprecipitated complexes (, E). Knocking-down DDX6 (with an siRNA) or inhibiting its activity with a helicase-dead transdominant mutant Citation27 abolished interaction between CPEB1 and AGO2 ().
The activity of CPEB1 and miR-15/RISC is coordinated during the cell cycle
WEE1 is regulated in a cell-cycle-dependent manner (), and the activity of CPEB1 is also cell cycle dependent in Xenopus embryo, CPEB1 acting alternatively as a repressor and an activator of gene translation.Citation31 We therefore investigated the importance of CPEB1 and miR-15b for WEE1 expression during the cell cycle, after releasing HeLa cells from a double thymidine block (). Surprisingly, we observed that when cells progressed toward G2/M, not only the CPE, but also the miR-15b sites lost their inhibitory activity (). Coincidentally, silencing granules that contained WEE1 mRNA disappeared in G2/M cells (Fig. S5A), implying that WEE1 mRNA is released into the cytoplasm and may be actively translated.
Figure 4. CPEB1 and miR-15b inhibitors switch to activators at G2/M. (A) Cell-cycle analysis (by FACS) of HeLa cells after release from a double thymidine block unsync, -unsynchronized cells. (B) HeLa cells analyzed in A were transfected after they were released from thymidine block with WEE1 3′UTR reporter constructs (described in ) and 20 h later, cell extracts were submitted to luciferase assays. Renilla luciferase activity was normalized to Firefly activity; the position of the majority of the cells in the cell cycle in the samples is indicated above the bars; Statistics: Student's t test; *: t = 0.05. (C) WEE1 expression along the cell cycle as monitored by Western blot; cyclin B1 is shown to document the position of the cells in the cell cycle. (D, E) Distribution of WEE1 3′UTR reporter mRNAs in polysomal gradients at the G0/G1 or G2/M phase of the cell cycle. Fractions 0–3 correspond to non polysomal fractions (NP), 4–7 to low polysomal fractions (LP), and 8–12 to heavy polysomal fractions (HP). GAPDH mRNA distribution among the polysomal fractions in 2 cell-lines that have been transfected by WEE1 3′UTR reporter wt and with CPE and miR-15b sites mutations were used as control.
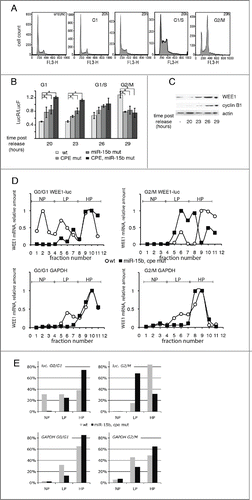
Moreover, WEE1 protein expression was maximal upon entering G2/M (). Both sites tend to have an opposite effect, which is to stimulate translation. Note that in this case, the activities of the 2 sites were synergistic rather than additive, both sites being required for activation. A more detailed time-course analysis (hourly, beginning 28 hours after transfection, Fig. S5B) showed that during G2/M, the CPE and miR-15b sequences in WEE1 3′UTR sequentially lost their inhibitory activity, then, when most of the cells had progressed into G2/M, together they became slightly stimulating for luciferase expression (Fig. S5C). When the cells returned to the G0/G1 cell-cycle stage, the inhibitory functions of CPE and miR-15b binding sites were restored (Fig. S5C).
Thus, CPEB1 and miR-15b seem to lose their inhibitory activity at the G2/M phase of the cell cycle. CPEB1 protein was down regulated during G2/M, but CPEB1 mRNA level was stable (Fig. S5D, E), similar to what was observed during Xenopus oocyte maturation. MiR-15b, were constitutively expressed throughout the cell cycle (Fig. S5F). Importantly, the WEE1 mRNA level did not change throughout the cell cycle (Fig. S5G), as observed earlier ().
Our results support the hypothesis that WEE1 translation is regulated in a cell-cycle-dependent manner. In order to further explore this hypothesis, we monitored WEE1 reporter translation throughout the cell cycle. Active translation of mRNAs can be monitored by their presence in the heavy fractions of a sucrose gradient, which is interpreted as association of mRNA with multiple ribosomes in polysomes. In a population of predominantly G0/G1 cells (83%), WEE1 wild-type 3′UTR was distributed into 3 peaks, with more than half in the monosomal or light polysomal fractions (, E, fractions 1 to 7), implying low translational activity. When only the CPE or, alternatively, the miR-15b sequence was mutated, the reporter mRNA shifted toward the heavier polysomal fractions (fractions 5 to 8) (Fig. S6A, C). Mutating both the miR-15b and CPE sites eliminated the 2 light peaks, and the reporter was then mostly associated with heavy polysomes (fractions 9 to 12), indicating high translational activity (, E). In a population with at least 50% of cells in G2/M, wt WEE1 3′UTR was mostly associated with the heavy polysomal fractions (fractions 8 to 12). However, when the miR-15b binding site, the CPE site, or both were mutated, the mRNA shifted to the lighter polysomal fractions (fractions 5 to 8) ( and S6A, C). This result correlates well with the reduced luciferase activity expressed by the same constructs in G2/M cells (). Moreover, also in this assay, mutating one of the 2 sites had the same effect in G2/M cells as mutating both of them ( and S6A, C), implying synergy between the 2 complexes. The control GAPDH mRNA was constitutively associated with heavy polysomal fractions in all samples ( and S6B). These data amount to strong support of the idea that CPEB1 and miR-15b activity on WEE1 mRNA depends on the cell cycle, with both regulators being inhibitors in the G1 and/or S phases and activators at G2/M.
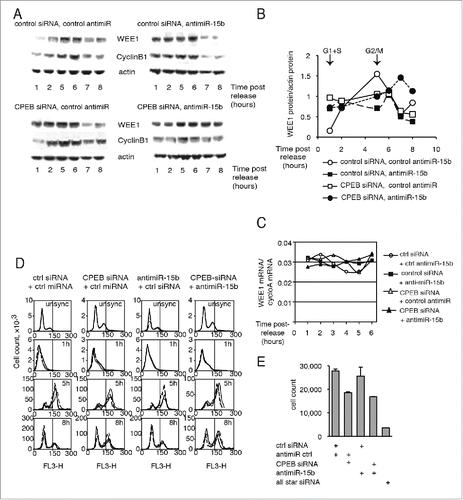
This idea was further supported by analysis of WEE1 protein accumulation during the cell cycle, since WEE1 protein was deregulated by CPEB1 and/or miRNA depletion (). In control cells, WEE1 protein was low in G1 or S cells (1h post-release) and increased at G2/M (5h post-release), whereas WEE1 mRNA levels remained quite stable throughout the cell cycle (). When both CPEB1 and miR-15b were depleted, WEE1 protein and, coincidently, Cyclin B1, used here as a cell-cycle marker, were deregulated, showing little variation in the time frame of the experiment (). Most interestingly, when miR-15b, CPEB1, or both were depleted, WEE1 protein was higher in G1/S cells than in control cells (), as expected if the 2 regulators are inhibitors. In sharp contrast, in G2/M cells (5 h post-release) depletion of CPEB1, miR-15b, or both resulted in reduced WEE1 expression, as expected for activators. Even though Western blot is not a fully quantitative technique, these data clearly indicate a tendency. Thus, the expression of endogenous WEE1 protein appears to be dependent on miR-15b and CPEB1 in G2/M cells, consistent with the data obtained using the WEE1 reporter construct (). Depletion of DDX6 also affected the expression of WEE1 and Cyclin B1 (Fig. S7). Together, these data indicate that CPEB1 and miR-15b/RISC are required for proper expression of WEE1 protein during the cell cycle.
Figure 5. Depletion of CPEB1 or miR-15b affects both the cell-cycle-dependent accumulation of WEE1 and cell-cycle progression. (A) Cells were transfected with a siRNA to CPEB1, an antisense to miR-15b, or control molecules as indicated and submitted to a double thymidine block as in ; WEE1 or Cyclin B1 expression was monitored by Western blot. (B) Quantification of the Western blots shown in (A) after standardization to actin. (C) WEE1 mRNA expression in the same cells, monitored by Q-RT-PCR. (D) Cell-cycle distribution (analyzed by FACS) of HeLa cells after depletion of CPEB1 and/or miR-15b. (E) HeLa cell-count after CPEB1 and/or miR-15b depletion.
Since WEE1 regulates the temporality of cell-cycle progression, affecting WEE1 expression, it should also impact cell-cycle progression and cell proliferation.
A significant change was observed in cell distribution during the cell cycle in CPEB1-depleted cells, indicating a delayed exit from the G2/M phase (). This effect was enhanced when CPEB1 and miR-15b were depleted simultaneously (). This result is consistent with the observation that depletion of WEE1 accelerates the G2/M phase in Xenopus. Here, depletion of CPEB1 and miR-15b increased WEE1 expression and resulted in delayed exit from G2/M.
Cell growth was also affected by CPEB1 depletion (), as documented by the decreased number of cells; however, inhibiting miR-15b had little effect on cell counts.
Taken together, these data show that WEE1 is regulated at the translational level by CPEB1 and miR-15b in a coordinated and cell-cycle-dependent manner.
Discussion
We have analyzed the regulation of WEE1, an oncogenic cell-cycle regulator, by the RNA binding complexes CPEB1 and miR-15b/RISC. We show that miR-15b/RISC and CPEB1 have a coordinated impact on WEE1 translation. There are few examples of coordination between RISC and other RNA binding proteins. Under stress conditions, the inactivation, storage, and reactivation of CAT-1 mRNA depend on the binding of both miR-122 and HuR, an AU-rich-element-binding protein, to the 3′UTR of CAT-1 mRNA.Citation28 Cooperation between miR-16 (a member of the miR-15/16 family of miRNPs) and an ARE-binding protein called TTP (tristetraprolin) was shown to occur through the association of TTP with AGO/eIF2C family members. MiR-16 assists TTP in targeting ARE, which appears to be an essential step in ARE-mediated mRNA degradation.Citation29 Our data not only constitute an additional example of such a coordinated action, but also demonstrate that such a coordinated action can be cell-cycle-dependent, the 2 sites being inhibitors during G1 and S and activators during G2/M. In G1/S, the 2 regulators are at least in part colocalized in cytoplasmic granules (), which may be silencing granules (since they include DDX6/p54, Fig. S2). These granules disappear at G2/M Citation30 and Fig. S5A. This could at least in part account for decreased inhibition by CPEB1 and miR-15b at G2/M. However, we observed that at G2/M the 2 sites not only lost their inhibitory activity, but also had a tendency to activate WEE1 mRNA translation, since their deletion decreased both the activity of a reporter construct ( and Fig. S5C) and the ribosome loading of the reporter mRNAs, as monitored by their distribution in heavy polysomal fractions (). Moreover, depletion of CPEB1 and miR-15b affected the expression of the endogenous protein in an opposite manner in G1/S or G2/M cells (). CPEB1 is known to act as a cell-cycle-dependent activator of translation by elongating mRNA poly(A) tails during Xenopus oocyte maturation and during the transition to M phase in Xenopus embryo.Citation9,31 But such a cell-cycle-dependent switch is much less expected for the RISC complex, and translational activation by RISC complexes is a rather controversial issue. There are, however, examples of such switches in the RISC complex in the literature. It has been shown that upon cell-cycle arrest, the ARE (AU-rich element) in tumor necrosis factor-α (TNFα) mRNA is transformed into a translation activation signal, recruiting AGO and fragile X mental retardation-related protein 1 (FXR1), factors associated with miRNPs. Human miRNA miR-369-3 directs the association of these proteins with the AREs, leading to translation activation (Vasudevan et al., 2008). Moreover, 2 well-studied miRNAs – let-7 and the synthetic miRNA miRcxcr4 – likewise induced translational up-regulation of target mRNAs upon cell-cycle arrest, yet they repressed translation in proliferating cells. It has been proposed that translation regulation by miRNPs oscillates between repression and activation during the cell cycle.Citation32 It is quite possible that switches of activity by the RISC complex depend on the switch in activity of other 3′UTR-binding proteins, such as CPEB1. Indeed, here, in the absence of CPE, miR-15b did not activate WEE1 3′UTR in a reporter construct.
CPEB1 activity is regulated by phosphorylation during the Xenopus embryo cell cycle.Citation10 It is likely that similar phosphorylation is involved in HeLa cells. Phosphorylation, possibly by the same enzyme, might also be responsible for the modification of miR-15b /RISC activity. In Xenopus embryo, CPEB1 phosphorylation results in the modification of the composition of the CPEB1 complex, which recruits a polymerase rather than a deadenylase. Thus, we speculate that at G2/M, CPEB1 and miR-15b/RISC recruit activating proteins rather than inhibitors, possibly increasing the size of the poly(A) tail of WEE1 mRNA. Most interestingly, whereas the inhibiting activities of the 2 complexes were acting independently of each other, activation required the integrity of each of the 2 sites, implying that the 2 complexes cooperate for this action. This was further supported by the observation that independently mutating each site also reduced the association of WEE1 mRNAs with the heavy polysomal fraction at G2/M, indicative of reduced translation. Thus, it appears that whereas each of the complexes can independently recruit translation inhibitors, they need to cooperate to recruit translation activators. The mechanism of this cooperation is elusive. The two complexes do not appear to interact directly in unsynchronized cells, which are mostly in G1/S, since most of the interaction is sensitive to RNase (). It is quite possible however, that the 2 complexes do interact in G2/M cells, which would account for the residual binding observed after RNase treatment (), since a small fraction of cells are in G2/M in the unsynchronized population. We will try to verify it in our future work.
In summary, here we demonstrate a cell cycle-dependent, coordinated action of a miRNA (miR-15b) and an RNA-binding protein (CPEB1) to inhibit WEE1 mRNA translation in G1/S and activate it in G2/M, providing an example of coordination between a miRNA/RISC complex and another RNA binding protein, as well as, even more importantly, of a cell cycle-dependent switch from inhibitor to activator of the 2 complexes.
Materials and methods
Cell lines and constructs
HeLa and HeLa S3 cells were maintained in DMEM media supplemented with 10% FCS, 100 mg/ml streptomycin, and 100 U/ml penicillin (Gibco®) cultured at 37°C and 5% CO2.
The WEE1 3′UTR sequence (nt 2274–2524, [GeneBank: NM 001143976.1]) was cloned into XbaI/NotI sites in the pRL-TK vector (Promega). An miR-15b site substitution mutation was introduced by replacing the 2316–2325 sequence in the WEE1 3′UTR by accacacgcga. CPE site mutation was accomplished by replacing ttttttaatt (2494–2504) with ggggggccgg in the WEE1 3′UTR. Double miR-15b and CPE mutations were done by simultaneous replacement of both sites.
CPEB1 transcript variant 1 [GeneBank: NM_030594.3] was cloned into XhoI/EcoRI sites of the pmCherry-N1 expression vector.
In transfection assays, pmCherry-N1 (632523, Clontech) was used as control.
MiRNA precursor molecules (Applied Biosystems) used were: miR-15b (PM17100); MiRNA inhibitors (Applied Biosystems) used were: miR-15b (AM17000) and Negative Control #1 (PM17110).
SiRNAs used were: siCPEB1 (5′ taggaggtgttccttgggata 3′, 5′cagccgaaggatgcgctgcaa 3′), and AllStars - negative control siRNA (Qiagen).
Transfection assays and luciferase measurements
All transfections were performed in triplicate in 96-well plates with 200ng (or 100 ng + 100 ng) DNA and lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Pre- and anti-miRNAs were used at 50 nM and siRNAs at 10 nM. Cells were lysed after 20 h of transfection using Passive Lysis Buffer (Promega), then Firefly and Renilla luciferase activities were measured using the Dual-Luciferase Reporter Assay System Kit (Promega). Results are expressed as Renilla luciferase values normalized to Firefly, and statistical analyses performed using Student's t test.
Quantitative PCR
Total RNA purification was performed using Trizol (Invitrogen) according to the manufacturer's instructions. 500 ng or 250 ng total RNA was used for CPEB1,WEE1 mRNA quantification, using QuantiTect SYBR-green RT-PCR kits (Qiagen) and a LightCycler Real-Time PCR System (Roche).
For miRNA analysis, 25 ng of total RNA was reverse-transcribed using the TaqMan® MicroRNA Reverse Transcription Kit (Applied Biosystems). QPCR was carried out in triplicate using the TaqMan Universal PCR Master Mix (Applied Biosystems).
Primers used were: WEE1 forward (5′ acctcggataccacaagtgctt 3′) reverse (5′ ggtcttctctctggatctggatga 3′), WEE1 3′UTR forward (5′ ttggatgttacaccagcctt 3′), WEE1 3′UTR reverse (5′ gatatacaagtctaattcaca 3′); CPEB1 forward (5′ tcccagatgcaaatgacttgtgcc 3′), CPEB1 reverse (5′ aacttgtccaccaagtcagaccca 3′); (5′ gacgatgcgctagatgtcaa 3′); GAPDH forward (5′ ggacctgacctgccgtctagaa 3′), GAPDH reverse (5′ ggtgtcgctgttgaagtcagag 3′); Cyclophiline A forward (5′-GTCAACCCCACCGTGTTCTT-3′); Cyclophilin A reverse (5′-CTGCTGTCTTTGGGACCTTGT-3′)
Immunoprecipitation assay
Four 15-cm dishes of HeLa S3 cells were transfected with Myc-FLAG-CPEB1, HA-FLAG-CPEB1 (or CPEB1 mutants), or HA-FLAG-AGO2 expression plasmids (OriGene). After 48 h of incubation, cells were collected, lysed in 150 ml/plate lysis buffer consisting of 100 mM KCl, 5 mM MgCl2, 20 mM Hepes-KOH (pH 7.8), 2 mM DTT, 0.25% NP-40, and protease inhibitor cocktail (Roche) for 30 min on ice.
Lysates were incubated with 100 ml anti-FLAG beads (SIGMA), washed in lysis buffer, and incubated overnight at 4°C with rotation. The beads were washed 5 times in 0.5 ml lysis buffer. Then CPEB1 complexes were eluted in 120 ml of lysis buffer with 100 mg/ml FLAG peptide at 4°C for 2 h with shaking. FLAG peptide elution were analyzed by Western blot.
SDS-PAGE and western Blot
Protein extracts were prepared in 100 mM KCl, 5 mM MgCl2, 20 mM Hepes-KOH (pH 7.8), 2 mM DTT, 0.25% NP-40, and protease inhibitor cocktail (Roche).
20 mg of protein was loaded onto NuPAGE 4–12% Bis-Tris Pre-Cast Gels and resolved using the NuPAGE System (Invitrogen), followed by Western blotting, using standard techniques. Primary antibodies and dilutions used were: CPEB1 1:1000 (NB100-1437, Novus Biological), b-actin 1:10000 (A5441, Sigma) and α-Tubulin 1:5000 (F2168, Sigma), WEE1 (sc-5285, Santa Cruz), Cyclin B1 (sc-752, Santa Cruz), Myc-tag (sc-40, Sigma), AGO1 (SAB4200084, SIGMA), AGO2 (SAB4200085, SIGMA). Secondary antibodies and dilutions used were anti-rabbit-HRP 1:25000 (A0545, Sigma), anti-mouse 1:25000 (A2304, Sigma), and anti-rat 1:25000 (A9037, SIGMA).
Protein levels determined by Western blot were quantified using BioImage software.
Ribosomal profile
15-cm plates of HeLa S3 cells were transfected with pRL-TK Vector containing a fragment of the WEE1 3′UTR (wt, or else mutated at the miR-15b and/or CPE sites) attached to the luciferase coding sequence. After 24 h, cells were lysed in 500 ml buffer containing 20 mM Tris-HCl (pH 8.5), 1.5 mM MgCl2, 140 mM KCl, 0.5 mM DTT, 0.5% NP-40, 1000U of RNAsin (Promega) per ml, and 0.1 mM cycloheximide. The lysate was centrifuged at 10,000g for 10 min, and supernatants were applied to linear 20–47% sucrose gradients in buffer containing 20 mM Tris-HCl (pH 8.0), 140 mM KCl, 5 mM MgCl2. Centrifugation was carried out at 40,000 rpm for 135 min in a Beckman SW41 rotor. 1 ml fractions were collected using a Teledyne ISCO Model 160 system with concomitant measurement of the absorbance at 260 nm followed by RNA precipitation by 1 ml of isopropanol and Q-PCR analysis with primers for Luciferase (forward: 5′ cgtggacaggctctggagcatc ’ and reverse: 5′ acaccgccagcaaacgcgagca 3′), with GAPDH (forward: 5′ ggacctgacctgccgtctagaa 3′and reverse: 5′ ggtgtcgctgttgaagtcagag 3′) as control.
Cell synchronization experiments were done in a similar way, but cells were transfected with plasmid after release from a double thymidine block. Cells were collected 29 h after transfection, lysed, and separated on a sucrose gradient. RNA were extracted and subjected to Q-PCR analysis.
Cell synchronization and FACS analysis
HeLa S3 cells (25–30% confluence) were incubated with 2 mM thymidine for 18 hours. Cells were then washed in PBS and incubated in DMEM (10% FBS) for 9 hours to release them from the first thymidine block. 2 mM thymidine was then added to DMEM for 17 hours (second thymidine block). After removal of thymidine by washing in PBS and adding fresh DMEM (10% FBS), cells were transfected with luciferase reporter constructs and 24 hours later some cells were used for the luciferase reporter assay and some were fixed in 70% EtOH at 4°C overnight. The next day, fixed cells were subjected to propidium iodide (PI) labeling. Cells were washed with PBS twice, resuspended in 50 mL PBS containing a final concentration of 1 mg/mL RNAse (DNAse-free, Qiagen), incubated at 4°C for 5 min, then added to 45 mL PBS containing 50 mg/mL PtdIns (final concentration), incubated at 4°C for 30 min in the dark and subjected to FACS analysis by FACScan (Becton Dickinson).
In situ hybridization
Antisense and control (sense) probes for in situ hybridization were prepared by incorporating fluorescein-12-UTP (11 427 857 910 Roche) into the luciferase mRNAs respectively transcribed in vitro from T7 and SP6 promoters, using the mMessage mMachine kit (Ambion); the coding sequence was obtained by PCR amplification of a DNA fragment using as primers:forward, 5′ taatacgactcactataggcgtggacaggctctggagcatc 3′;reverse, 5′ atttaggtgacactatagaacaccgccagcaaacgcgagca 3′.
HeLa cells were fixed for 15 min in 3.7 % PFA in PBS at RT, washed in PBT (0.1% Tween-20 in PBS) and stored in 70% EtOH at 4°C overnight. After rehydration in PBT, cells were treated for 3 min with 1 mg/mL Proteinase K in PBT, washed in 2 mg/mL glycine in PBT and post-fixed for 10 min in 3.7% PFA in PBT. Cells were initially incubated for 1 hour in hybridization mix (HM; 50% formamide, 5xSSC, 1x Denhardt's solution, 200 mg/mL yeast tRNA, 500 mg/mL salmon sperm DNA, 2% blocking reagent (Roche)) at 37°C, and then hybridized overnight with 40 nM probe at 37° C. Post-hybridization washes were carried out for 3 × 15 min in 30% formamide/2x SSC at 50° C, 1 × 15 min in 2x SSC at RT, and 1 × 15 min in 0.2x SSC at RT. Cells were rinsed 3 times with PBS then rinsed once for 2–5 min with Hoechst 33258 (the 16 mM stock solution was diluted 1:8000 final concentration 2 mM), mounted on glass slides with Dako mounting medium, and analyzed by fluorescence microscopy.
Co-transfection of labeled molecules and confocal microscopy
FITC-labeled WEE1 3′UTR mRNAs were prepared by incorporation of fluorescein-12-UTP (Roche) into the in-vitro transcript obtained from the T7 promoter using the mMessage mMachine kit (Ambion); the coding sequence was obtained by PCR amplification of a DNA fragment from Rluc-WEE1 3′UTR constructs using as primers:forward, 5′ taatacgactcactatacgttcgttgagcgagttctc 3′;reverse, 5′ atttaggtgacactatacactgcattctagttgtggt 3′.
Mir-15b precursor (Applied Biosystems) was labeled with Cy5 (Label IT siRNA Tracker Cy5 Kit, Mirus) according to the company protocol.
FITC-labeled WEE1 3′UTR (wt, CPE and/or miR-15b site mutations), Cy5-labeled miR-15b precursor and/or CPEB1-Cherry DNA were cotransfected into HeLa cells with a Lipofectamin 2000 Kit (Invitrogen) according to the company protocol. Cells were fixed 48 hours later in 4% paraformaldehyde, labeled, and visualized by confocal microscopy.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
2015CC6841R-f12-z-bw.pdf
Download PDF (437.7 KB)2015CC6841R-f11-z-bw.pdf
Download PDF (290.4 KB)2015CC6841R-f10-z-4c.pdf
Download PDF (910.8 KB)2015CC6841R-f09-z-bw.pdf
Download PDF (413.9 KB)2015CC6841R-f08-z-4c.pdf
Download PDF (1.5 MB)2015CC6841R-f07-z-4c.pdf
Download PDF (1 MB)2015CC6841R-f06-z-4c.pdf
Download PDF (335.2 KB)Acknowledgments
We thank Muriel David for help with microscopy analyses; Dominique Weil for helpful discussion and for providing the CPEB1 antibody and CPEB1 siRNA at the initial step of the work as well as plasmid with RFP-DDX6 wt and one amino acid substitution mutant (DQAD); Noah Hardy and Vladimir Zakon for technical support; Anna Polesskaya for critical reading of manuscript.
Funding
This work was supported in part by grants to IG from the Ligue contre le Cancer (Comité du Val de Marne) and the Institut des Sciences Biologiques CNRS (Projet Exploratoire Premier Soutien).
Related Research Data
References
- Hake LE, Richter JD. CPEB is a specificity factor that mediates cytoplasmic polyadenylation during Xenopus oocyte maturation. Cell 1994; 79:617-27; PMID:7954828; http://dx.doi.org/10.1016/0092-8674(94)90547-9
- Costa-Mattioli M, Sonenberg N, Richter JD. Translational regulatory mechanisms in synaptic plasticity and memory storage. Prog Mol Biol Transl Sci 2009; 90:293-311; PMID:20374745; http://dx.doi.org/10.1016/S1877-1173(09)90008-4
- Alarcon JM, Hodgman R, Theis M, Huang YS, Kandel ER, Richter JD. Selective modulation of some forms of schaffer collateral-CA1 synaptic plasticity in mice with a disruption of the CPEB-1 gene. Learn Mem 2004; 11:318-27; PMID:15169862; http://dx.doi.org/10.1101/lm.72704
- Burns DM, Richter JD. CPEB regulation of human cellular senescence, energy metabolism, and p53 mRNA translation. Genes Dev 2008; 22:3449-60; PMID:19141477; http://dx.doi.org/10.1101/gad.1697808
- Groisman I, Huang YS, Mendez R, Cao Q, Richter JD. Translational control of embryonic cell division by CPEB and maskin. Cold Spring Harb Symp Quant Biol 2001; 66:345-51; PMID:12762037; http://dx.doi.org/10.1101/sqb.2001.66.345
- Groisman I, Ivshina M, Marin V, Kennedy NJ, Davis RJ, Richter JD. Control of cellular senescence by CPEB. Genes Dev 2006; 20:2701-12; PMID:17015432; http://dx.doi.org/10.1101/gad.1438906
- Novoa I, Gallego J, Ferreira PG, Mendez R. Mitotic cell-cycle progression is regulated by CPEB1 and CPEB4-dependent translational control. Nat Cell Biol 2010; 12:447-56; PMID:20364142; http://dx.doi.org/10.1038/ncb2046
- Mendez R, Richter JD. Translational control by CPEB: a means to the end. Nat Rev Mol Cell Biol 2001; 2:521-9; PMID:11433366; http://dx.doi.org/10.1038/35080081
- Hake LE, Mendez R, Richter JD. Specificity of RNA binding by CPEB: requirement for RNA recognition motifs and a novel zinc finger. Mol Cell Biol 1998; 18:685-93; PMID:9447964; http://dx.doi.org/10.1128/MCB.18.2.685
- Mendez R, Hake LE, Andresson T, Littlepage LE, Ruderman JV, Richter JD. Phosphorylation of CPE binding factor by Eg2 regulates translation of c-mos mRNA. Nature 2000; 404:302-7; PMID:10749216; http://dx.doi.org/10.1038/35005126
- Barnard DC, Ryan K, Manley JL, Richter JD. Symplekin and xGLD-2 are required for CPEB-mediated cytoplasmic polyadenylation. Cell 2004; 119:641-51; PMID:15550246; http://dx.doi.org/10.1016/j.cell.2004.10.029
- Rouhana L, Wang L, Buter N, Kwak JE, Schiltz CA, Gonzalez T, Kelley AE, Landry CF, Wickens M. Vertebrate GLD2 poly(A) polymerases in the germline and the brain. RNA 2005; 11:1117-30; PMID:15987818; http://dx.doi.org/10.1261/rna.2630205
- Sarkissian M, Mendez R, Richter JD. Progesterone and insulin stimulation of CPEB-dependent polyadenylation is regulated by Aurora A and glycogen synthase kinase-3. Genes Dev 2004; 18:48-61; PMID:14724178; http://dx.doi.org/10.1101/gad.1136004
- Kwak JE, Wang L, Ballantyne S, Kimble J, Wickens M. Mammalian GLD-2 homologs are poly(A) polymerases. Proc Natl Acad Sci U S A 2004; 101:4407-12; PMID:15070731; http://dx.doi.org/10.1073/pnas.0400779101
- Kahvejian A, Svitkin YV, Sukarieh R, M'Boutchou MN, Sonenberg N. Mammalian poly(A)-binding protein is a eukaryotic translation initiation factor, which acts via multiple mechanisms. Genes Dev 2005; 19:104-13; PMID:15630022; http://dx.doi.org/10.1101/gad.1262905
- Piao X, Zhang X, Wu L, Belasco JG. CCR4-NOT deadenylates mRNA associated with RNA-induced silencing complexes in human cells. Mol Cell Biol 2010; 30:1486-94; PMID:20065043; http://dx.doi.org/10.1128/MCB.01481-09
- Fabian MR, Cieplak MK, Frank F, Morita M, Green J, Srikumar T, Nagar B, Yamamoto T, Raught B, Duchaine TF, et al. miRNA-mediated deadenylation is orchestrated by GW182 through two conserved motifs that interact with CCR4-NOT. Nat Struct Mol Biol 2011; PMID:21984185
- Wu L, Fan J, Belasco JG. MicroRNAs direct rapid deadenylation of mRNA. Proc Natl Acad Sci U S A 2006; 103:4034-9; PMID:16495412; http://dx.doi.org/10.1073/pnas.0510928103
- Eulalio A, Huntzinger E, Nishihara T, Rehwinkel J, Fauser M, Izaurralde E. Deadenylation is a widespread effect of miRNA regulation. RNA 2009; 15:21-32; PMID:19029310; http://dx.doi.org/10.1261/rna.1399509
- Chu CY, Rana TM. Translation repression in human cells by microRNA-induced gene silencing requires RCK/p54. PLoS Biol 2006; 4:e210; PMID:16756390; http://dx.doi.org/10.1371/journal.pbio.0040210
- Minshall N, Thom G, Standart N. A conserved role of a DEAD box helicase in mRNA masking. RNA 2001; 7:1728-42; PMID:11780630; http://dx.doi.org/10.1017/S135583820101158X
- McGowan CH, Russell P. Cell cycle regulation of human WEE1. EMBO J 1995; 14:2166-75; PMID:7774574
- Charlesworth A, Welk J, MacNicol AM. The temporal control of Wee1 mRNA translation during Xenopus oocyte maturation is regulated by cytoplasmic polyadenylation elements within the 3′-untranslated region. Dev Biol 2000; 227:706-19; PMID:11071785; http://dx.doi.org/10.1006/dbio.2000.9922
- Pouliot LM, Chen YC, Bai J, Guha R, Martin SE, Gottesman MM, Hall MD. Cisplatin sensitivity mediated by WEE1 and CHK1 is mediated by miR-155 and the miR-15 family. Cancer Res 2012; 72:5945-55; PMID:22942255; http://dx.doi.org/10.1158/0008-5472.CAN-12-1400
- Serman A, Le Roy F, Aigueperse C, Kress M, Dautry F, Weil D. GW body disassembly triggered by siRNAs independently of their silencing activity. Nucleic Acids Res 2007; 35:4715-27; PMID:17604308; http://dx.doi.org/10.1093/nar/gkm491
- Naguibneva I, Ameyar-Zazoua M, Nonne N, Polesskaya A, Ait-Si-Ali S, Groisman R, Souidi M, Pritchard LL, Harel-Bellan A. An LNA-based loss-of-function assay for micro-RNAs. Biomed Pharmacother 2006; 60:633-8; PMID:16962735; http://dx.doi.org/10.1016/j.biopha.2006.07.078
- Minshall N, Kress M, Weil D, Standart N. Role of p54 RNA helicase activity and its C-terminal domain in translational repression, P-body localization and assembly. Mol Biol Cell 2009; 20:2464-72; PMID:19297524; http://dx.doi.org/10.1091/mbc.E09-01-0035
- Bhattacharyya SN, Habermacher R, Martine U, Closs EI, Filipowicz W. Relief of microRNA-mediated translational repression in human cells subjected to stress. Cell 2006; 125:1111-24; PMID:16777601; http://dx.doi.org/10.1016/j.cell.2006.04.031
- Jing Q, Huang S, Guth S, Zarubin T, Motoyama A, Chen J, Di Padova F, Lin SC, Gram H, Han J. Involvement of microRNA in AU-rich element-mediated mRNA instability. Cell 2005; 120:623-34; PMID:15766526; http://dx.doi.org/10.1016/j.cell.2004.12.038
- Sivan G, Kedersha N, Elroy-Stein O. Ribosomal slowdown mediates translational arrest during cellular division. Mol Cell Biol 2007; 27:6639-46; PMID:17664278; http://dx.doi.org/10.1128/MCB.00798-07
- Groisman I, Jung MY, Sarkissian M, Cao Q, Richter JD. Translational control of the embryonic cell cycle. Cell 2002; 109:473-83; PMID:12086604; http://dx.doi.org/10.1016/S0092-8674(02)00733-X
- Vasudevan S, Tong Y, Steitz JA. Cell-cycle control of microRNA-mediated translation regulation. Cell Cycle 2008; 7:1545-9; PMID:18469529; http://dx.doi.org/10.4161/cc.7.11.6018