
ABSTRACT
Mesenchymal stem-like/claudin-low (MSL/CL) breast cancers are highly aggressive, express low cell-cell adhesion cluster containing claudins (CLDN3/CLDN4/CLDN7) with enrichment of epithelial-to-mesenchymal transition (EMT), immunomodulatory, and transforming growth factor-β (TGF-β) genes. We examined the biological, molecular and prognostic impact of TGF-β upregulation and/or inhibition using in vivo and in vitro methods. Using publically available breast cancer gene expression databases, we show that upregulation and enrichment of a TGF-β gene signature is most frequent in MSL/CL breast cancers and is associated with a worse outcome. Using several MSL/CL breast cancer cell lines, we show that TGF-β elicits significant increases in cellular proliferation, migration, invasion, and motility, whereas these effects can be abrogated by a specific inhibitor against TGF-β receptor I and the anti-diabetic agent metformin, alone or in combination. Prior reports from our lab show that TNBC is exquisitely sensitive to metformin treatment. Mechanistically, metformin blocks endogenous activation of Smad2 and Smad3 and dampens TGF-β-mediated activation of Smad2, Smad3, and ID1 both at the transcriptional and translational level. We report the use of ID1 and ID3 as clinical surrogate markers, where high expression of these TGF-β target genes was correlated to poor prognosis in claudin-low patients. Given TGF-β's role in tumorigenesis and immunomodulation, blockade of this pathway using direct kinase inhibitors or more broadly acting inhibitors may dampen or abolish pro-carcinogenic and metastatic signaling in patients with MCL/CL TNBC. Metformin therapy (with or without other agents) may be a heretofore unrecognized approach to reduce the oncogenic activities associated with TGF-β mediated oncogenesis.
Introduction
Triple negative breast cancers (ER−/PR−/HER2−; TNBC), occur infrequently (10–15% of all breast cancer) and are phenotypically aggressive, often resulting in significant morbidity and mortality.Citation1-3 TNBC have been further subdivided on the basis of reproducible molecular differences into basal (BL1 and BL2), immunomodulatory (IM), mesenchymal (M), mesenchymal stem-like/claudin-low (MSL/CL) and luminal androgen receptor (LAR).Citation4-6 Of these, MSL/CL breast cancers reportedly have the worst prognosis.Citation1-4,7 MSL/CL characteristically show a very highly proliferative rate, are histologically diverse (invasive ductal not otherwise specified, medullary, or metaplastic) and show a high rate of primary and/or secondary chemoresistance.Citation2,4,5,8-11 There are no currently available treatment protocols that have shown long term efficacy against metastatic, chemoresistant TNBC or more specifically, MSL/CL cancers.Citation2,8,12
TNBCs typically show a loss of BRCA1 expression, reduced P53 and claudin-3,-4,-7 gene function and exhibit high expression of epidermal growth factor receptor (EGFR) and basal cytokeratins (CK-5/6, −14 and −17).Citation1-4,7 In addition, TNBCs of the MSL/CL subtype demonstrate proportionally higher stem/progenitor cell subpopulations (CD44+/CD24low/−/ALDH1+), with enrichment of cells expressing epithelial-to-mesenchymal transition (EMT) markers, and upregulation of immunomodulatory response genes.Citation1-4,7 The M and MSL/CL cancers are notable for higher expression of a number of motility genes (modulation of actin by Rho Pathway), extra cellular matrix (ECM) receptor interaction, cell differentiation and growth factor pathways (regulated by Wnt pathway), anaplastic lymphoma kinase (ALK) and TGF-β signaling pathway.Citation4 The TGF-β pathway modulates genes highly enriched in EMT and immuno-regulatory mechanisms in M and MSL/CL breast cancers.Citation4-6
The TGF-β pathway has unique paradoxical roles in different cell types and tissues.Citation13 A marked increase in TGF-β production has previously been correlated with increased invasiveness and poor prognosis in breast cancer patients.Citation14-16 However, studies that have implicated TGF-β in tumor progression were typically complex and context-dependent.Citation17-19 The TGF-β superfamily members (TGF-βs, Activins, Bone Morphogenetic Proteins (BMPs), among others) act as multifunctional cytokines, regulating cell growth, adhesion, proliferation, motility, differentiation, and cell death.Citation13,19,20 TGF-β signaling is typically initiated upon ligand binding to a heterodimeric receptor complex (TβRI/TβRII), with activation of R-Smads (Smad2/3), which couple to Smad4 and translocate to the nucleus to regulate gene expression of specific TGF-β target genes. Through this mechanism, TGF-β signaling has been implicated in the pathogenesis of breast cancerCitation9,21-23 and metastasis to the lungCitation24 or brain.Citation25-27 The mechanisms underlying its dysregulation in distinct molecular subtypes of breast cancer are not well defined. In MSL/CL tumors, gene expression profiling has identified enrichment of immunomodulatory and EMT transcribed genes.Citation4,5,7 Other studies have highlighted TGF-β's role in cancer associated immunosuppression, where gain of TGF-β function enables cancerous cells to evade immune-surveillance and enhance metastasis through direct regulation of helper and effector T-cell differentiation.Citation18,19,28 TGF-β has also been shown to suppress T-cell proliferation through IL-2 dependent and independent mechanisms.Citation29
The TGF-β1 ligand is overexpressed and associated with an increased risk of tumor progression, distant metastasis, and a shortened survival in breast cancer (reviewed inCitation30-32).Citation33 Targeted therapeutic strategies have been developed to directly abrogate TGF-β signaling, including: 1) neutralizing antibodies and soluble decoy receptor proteins (GC-1008, PF-03446962); 2) antisense oligonucleotides (ASOs, AP-12009, NovaRx); 3) TGF-β receptor kinase inhibitors (TβRI, TβRII and ALK5; LY2157299, LY2109761, and SB431542); 4) immune response-based strategies (SB-431542, SD-108, SX-007); and 5) other inhibitors of the TGF-β pathway (e.g. nano-based liposomal polymeric gels releasing TGF-β inhibitors, P144 and P17, synthetic small peptides inhibiting TGF-β ligands).Citation34-36 Pre-clinical studies have shown that TβRI-KI antagonists promote conversion of basal-like cells to a more epithelioid phenotype with less potent metastatic capacity.Citation35,36 We have evaluated TβRI-KI (LY2157299 or SB431542) in MSL/CL TNBC, to determine the effects of direct inhibition of TGF-β signaling in this unique cancer subtype.
In vivo and in vitro data from our lab, as well as epidemiological and retrospective clinical studies support the use of metformin, which belongs to the biguanide class of antidiabetic drugs, to reduce the incidence of breast cancer and improve the outcome for diabetic patients with the disease.Citation37-39 The efficacy and safety of this agent in non-diabetics is currently being evaluated in phase III prospective trials of breast cancer patients (not stratified by molecular subtype or tumor profiling). We have shown that TNBC cells are exquisitely sensitive to the anti-cancer activity of metformin, consistent with data from the laboratory of Dr. Kevin Struhl showing that it is especially potent against stem cells which are significantly upregulated in breast cancers.Citation40-42 The Struhl lab also identified that metformin is a potent inducer of the immune system through direct activation of early inflammatory transcription factor NF-κβ in a SRC-inducible model of transformation.Citation40 Metformin induces unique molecular activity in TNBC, including S phase cell cycle arrest and apoptosis.Citation43-45 Based on the prominent biological activity and dominance of TGF-β signaling in MSL/CL TNBC, further interrogation of specific mechanisms of metformin action in this subtype may have great clinical utility.
We have previously demonstrated that metformin is more efficacious against TNBC as compared to non-TNBC, with blockade of cell cycle progression the induction of cell death through apoptosis.Citation42-44 In this study we focus on the TGF-β pathway in one subtype of TNBC, MSL/CL. We show that TGF-β activation is critical for the development and progression of this highly aggressive TNBC subtype. Mechanistically, a number of TGF-β transcriptional regulators known as inhibitors of differentiation (ID1 and ID3) are downstream effectors, contributing to worse outcomes for these patients. These TGF-β target genes may serve as clinical surrogate markers to identify MSL/CL patients whose disease will have a poorer outcome. We also demonstrate that metformin attenuates pro-carcinogenic and pro-metastatic cues induced by TGF-β in MSL/CL subtype. Metformin may represent a robust, novel and low toxicity therapeutic option in patients with this highly aggressive subtype of TNBC.
Results
TGF-β signature is highly expressed in MSL/CL TNBC and is associated with prognosis
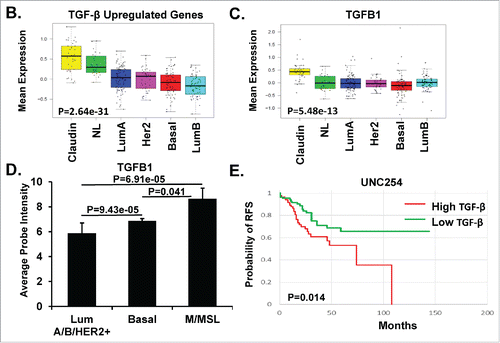
Prior publications have associated TGF-β signaling with tumor suppressor activity in some breast cancer molecular subtypes, whereas in others it promotes carcinogenesis.Citation4,13-16,18,19 In breast cancers as a whole, TGF-β signaling has been widely demonstrated to correlate with lymph node metastasis and poor prognosis.Citation13,16,46 We hypothesized that a TGF-β gene signature could be used as a surrogate marker for signaling activity, and generated a list of TGF-β-regulated genes involved breast carcinogenesis (see Supplemental Table 1). We utilized publically accessible collections of TGF-β gene sets, maintained by the GSEA (http://www.broadinstitute.org/gsea) and the molecular signature database (MSigDb), to evaluate which subtypes of breast cancer show the strongest associations with TGF-β signaling. We then used a number of public available datasets of breast cancers (UNC337: GSE10886 and GSE18229,Citation7,47 UNC855Citation48), as well as a panel of 51 breast cancer cell linesCitation49 to evaluate TGF-β signaling/activation by molecular breast cancer subtype. A total of 1,192 breast cancers were queried for subtype-specific distribution of our TGF-β gene signature outlined in Supplemental Table 1 ( and Supplemental Fig. 1 and 2). Each colored square in the heat map represents the relative mean transcript abundance (log2 ratio) of the TGF-β differentially expressed genes (P < 0.05) in each intrinsic molecular subtype of breast cancer using the UNC337 data set. Heat maps of the gene expression microarray data from UNC337 showed that the MSL/CL (also referred to as Claudin-low) TNBC had the highest levels of TGF-β-specific gene expression as compared to other breast cancer subtypes ( and Supplemental Fig. 1). TGF-β associated genes expressed by the MSL/CL TNBC include: TGF-β receptors and other receptors (TGFBR2, TGFBR3, ACVR1, ACVR2, IL7R, IL10RA, CXCR4), TGF-β superfamily ligands and other ligands (TGFB1, TGFB2, TGFB3, BMP2, LTBP1, SERPINE, IGF1, IL6), TGF-β responsive genes (ID1, ID2, ID3, HMOX1, MMP1, MMP2, MMP3, PGST2, CRYAB), EMT responsive genes (SNAI1, VIM, TWIST, ZEB1, ZEB2), and other transcription factors (BACH1, TXNIP, CREB1, COL5A2, SPARC, THY1, SPOCK1). Similar findings were observed using a second larger dataset (UNC855 data set; Supplemental Fig. 2). Both datasets confirmed that the TGF-β upregulated genes were highest in the MSL/CL TNBC as compared to other breast cancer subtypes (P = 2.64e-31, ). Similarly, examination of a conserved panel of TGF-β upregulated genes from three independent studiesCitation4,7,10 (Supplemental Table 1) was highest in MSL/CL tumors (P = 4.87e-26, Supplemental Fig. 3A). Target genes like TGFB1 and TGFBR2 were also shown to be highly expressed in MSL/CL tumors relative to the other subtypes (, P = 5.48e-13; Supplemental Fig. 3B, P = 1.62e-35). Conversely, genes downregulated by TGF-β (Supplemental Table 1) were significantly lower in MSL/CL TNBC as compared to the other subtypes (P = 6.01e-69, Supplemental Fig. 3C).
Figure 1. TGF-β Gene Expression Signature Upregulated in MSL/CL Subtype of TNBC. A. Heat maps showing relative gene expression of the TGF-β differentially expressed genes (P < 0.05) in each intrinsic subtype of breast cancer using UNC337 data set.Citation7,47 Colored squares in the heat map are the relative mean transcript abundance (log2, –3 to 3) for each subtype with highest expression in red, average expression in black, and lowest expression in green. B. Box-and-whisker plots are representative of the average expression of the TGF-β upregulated gene signatures across the intrinsic breast cancer subtypes. C. Box-and-whisker plots are representative of the average expression TGFB1 in the different breast cancer subtypes (P = 5.48e-13). D. Average probe intensity for TGFB1 in each of the defined intrinsic subtypes of breast cancer as was extrapolated from.Citation49 Bar graph is representative of luminal A/B and HER2 (LumA/B/HER2+), Basal, and Mesenchymal/Mesenchymal Stem-like/Claudin-low (M/MSL) cell lines. Standard deviations between examined cell lines are identified. E. Kaplan-Meier plot for relapse free survival (RFS) and log-rank test P values. Tumors were independently ranked from low to high signature score for TGF-β expression utilizing the UNC254 tumors with survival data. The Kaplan-Meier plot and log rank test P value compares the tumors with the lowest TGF-β signature (TGF-β downregulated genes) expression relative to TGF-β-high (TGF-β upregulated genes) expression in all intrinsic breast cancer subtypes, P = 0.014. Statistics were performed using a two-tailed t-test using excel. P in box-whisker plots were calculated by comparing gene expression means across all subtypes.
Similar analyses were performed using an alternate data set of 855 human breast tumors. MSL/CL tumors had the highest expression of the TGF-β upregulated genes (P = 8.83e-33, Supplemental Fig. 4A), conserved TGF-β genes (P = 4.08e-44, Supplemental Fig. 4B), KEGG TGF-β pathway genes (P = 2.14e-33, Supplemental Fig. 4C), and Reactome TGF-β pathway genes (http://www.reactome.org/) (P = 2.34e-13, Supplemental Fig. 4D) relative to other intrinsic molecular subtypes of breast cancers. MSL/CL cancers also showed lower levels of TGF-β down-regulated genes (P = 2.59e-93, Supplemental Fig. 4E) relative to the other intrinsic subtypes.
We next sought to validate our data with those reported by Neve et. al,Citation49 who studied gene expression in 51 breast cancer cell lines. Using their publically available database, we examined the relative gene expression in 17 different breast cancer cell lines categorized into distinct subtypes. The identified 17 cell lines were selected based on available cell lines in the lab that were used to perform secondary analysis. MCF10A was also selected as a immortalized control cell line. TGFBR2 and SMAD3 were more often expressed in the MSL/CL cohort relative to other tumor subtypes (Supplemental Fig. 5A & B). TGF-β regulated genes, such as: TGFB1 (P = 6.91e-05, ), TGF-β receptor II (TGFBR2, P < 0.0001), SMAD3 (P = 0.02), HMOX1 (P = 0.037), ID1 (P = 0.007) and ID3 (P = 0.001) were higher in the MSL/CL relative to luminal A/B or HER2+ cells (Supplemental Fig. 5C). Only the TGF-β1 ligand showed significant upregulation in the MSL/CL cohort relative to basal cell lines ().
Finally, we tested whether high expression of the TGF-β gene signature, as outlined in Supplemental Table 1, was associated with a poor prognostic outcome in 337 breast cancer patients. Kaplan-Meier analyses showed that high TGF-β gene expression was associated with a decrease in relapse free survival (RFS) (P = 0.014, ) for patients with MSL/CL cancers. Breast tumors with high expression of the TGF-β gene signature, as compared to a low TGF-β gene signature at diagnosis had a shorter RFS.
ID1 and ID3 expression is a surrogate marker for TGF-β signature in MSL/CL tumors
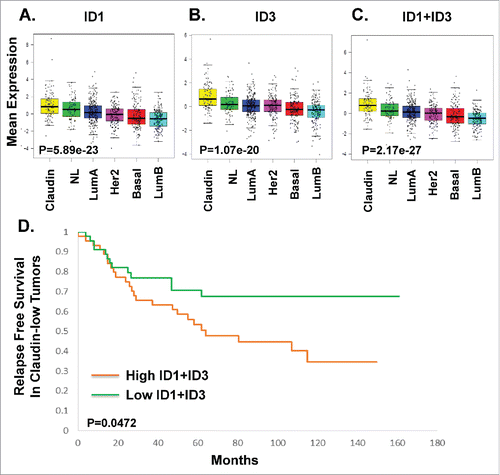
The inhibitor of DNA binding genes (ID1, ID2, ID3, and ID4) are TGF-β-specific transcriptional regulators that bind to basic helix-loop-helix (bHLH) and non-bHLH factors are reported to play critical roles in breast cancer metastasis.Citation50-52 ID1 proteins are thought to be predominantly expressed in metastatic TNBC and have been shown to correlate with a poor prognosis.Citation50 However, the mechanism by which ID1 and ID3 induce metastasis is not clearly understood.Citation53,54 Given the TGF-β signature data presented above, we sought to determine if ID1 and ID3 data alone could be used as an independent biomarker indicative of TGF-β activation in MSL/CL subtype of breast cancer. Using UNC855 dataset, we show that MSL/CL highly express ID1 and ID3 as an independent gene expression signature (, P = 5.89e-23; , P = 1.70e-20). The combined average expression of ID1 and ID3 was found to be statistically higher in MSL/CL tumors (; P = 2.17e-27) relative to the alternate subtypes of breast cancer. For these MSL/CL cancer patients, high expression of both ID1 and ID3 was associated with a shortened relapse free survival (; P = 0.0472). The average expression of ID1 and ID3 could be attributed to the high degree in sequence similarities shared between these two transcription factors.
Figure 2. Prognostic Significance of ID1 and ID3 TGF-β Gene Expression in Relapse Free Survival. Box-and-whisker plots are representative of the expression ID1 gene expression (A, P = 5.89e-23), ID3 gene expression (B, P = 1.07e-20), and the average expression of ID1and ID3 genes (C, P = 2.17e-27) signatures were defined in the intrinsic breast cancer subtypes from UNC855 dataset.Citation48 D. Kaplan-Meier plot for relapse free survival in claudin-low tumors with log-rank test p-values. The P value compared high ID1 and ID3 expression to low ID1 and ID3 gene expression within claudin-low tumors using the UNC855 data set Citation48 (P = 0.0472).
TGF-β enhanced proliferation and activation of SMADs in M and MSL/CL cell lines
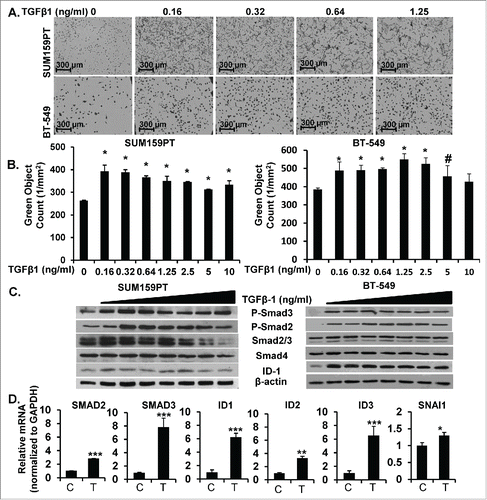
To study the mechanisms of TGF-β in pro-oncogenic signaling we used a number of M and MSL/CL cell lines (M: BT-549; MSL/CL: SUM159PT, MDA-MB-231, HS578T) that were previously labeled with a nuclear-GFP tag (described in methods). M and MSL/CL cells were treated with increasing doses of TGF-β1 and monitored for changes in proliferation over time using a live cell kinetic assay IncuCyte Zoom™. SUM159PT and BT-549 cells treated with increasing picomolar doses of TGF-β1 exhibited a marked-induction of proliferation (). Levels of TGF-β1 over 5 ng/ml, in contrast, suppressed proliferation or had no significant effect (as previously reported by others utilizing different assays such as [3H] thymidine incorporation experiments or cell count).Citation55,56 Control cell lines lacking TGF-β receptors (MCF-7, luminal A/B) or Smad4 (MDA-MB-468, basal 1) did not show significant changes in cell proliferation with TGF-β1 treatment (data not shown). M and MSL/CL cells (SUM159PT, BT-549, and MDA-MB-231) treated with low doses of TGF-β1 ligand showed a marked increase in phospho-Smad2 (P-Smad2), phospho-Smad3 (P-Smad3) and ID1 protein expression as detected by Western blot assay (, Supplemental Fig. 6). These data were confirmed by analyzing mRNA levels in TGF-β1 treated, as compared to untreated, cells. TGF-β1 enhanced mRNA activation of a number of TGF-β target genes in SUM159PT cells cultured under physiological glucose conditions (glucose 5 mM), including: Smad2 (P < 0.0001), Smad3 (P < 0.0001), ID1 (P < 0.0001), and ID3 (P < 0.0001), ID2 (P < 0.001) and SNAI1 (P < 0.01; ). Collectively, this data suggests that low dose TGF-β1 enhanced proliferation and increased TGF-β-specific gene expression in MSL/CL breast cancer cell lines.
Figure 3. TGF-β Increases Proliferation, Signal Transduction, and Gene Expression of Downstream Target Genes in MSL/CL Cell Lines. A. SUM159PT-Nuc-GFP and BT-549 parental cells were treated with increasing concentrations of TGF-β1 (0, 0.16, 0.32, 0.64, 1.25 ng/ml) monitored for proliferation using IncuCyte Zoom. Images are representative to 72 hr, n = 12 of three independent experiments. GFP transposed to black and phase for visualization. B. Bar graph quantitation of green object count (1/mm2) of live proliferation over time at 72 hr (*P < 0.001, #P < 0.01). C. SUM159PT and BT-549 cells were treated with increasing concentrations of TGF-β1 (0, 0.16, 0.32, 0.64, 1.25, 2.5, 5, 10 ng/ml) for 4–6 hours then harvested for WB and probed with TGF-β signaling pathway proteins. D. SUM159PT cells were treated TGF-β1 (T;1.25 ng/ml) or vehicle control (C) for 4–6 hours then harvested for mRNA analysis of TGF-β gene targets (***P < 0.0001, **P < 0.001, *P = 0.01). Experiments are representative triplicate experiments.
Metformin alone or in combination with TGF-β kinase inhibitor block TGF-β-induced proliferation and activation of TGF-β signaling pathway
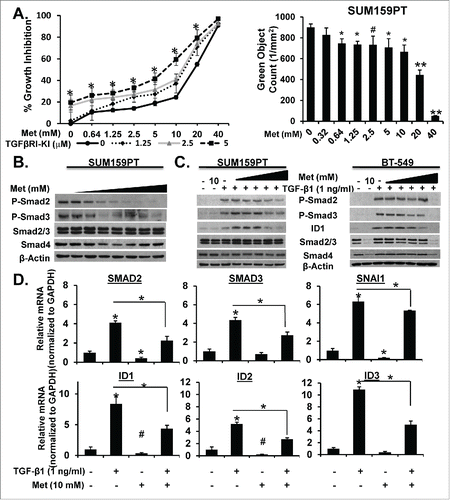
To clarify the importance of TGF-β in the MSL/CL breast cancers, we studied the effects of its inhibition. A selective TGF-β Receptor I-Kinase Inhibitor (TβRI-KI; LY2197299), in development and clinical use, was used to inhibit TGF-β signaling. We also evaluated the ability of metformin, a biguanide derivative, to inhibit proliferation and/or TGF-β induced signaling or proliferation. MSL/CL cell lines expressing Nuclear-GFP were seeded in media with 5 mM glucose, then treated with TβRI-KI alone or in combination with metformin. SUM159PT and BT-549 cells were monitored for percent growth inhibition by examining proliferation using IncuCyte Zoom™ for 6 days. (, left; Supplemental Fig. 7A, 7C, and 8). BT-549 cells were monitored for 4 days. This assay was also repeated with HS578T cells and generated similar data (not shown). Metformin alone significantly attenuated cellular proliferation of all MSL/CL cells in a dose-dependent fashion (at concentrations from 2.5 to 5 mM), whereas the TβRI-KI induced only a marginal decrease in cellular proliferation (, right; Supplemental Fig. 7A, 7C, 8A-E). We then studied these agents in combination, using the method of Chou-TalalayCitation57 to define synergistic interactions. Metformin and the TβRI-KI were synergistic in inhibiting cell proliferation in these cell lines (Supplemental Fig 7B).
Figure 4. Metformin Alone or in Combination with TGF-β-KI Attenuates TGF-β-induced Proliferation and Activation of TGF-β Signaling Proteins and mRNA. A. SUM159PT-Nuc-GFP cells were treated with increasing concentrations of metformin (0–40 mM) in the presence of increasing concentrations of TGF-β-KI (LY2157299; 0, 1.25, 2.5, and 5.0 µM) and monitored for proliferation over time using IncuCyte Zoom™ for 6 days, (n = 4, *P < 0.001). Bar graph represents metformin dose-response (0–40 mM) mediated inhibition of proliferation at 72 hrs in SUM159PT cells (**P < 0.0001, *P < 0.001, #P < 0.01). B. SUM159PT cells were treated with metformin dose response (0, 0.64, 1.25, 2.5, 5, 10, 20, 40mM) for 24 h then harvested for WB analysis of TGF-β-protein targets. C. SUM159PT or BT-549 cells were treated with metformin (0, 0.64, 1.25, 2.5, 5, 10 mM) for 20 h prior to TGF-β1 (1 ng/ml) stimulation for 4 hours then harvested for WB examination of TGF-β signaling proteins. D. SUM159PT were treated with metformin (10 mM) for 20 hours prior to TGF-β1 (1ng/ml) stimulation for 4 hours then mRNA was isolated and purified for qRT-PCR examination of TGF-β-specific gene targets (n = 3, *P < 0.0001, #P < 0.001). Experiments are representative at least three experiments.
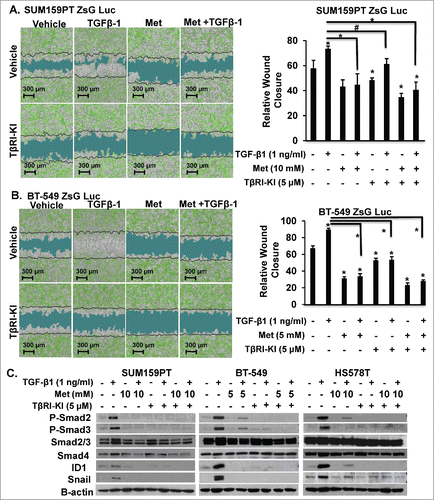
We then tested whether metformin could attenuate endogenous expression or activation of Smads in both BT-549 (M) and SUM159PT (MSL/CL) cell lines. Increasing concentrations of metformin (in the 1.5 to 2.5 mM range) blocked endogenous activation of phospho-Smad2 and phospho-Smad3 in SUM159PT and BT-549 cells by Western blot analysis ( and Supplemental Fig. 8F). Similarly, treating BT-549 (M) or SUM159PT (MSL/CL) cells with increasing concentrations of metformin for 20 h prior to TGF-β1 (1 ng/ml) for 4 h attenuated phospo-Smad2, phospho-Smad3, and ID1 expression in Western blot analysis (). Metformin also blocked TGF-β downstream signaling protein expression in a number of M and MSL/CL cell lines (BT-549, MDA-MB-436, MDA-MB-231), but had no effect in MCF7 cells (that lack TGF-β receptor I) (Supplemental Fig. 9). Metformin also attenuated TGF-β-mediated transcriptional activation of a number of TGF-β specific target genes including: SMAD2, SMAD3, SNAI1, ID1, ID2, and ID3 in MSL/Cl cell line SUM159PT (). These results show that metformin attenuates TGF-β-induced proliferation and signaling in the M and MSL/CL cells examined. In summary, the TGF-β pathway is an important target of metformin in this subtype of breast cancer cells. Metformin may represent a low toxicity option for patients with MSL/CL TNBC that have upregulation/activation of the TGF-β pathway.
Metformin alone or with TβRI-KI abrogates TGF-β-induced motility and invasion in MSL/CL subtype
To directly investigate whether metformin alone or in combination with TβRI-KI could inhibit TGF-β1 effects on motility in MSL/CL cells, we used a live kinetic assay to induce a wound and monitored relative wound closure after single agent or combination agents for 48 h. Metformin alone or in combination with TβRI-KI significantly attenuated motility in MSL/CL cell lines (SUM159PT, BT-549, MDA-MB-231, and HS578T) (, Supplemental Fig. 10). To study the influence of metformin treatment combined with TβRI-KI on MSL/CL cell invasion, we used an assay similar to the scratch assay; however, we filled the wound with BD Matrigel™ mimicking an invasive ECM boundary. Metformin alone or in combination with TβRI-KI abrogated invasion induced by TGF-β1 ligand in SUM159PT and BT-549 cells (), but not in MCF7 cells lacking TGF-βRI (Supplemental Fig. 11). In addition, metformin alone or in combination with TRI-KI reduced expression of TGF-β down-stream signaling modulators including phospho-Smad2, phospho-Smad3, ID1, and Snail1 in Western blot assays of different MSL/CL cell lines (). These data provide strong preclinical evidence that metformin alone, or in combination with TβRI-KI, may attenuate cell growth, invasion, and motility of M and MSL/CL breast cancer cells that highly express the TGF-β-mediated pro-oncogenic signature in MSL/CL examined.
Figure 5. Metformin blocks TGF-β-mediated Motility in MSL/CL Cells. SUM159PT expressing ZsG-Luc and BT-549 parental cells were seeded in a monolayer prior to infliction of wound then treated with metformin (10 mM), TβRI-KI (5 µM), TGF-β1 (1 ng/ml), vehicle control or defined combinations. Cells were monitored for motility indicated by relative wound closure percentage for 6 days using IncuCyte Zoom™ in SUM159PT (A), BT-549 (B) cell lines. Representative images were taken at 24 hr time point, with black time indicating t = 0. Bar graph is represents relative wound closure at 24 hr time point for each cell line treated as indicated. Experiments were done n = 8 for each assay. Each assay was repeated at least three times for each cell line. Statistics were performed using a two-tailed t-test using excel and GraphPad Prism 6® to generate comparisons in bar graphs quantitation of relative wound closure (*P < 0.001 and #P < 0.01).
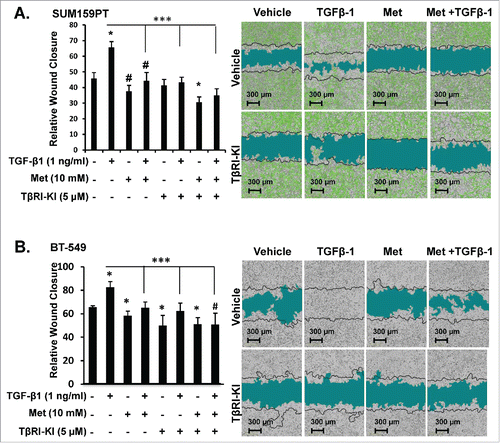
Figure 6. Metformin Retards TGF-β-induced Invasion in MSL/CL Cells. SUM159PT-Luc-ZsG (A) and BT-549-Luc-ZsG (B) cells were seeded in a monolayer prior to infliction of wound followed by filling the void with Matrigel® to mimic an invasive boundary for cells to intravasate through. Cells were treated with metformin (5 or 10 mM), TβRI-KI (5 µM), TGF-β1 (1 ng/ml), vehicle control or defined combinations. Cells were monitored for relative wound closure using IncuCyte Zoom™ for 6 days. Images are representative of 48 hr time point, with t = 0 represented as a black line. Relative scratch wound mask is indicated in blue. Bar graph quantitation of relative wound closure is shown to the right of the images for each cell line at 48 hr time point. Each experiment is representative of n = 8 and performed as three independent experiments, (*P < 0.0001, #P < 0.001). C. MSL/CL cell lines were treated with 5 mM or 10 mM metformin for TβRI-KI (5 µM) for 20 hrs prior to TGF-β1 (1 ng/ml) stimulation for 4 hrs. Cells were harvested for WB and probed for TGF-β protein targets.
Discussion
TGF-β signaling has complex, and often opposing, context-specific effects on cellular targets which are compounded with cross-talk to a number of different signaling pathways. Various independent and interrelated events are altered in response to TGF-β during tumor progression. Such factors include: alterations in receptor expression, dampening of downstream TGF-β signaling components, evasion of immune response, activation of inflammation, presence of local and systemic factors (autocrine, endocrine, paracrine interactions), and recruitment of cell types that enhance tumor growth or promote angiogenesis. We sought to resolve the controversial role of TGF-β in breast cancer by focusing on its activation in specific molecular subtypes of breast cancer. Mesenchymal (M) and mesenchymal stem-like/claudin low (MSL/CL) breast cancers within the triple negative cohort share similar ontologies and gene expression profiles involving: TGF-β, Wnt/β catenin, FGFR, PDGFR, mTOR, and VEGF signaling pathways.Citation4 Interestingly, both the M and MSL/CL subtype shows enrichment for epithelial-to mesenchymal transition (EMT) markers, immune response genes, cancer stem cell-like features, and higher activity of TGF-β pathways. Enrichment of EMT, immunomodulatory complexes, and stem-cells are thought to be driven by TGF-β. Increased activation of TGF-β, as well as other pathways, can enhance tumor progression and eventually aid in the metastatic process via increase in invasion and migratory capacity.
The present study demonstrates that the TGF-β signaling pathway is highly expressed in mesenchymal stem-like/claudin-low (MSL/CL) a subtype of TNBC. Using gene expression microarray databases representing 1,192 breast cancers we find that high activity of TGF-β (determined by TGF-β gene signatures) was most frequent in MSL/CL cancers and for these patients, this tumor signature correlated with worse outcome. MSL/CL cancers strongly expressed components of the TGF-β signaling pathway, including the TGF-β receptor 2, Smad2, Smad3, ID1, and ID3. Utilizing the UNC337Citation7,47 and combined 855 publically available platforms, we confirmed that the MSL/CL tumors have upregulation of the TGF-β receptors, TGF-β superfamily ligands, TGF-β responsive genes, and EMT associated genes (outlined in Supplemental Fig. 1). Additionally, we saw a prominent down regulation of growth factor receptors (FGFR2 and FGFR3), hormone receptors (AR, ESR1, and PGR), and claudin integral membrane proteins of tight junctions (CLDN3, CLDN4, and CLDN7) in the MSL/CL cohort. Our in vitro assays demonstrate that MSL/CL cells stimulated with low doses of TGF-β1 ligand (below 1 ng/ml) displayed a marked increase in proliferative response and enhanced migration and invasion. Induction of TGF-β1 signaling enhanced phosphorylation of Smad2 and Smad3 and increased translational expression of downstream signaling intermediates such as ID1 and Snail. This response could be abrogated by the anti-diabetic drug metformin, which inhibited TGF-β mediated signaling, migration, and invasion. Novel data presented here establish that metformin can directly block phospho-activation of both Smad2 and Smad3. The results with metformin were often greater than effects observed using selective TGF-β kinase inhibitors alone (TβRI-KIs; LY2197299 or SB431542) (data not shown). These results, and the demonstrated synergistic interactions between this selective inhibitor and the more broadly acting drug metformin, were unexpected. Metformin alone or in combination with a selective anti-TGF-β inhibitor may enhance anti-cancer activity. Finally, our data suggests that targeting TGF-β signaling may be a valuable new approach for a molecular subset of patients with aggressive, chemo-refractory breast cancer that falls into the MSL/CL subtype.
Utilizing a selective and potent inhibitor of TGF-β, as well as other broad inhibitory agents like metformin, may serve as robust therapeutic agents in preclinical models. Since TGF-β plays a key role in EMT, the use of TGF-β-neutralizing antibodies and TGF-β receptor inhibitors have been studied to abrogate EMT.Citation58 More recently, studies have employed metformin to disrupt EMT in breast cancer.Citation59,60 However the direct mechanism by which metformin can attenuate key EMT markers remains unknown. In this study, we demonstrate that use of the broad acting inhibitor metformin can attenuate TGF-β-mediated oncogenic signaling to a similar or more potent degree as compared to a selective TβRI-KI. In fact, we believe that metformin is a more robust inhibitor of TGF-β in tumor cells and may enhance immune surveillance in surrounding stromal tissues. This study is the first to show that metformin can directly attenuate TGF-β signaling through suppression of endogenous Smad2 and Smad3. Further, we found that metformin can not only retard TGF-β signaling in MSL/CL subtype, but it also reduced migration and invasion of MSL/CL cell lines. Importantly, we found that metformin reduced TGF-β-induced EMT through reduction of Snail expression in MSL/CL cell lines. Thus, the inhibitory effect of metformin on tumor metastasis might be due to a reduction in migratory or invasive tumor cells. However, the relationship between the effect of metformin on tumor metastasis or on tumor growth still needs further investigation.
The regulation of tumor cell survival, proliferation, and EMT are significant factors that contribute to TGF-β mediated regulation of tumorigenesis and metastasis. During advance states in tumor progression many carcinoma cell populations find ways to circumvent the cytostatic and apoptotic responses attributed to TGF-β stimulation. In the midst of TGF-β mediated stimulation of tumor cells, adjacent immune effects are also prevented from destroying the invasive tumor cells.Citation61 In understanding the MSL/CL subtype of breast cancer, the interconnection between high TGF-β signaling and the direct up regulation of immunomodulatory genes are of high interest. Growing evidence suggests that inflammatory mediators produced by TGF-β-induced tumor-infiltrating leukocytes and cancer-related fibroblast can enhance tumor metastasis, in part through upregulation of EMT.Citation21,62 Direct stimulation of the TGF-β pathway, via activation of Smad2, in tumor cells may promote tumor growth through increasing the capacity of tumor cells to evade the immune system.Citation61 Some highly metastatic breast cancer cells are thought to directly recruit of pro-metastatic myeloid derived suppressor cells (MDSCs) to the tumor microenvironment at the leading invasive edge. The presence of MDSCs can enhance angiogenesis, promote immune tolerance, and recruit matrix degrading enzymes that enhance tumorigenesis and metastasis. MDSCs harbor an abundant source of TGF-β production and recruitment of MDSCs possibly enhances the classical role of TGF-β in immune suppression and tumor evasion. The influence of inflammatory cells and secreted cytokines, such as TGF-β and IL-6, on tumor promotion and metastasis has not been well defined in human breast cancer. Given this context, it is clear that tumor-immune cell cross talk initiated by TGF-β within the tumor cell is a significant determinant worth considering when designing therapeutic strategies to manage tumor progression and metastasis.
Understanding the role of tumor-immune signaling is integral in offering promising therapeutic strategies that will block pro-invasive and metastatic cues that enable a tumor cell to evade immune surveillance. Inflammatory cytokines, such as TGF-β and IL-6, which enable tumor cells to become immunologically invisible by passive avoidance of immune surveillance could be suppressed with use of broad acting inhibitor such as metformin. We previously showed that metformin can attenuate STAT3 activation a downstream signaling mediator of the IL-6 signaling pathway, and this study shows we can now retard TGF-β mediated tumorigenesis in the MSL/CL subtype of breast cancer. Attenuation of TGF-βs role in tumor-induced immunosuppression may be the key to decreasing angiogenesis and metastasis in the high risk mesenchymal subtypes such as MSL/CL breast cancers. Further, our prior studies revealed that metformin can directly block the CD44-moesin-ezrin complex found at the leading edge of migrating cells.Citation41,42 The ability of metformin to attenuate moesin expression can possibly serve to diminish MDSC populations located on the invasive edge. Therefore this strategy can directly diminish recruitment of matrix degrading enzymes and reduce tumor formation and metastasis of tumor cells. Knowledge of key signaling mechanisms that contribute to the highly aggressive nature of MSL/CL subtype will ultimately allow us to define therapeutic strategies to counteract tumor- immunosupression and reduce tumor progression.
Collectively, our data shows that TGF-β induces activation of critical signaling pathways and upregulation of TGF-β target genes in the most aggressive molecular forms of TNBC. Activation of these pathways induces proliferation, motility and invasion. Human breast tumors that harbor high expression of TGF-β genes have shortened relapse free survival. We have demonstrated that the treatment of MCL/CL breast cancer with specific inhibitors of TGF-β signaling or metformin, can provide potent anti- TGF-β activity and can markedly abrogate its pro-carcinogenic effects. In this study, we extend our knowledge of metformin's mechanism of action to include attenuation of TGF-β signaling, inhibition of down steam Smad activation, and decrease in TGF-β-mediated invasion and motility in MSL/CL cells. In addition, metformin may attenuate the expression of TGF-β in MDSCs located in the leading edge of invasive filopodial extensions during metastasis. This may further inhibit TGF-β-induced immunosupression in selected subtypes of TNBC. Use of metformin to target a multiplex of different signaling machinery in highly metastatic and migratory MSL/CL cells may provide significant clinical benefit for these patients.
In conclusion, TGF-β plays a prominent role in the MSL/CL breast cancer subtype leading to enhanced invasion and motility. Increased TGF-β signaling in the tumor and tumor microenvironment promote EMT, reprograming of immune surveillance, and direct facilitation of tumor proliferation in MSL/CL cells, thereby making it a critical druggable target. Use of current TGF-β therapeutic strategies used to block TGF-β signaling including; TGF-β antibodies, antisense oligonucleotides, and receptor kinase inhibitors, can often provide limitation with respect to delivery and toxicity profiles. Combination of immunomodulatory agents with specific inhibitors that directly enhance tumor surveillance and augment tumor metastasis hold promise for enhancing treatment for breast cancer patients. We believe that metformin can directly attenuate TGF-β-mediated pro-metastatic signaling and enhance tumor immunity specifically in MSL/CL patients. The ease of metformin delivery and low toxicity profile provide a strong case that it can be used within a combinatorial therapy setting for oncology, specifically for high grade MSL/CL breast cancer patents.
Methods
Cell lines
Human breast cancer derived cell lines MCF7 (luminal A), MDA-MB-468 (basal1), BT-549 (Mesenchymal; M), SUM159PT, HS578T, MDA-MB-436 and MDA-MB-231 (Mesenchymal Stem-Like/Claudin-low; MSL/CL) were obtained from the American Type Culture Collection (ATCC) or the University of Colorado Cancer Center (UCCC) Tissue Culture shared resource, Aurora CO. The identity of each cell line was demonstrated to be authentic by analysis of short tandem repeats by the University of Colorado Cancer Center DNA Sequencing and Analysis Shared Resource before the start of this project. All cell lines were confirmed to be free of mycoplasma contamination (using a MycoAlert detection kit, Lonza Walkersville Inc., Walkersville, MD) and tested every three months. All cells, with the exception of SUM159PT and BT549 cells, were maintained in DMEM: Nutrient Mix F-12 (D-MEM/F-12 1:1; Invitrogen Corp.), supplemented with 5–10% fetal bovine serum (FBS; Sigma Chemical Co.) with 2.50 mM L-glutamine and antibiotic/antimycotic solution containing 10,000 U/mL penicillin and 10,000 µg/mL streptomycin and 25 μg/mL amphotericin B (Hyclone™, Inc. ). Media for BT-549 were additionally supplemented with 100 ng/mL insulin. SUM159PT cells were maintained in Ham's F-12 (Hyclone™, Inc.) supplemented with 5% FBS, HEPES (10 mM) (Hyclone™, Inc.), Hydrocortisone (1 μg/ml) (Stem Cell Technologies, Vancouver, BC, Canada) and insulin (5 μg/ml) (Sigma Chemical Co.) as recommended by Asterland (http://www.asterandbio.com). Cells were maintained at 37°C in a humidified atmosphere containing 95% air and 5% CO2. Cells were plated in 5–10% FBS, starved for 6 hours or 6 days depending on described assay in serum free medium devoid of insulin or growth factors then replenished with low serum conditions (0.5–1% FBS) in assigned medium prior to treating cells as described. Cells were treated in either 5 or 17 mM glucose (Sigma Chemical Co.) glucose depending on treatment conditions. All other experiments were performed in low serum (0.5%–1.0%), with the exception of migration assays that were performed in serum-free conditions to prevent cellular proliferation.
Cellular assays and reagents
Cells were treated metformin 1,1-dimethylbiguanide hydrochloride (MP Biomedicals, LLC), recombinant TGF-β1 (R & D System, Inc.), LY2157299 (Selleck, Chemicals). IC50 for each assigned agent were tested in each of the described cell lines that were examined (data not shown). Puromycin (Life Technologies, Carlsbad, CA; A1113803) was used as a selection reagent to derive stable transfected cell lines. Hexadimethrine Bromide, aka Polybrene® (2 mg/mL stock solution; Sigma Chemical Co: H9268) was used as a transduction reagent.
Proliferation
The IncuCyte Zoom™, kinetic live cell imaging system (Essen BioSciences, Inc.) was used to track proliferating cells. Described cells were transfected with 3rd generation HIV-based, VSV-G pseudotyped lentiviral particles (Essen BioSciences, Inc.) encoding a nuclear restricted GFP under promoter EF-1 α. Selection of transduced cells was maintained using puromycin as per manufacturer's description. Stably expressing GFP cells were starved from insulin or alternate growth factors 6 day prior to use for proliferation assay. Nuclear-GFP expressing cells were seeded 1000 cell per 96 well plate in normal serum conditions overnight without the presence of insulin. The next day medium was changed medium containing glucose 5 mM (G5) or glucose 17 mM (G17, standard medium) and low serum conditions (0.5% to 1% serum depending on cell line) along with described treatment conditions. Plates were placed in an IncuCyte Zoom™ and images were recorded every 4 hours using 10× ocular lens. Individual cell division and proliferation was monitored for 24–196 hours. Visualization of cells was exported using the IncuCyte Zoom™ program, where live image of GFP expressing cells were visualized. For clarity, translocation of colors from green to black was done for ease of visualization of proliferating cells. Green object count (1/mm2) were quantified for all described cell lines.
Migration and invasion wound assay
To track motility and invasion, cells were tagged with ZsGreen (ZsG) (gift Kathryn Horwitz, University of Colorado) by retroviral infectionCitation63 Lentiviral mediated transfected cells were also modified to express luciferase. In brief, pMSCV-Luciferase PGK-hygro (gift of Heide Ford, University of Colorado) was transfected into PT-67 packaging cells (gift of Kathryn Horwitz, University of Colorado). ZsG-tagged cells were incubated in filtered, virus-containing supernatants and selected for hygromycin resistance. Luciferase expression was confirmed using D-luciferin (Promega, Fitchburg, WI, USA). Cells expressing both ZsGreen and Luciferase were denoted at (ZsG-Luc) cells. Prior to using cells for a migration or invasion assay, cells were starved from insulin or alternate growth factors for 4–6 days prior to use in a Essen BioScience's CellPlayer™ 96-Well cell migration and invasion assay. Essen BioScience's CellPlayer™ 96-Well cell migration and invasion assay was used for a kinetic quantification of cell migration in vitro. In brief, parental or transfected cells retaining ZsG-Luc expression vector were plated 10–30,000 cells (depending on cell line) to achieve confluence in a 96-well ImageLock plate overnight. Prior to wound generation, cells were treated with 10 μg/ml mitomycin C in serum free conditions (Sigma Chemical Co.) for 3–4 h to prevent wound closure due to cellular proliferation. A mechanical WoundMaker™ (Essen BioSciences, Inc.) was used to generate precise and reproducible wounds in all wells of 96-well ImageLock plate. Once a wound was generated, media was aspirated from each well and gently washed at least 3 times with serum-free medium or chilled PBS to prevent dislodged cells from settling or reattaching. After washing, 150 µl of serum-free media containing drug or test material was added at the described concentration. All bubbles were removed and the plate was placed into the IncuCyte Zoom™ for scheduled repeating scanning at 4 hour intervals. Analysis of relative wound density (RWD) (%) was calculated using IncuCyte Zoom™ software. RWD measures the spatial cell density in the wound area relative to the spatial cell density outside of the wound area at every time point. It is designed to be 0% at time = 0, and 100% when the cell density inside the wound is the same as the cell density outside of the initial wound. RWD metric is a robust across multiple cell types as it does not rely on finding cell boundaries.
The Essen BioScience's CellPlayer™ 96-Well cell invasion assay was used for kinetic quantification of cell migration in vitro. Cells were devoid of insulin or growth factors for 4–6 days prior to seeding cells in a Essen BioScience's CellPlayer™ 96-Well cell invasion assay. A similar procedure as described above was used to generate wounds. A coat of BD Biosciences Matrigel™ (Corning Life Sciences) was used to generate an invasive boundary for cells to intravasation into based on chemokine stimulation. The set up for this assay is similar to that described above, however BD Matrigel™ was diluted (as per manufacturers protocol) and added to cells that were placed in a CoolSink then placed in the incubator (37° C) for 15–20 mins to allow solidification of the BD Matrigel™ that has generated a matrix in the generated mechanical wound. Once solid, additional serum-free media with combinations of drug treatments were added. Relative wound closure over time (based on 4 hr scanning intervals) was monitored. Data was generated using the IncuCyte Zoom™ Software program. Three independent experiments were performed (n = 6) per treatment condition.
Western blot
Whole cell protein extracts were prepared using RIPA lysis buffer. Protein concentrations were determined using the Coomassie Plus protein assay reagent (Pierce Chemical Co.). Equal amounts of cell protein lysates (30–50 μg total protein) were combined with SDS-PAGE loading buffer containing 5% 2-mercaptoethanol, and proteins resolved by gel electrophoresis using various percent gels, and then transferred to a nitrocellulose membrane (BioRad Laboratories). Blots were probed with antibodies from the following sources: to anti-phospho-Smad2, anti-phospho-Smad3, Smad2/3, Smad4, Snail 1, (Cell Signaling Technology, Inc.), ID-1 (Santa Cruz, Dallas, TX), β-actin (AC-75), and rabbit anti-mouse HRP were obtained from Sigma Chemical Co.
Real time quantitative reverse Transcriptase-PCR (qRT-PCR)
SUM159PT cells were devoid of insulin or growth factors for 6 days prior to seeding cells. Cells were seeded in 5 mM glucose with 1% FBS for 24 h then treated with 1.25 ng/ml TGF-β for 6 hours. RNA was harvested using Buffer RLT RNeasy Mini kit (Qiagen). For qRT-PCR analysis of described human genes cDNA was created using Verso cDNA Synthesis Kit (Cat #AB-1453/A, Thermo Scientific, Houston, TX) and 1 μg of total RNA. Predesigned gene specific primer and probe sets were obtained from (SA Biosciences), for human genes: Smad2 (Cat No. PPH01949F, NM 005901), Smad3 (Cat No. PPH01921C; NM 005902), ID1 (PPH003178B, NM 002165), ID2 (PPH00414A; NM 002166), ID3 (PPH00413F, NM 002167), Snail (PH02475A; NM 003068), GAPDH (PH00150F; NM 002046). qRT-PCR synthesis of assigned genes was performed using DyNAmo Flash SYBR Green qPCR kit (Cat No#F-415L, Life Technologies Inc.) according to the manufacturer's protocol on a ABI 7500 Fast Real Time PCR System. The relative mRNA levels were calculated using the comparative Ct method (ΔΔCt). Briefly, the Ct (cycle threshold) values for the BACTIN were subtracted from Ct values of the FASN mRNA to achieve the ΔCt value. The 2−ΔCt was calculated and then divided by a control sample to achieve the relative mRNA levels (ΔΔCt). Reported values are the means and standard errors of three biological replicates.
Gene array, microarray signatures, and microarray data processing
Gene expression microarrays were performed according to established protocols previously published.Citation7,47,64 Human breast tumor microarray data was derived from two data sets that are available to the public at the UNC microarray database (UNCMD; https://genome.unc.edu/pubsup/breastGEO/clinicalData.shtml). One of the datasets used is defined as UNC337.Citation7,47 The other microarray data set is defined as combined UNC855 dataset.Citation48 All human tumor and normal tissue samples were collected and preserved using IRB-approved protocol that are maintained under UNC microarray database as previously described.Citation7,47,48 All probes/genes were normalized as previously described.Citation48 All probes for each gene were averaged to generate independent expression estimates for any one gene. Similar methods as defined inCitation7,48 were used for hierarchical clustering and survival analyses from the UNC337 and Combined 855 data set.
Breast cancer cell line microarray dataset
Using an Affymetrix U133A gene expression microarrays that included 51 breast cancer cell lines raw data were normalized using the robust multiarray analysis (RMA) normalization approach.Citation49 In each data set, genes were median-cantered within each dataset and samples were standardized to zero mean and unit variance before other analyses were performed. Among the 52 breast cancer cell lines examined, we specifically focused on luminal A/B and Her2 subtypes (SUM185P, MDA-MB-453, SKBR3, BT474, T47D, and MCF7), basal A (BT20, HCC70, MDA-MB-468), normal-like transformed (MCF-10A), mesenchymal (BT-549, SUM149PT), mesenchymal stem-like (claudin-low; SUM159PT, HS578T, MDA-MB-436, MDA-MB-231, MDA-MB-157). We combined average probe intensities for luminal A/B, Her2 (luminal), basal (basal), mesenchymal and mesenchymal-stem-like (M/MSL) cell lines and derived the average probe intensities for each defined subgroup.
Statistics and survival analysis
Statistical analyses of the experimental data were performed using a two-sided Student's t test. Significance was set at P<0.05. All statistical analysis were additionally peformed using Prism5 program (GraphPad Software Inc.). Box-and-whisker plots were used to observe the relationship of the intrinsic subtypes with the organ-specific metastatic signatures and were performed in R. Kaplan-Meier univariate survival analyses were performed in WinStat and were used to determine the significance of gene signature expression with survival outcome.
Supplementary Files
Download MS Word (61 KB)Funding
This study was supported by finds from the Colorado Cancer League 2011 (PI: AT/RSW). Support for the UC Denver Cancer Center DNA Sequence, University of Colorado Cancer Center (UCCC) Tissue Culture shared resource, and Analysis Shared Resource Core for cell line authentication is provided in part by Colorado's NIH/NCI Cancer Center Support Grant P30CA043934.
References
- Cheang MC, Voduc D, Bajdik C, Leung S, McKinney S, Chia SK, Perou CM, Nielsen TO. Basal-like breast cancer defined by five biomarkers has superior prognostic value than triple-negative phenotype. Clin Cancer Res 2008; 14(5):1368-76; PMID:18316557; http://dx.doi.org/10.1158/1078-0432.CCR-07-1658
- Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. N Engl J Med 2010; 363(20):1938-48; PMID:21067385; http://dx.doi.org/10.1056/NEJMra1001389
- Lehmann BD, Pietenpol JA. Identification and use of biomarkers in treatment strategies for triple-negative breast cancer subtypes. J Pathol 2014; 232(2):142-50; PMID:24114677; http://dx.doi.org/10.1002/path.4280
- Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, Pietenpol JA. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest 2011; 121(7):2750-67; PMID:21633166; http://dx.doi.org/10.1172/JCI45014
- Perou CM. Molecular stratification of triple-negative breast cancers. Oncologist 2010; 15 Supp l5:39-48; PMID:21138954; http://dx.doi.org/10.1634/theoncologist.2010-S5-39
- Sabatier R, Finetti P, Guille A, Adelaide J, Chaffanet M, Viens P, Birnbaum D, Bertucci F. Claudin-low breast cancers: clinical, pathological, molecular and prognostic characterization. Mol Cancer 2014; 13:228; http://dx.doi.org/10.1186/1476-4598-13-228
- Prat A, Parker JS, Karginova O, Fan C, Livasy C, Herschkowitz JI, He X, Perou CM. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res 2010; 12(5):R68; PMID:20813035; http://dx.doi.org/10.1186/bcr2635
- Cleator S, Heller W, Coombes RC. Triple-negative breast cancer: therapeutic options. Lancet Oncol 2007; 8(3):235-44; PMID:17329194; http://dx.doi.org/10.1016/S1470-2045(07)70074-8
- Ding L, Ellis MJ, Li S, Larson DE, Chen K, Wallis JW, Harris CC, McLellan MD, Fulton RS, Fulton LL, et al. Genome remodelling in a basal-like breast cancer metastasis and xenograft. Nature 2010; 464(7291):999-1005; PMID:20393555; http://dx.doi.org/10.1038/nature08989
- Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, Hastie T, Eisen MB, van de Rijn M, Jeffrey SS, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A 2001; 98(19):10869-74
- Weigman VJ, Chao HH, Shabalin AA, He X, Parker JS, Nordgard SH, Grushko T, Huo D, Nwachukwu C, Nobel A, et al., Basal-like Breast cancer DNA copy number losses identify genes involved in genomic instability, response to therapy, and patient survival. Breast Cancer Res Treat 2011; 133(3):865-80.; PMID:22048815
- Network TCGA. Comprehensive molecular portraits of human breast tumours. Nature 2012; 490(7418):61-70; PMID:23000897; http://dx.doi.org/10.1038/nature11412
- Massague J. TGFbeta in Cancer. Cell 2008; 134(2):215-30; PMID:18662538; http://dx.doi.org/10.1016/j.cell.2008.07.001
- Blobe GC, Schiemann WP, Lodish HF. Role of transforming growth factor β in human disease. N Engl J Med 2000; 342(18):1350-8; PMID:10793168; http://dx.doi.org/10.1056/NEJM200005043421807
- Picon A, Gold LI, Wang J, Cohen A, Friedman E. A subset of metastatic human colon cancers expresses elevated levels of transforming growth factor beta1. Cancer Epidemiol Biomarkers Prev 1998; 7(6):497-504.; PMID:9641494
- Meulmeester E, Ten Dijke P. The dynamic roles of TGF-β in cancer. J Pathol 2011; 223(2):205-18; PMID:20957627; http://dx.doi.org/10.1002/path.2785
- Padua D, Massague J. Roles of TGFbeta in metastasis. Cell Res 2009; 19(1):89-102; PMID:19050696; http://dx.doi.org/10.1038/cr.2008.316
- Wakefield LM, Roberts AB. TGF-β signaling: positive and negative effects on tumorigenesis. Curr Opin Genet Dev 2002; 12(1):22-9; PMID:11790550; http://dx.doi.org/10.1016/S0959-437X(01)00259-3
- Derynck R, Akhurst RJ, Balmain A. TGF-β signaling in tumor suppression and cancer progression. Nat Genet 2001; 29(2):117-29; PMID:11586292; http://dx.doi.org/10.1038/ng1001-117
- Massague J. TGF-β signal transduction. Annu Rev Biochem 1998; 67:753-91; PMID:9759503; http://dx.doi.org/10.1146/annurev.biochem.67.1.753
- Bierie B, Stover DG, Abel TW, Chytil A, Gorska AE, Aakre M, Forrester E, Yang L, Wagner KU, Moses HL. Transforming growth factor-β regulates mammary carcinoma cell survival and interaction with the adjacent microenvironment. Cancer Res 2008; 68(6):1809-19; PMID:18339861; http://dx.doi.org/10.1158/0008-5472.CAN-07-5597
- Moses HL. Serra R. Regulation of differentiation by TGF-β. Curr Opin Genetics Dev 1996; 6(5):581-6; PMID:8939725; http://dx.doi.org/10.1016/S0959-437X(96)80087-6
- Muraoka-Cook RS, Dumont N, Arteaga CL. Dual role of transforming growth factor β in mammary tumorigenesis and metastatic progression. Clin Cancer Res 2005; 11(2 Pt 2):937s-43s.; PMID:15701890
- Ganapathy V, Ge R, Grazioli A, Xie W, Banach-Petrosky W, Kang Y, Lonning S, McPherson J, Yingling JM, Biswas S, et al. Targeting the Transforming Growth Factor-β pathway inhibits human basal-like breast cancer metastasis. Mol Cancer 2010; 9:122; PMID:20504320; http://dx.doi.org/10.1186/1476-4598-9-122
- Tan AR, Alexe G, Reiss M. Transforming growth factor-β signaling: emerging stem cell target in metastatic breast cancer? Breast Cancer Res Treat 2009; 115(3):453-95.; PMID:18841463
- Bruna A, Darken RS, Rojo F, Ocaña A, Peñuelas S, Arias A, Paris R, Tortosa A, Mora J, Baselga J, et al. High TGFbeta-Smad activity confers poor prognosis in glioma patients and promotes cell proliferation depending on the methylation of the PDGF-B gene. Cancer Cell 2007; 11(2):147-60; PMID:17292826; http://dx.doi.org/10.1016/j.ccr.2006.11.023
- Kjellman C, Olofsson SP, Hansson O, Von Schantz T, Lindvall M, Nilsson I, Salford LG, Sjögren HO, Widegren B. Expression of TGF-β isoforms, TGF-β receptors, and SMAD molecules at different stages of human glioma. Int J Cancer 2000; 89(3):251-8; PMID:10861501; http://dx.doi.org/10.1002/1097-0215(20000520)89:3<251::AID-IJC7>3.0.CO;2-5
- Duffy MM, Ritter T, Ceredig R, Griffin MD. Mesenchymal stem cell effects on T-cell effector pathways. Stem Cell Res Ther 2011; 2(4):34; PMID:21861858; http://dx.doi.org/10.1186/scrt75
- Li MO, Sanjabi S, Flavell RA. Transforming growth factor-β controls development, homeostasis, and tolerance of T cells by regulatory T cell-dependent and -independent mechanisms. Immunity 2006; 25(3):455-71; PMID:16973386; http://dx.doi.org/10.1016/j.immuni.2006.07.011
- Massague J. TGF-β signaling in development and disease. FEBS Lett 2012; 586(14):1833; PMID:22651913; http://dx.doi.org/10.1016/j.febslet.2012.05.030
- Massague J. TGFbeta signalling in context. Nat Rev Mol Cell Biol 2012; 13(10):616-30; PMID:22992590; http://dx.doi.org/10.1038/nrm3434
- Zarzynska JM. Two faces of TGF-beta1 in breast cancer. Mediators Inflamm 2014; 2014:141747; PMID:24891760; http://dx.doi.org/10.1155/2014/141747
- Panis C, Herrera AC, Victorino VJ, Aranome AM, Cecchini R. Screening of circulating TGF-β levels and its clinicopathological significance in human breast cancer. Anticancer Res 2013; 33(2):737-42; PMID:23393376
- Buijs JT, Stayrook KR, Guise TA. The role of TGF-β in bone metastasis: novel therapeutic perspectives. Bonekey Rep 2012; 1:96; PMID:23951484; http://dx.doi.org/10.1038/bonekey.2012.96
- Ikushima H, Miyazono K. TGFbeta signalling: a complex web in cancer progression. Nat Rev Cancer 2010; 10(6):415-24; PMID:20495575; http://dx.doi.org/10.1038/nrc2853
- Maier A, Peille AL, Vuaroqueaux V, Lahn M. Anti-tumor activity of the TGF-β receptor kinase inhibitor galunisertib (LY2157299 monohydrate) in patient-derived tumor xenografts. Cell Oncol (Dordr) 2015; 38(2):131-44; PMID:25573078; http://dx.doi.org/10.1007/s13402-014-0210-8
- Goodwin PJ, Ligibel JA, Stambolic V. Metformin in breast cancer: time for action. J Clin Oncol 2009; 27(20):3271-3; PMID:19487373; http://dx.doi.org/10.1200/JCO.2009.22.1630
- Iliopoulos D, Hirsch HA, Struhl K. Metformin decreases the dose of chemotherapy for prolonging tumor remission in mouse xenografts involving multiple cancer cell types. Cancer Res 2011; 71(9):3196-201; PMID:21415163; http://dx.doi.org/10.1158/0008-5472.CAN-10-3471
- Jiralerspong S, Palla SL, Giordano SH, Meric-Bernstam F, Liedtke C, Barnett CM, Hsu L, Hung MC, Hortobagyi GN, Gonzalez-Angulo AM. Metformin and pathologic complete responses to neoadjuvant chemotherapy in diabetic patients with breast cancer. J Clin Oncol 2009; 27(20):3297-302; PMID:19487376; http://dx.doi.org/10.1200/JCO.2009.19.6410
- Hirsch HA, Iliopoulos D, Tsichlis PN, Struhl K. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res 2009; 69(19):7507-11; PMID:19752085; http://dx.doi.org/10.1158/0008-5472.CAN-09-2994
- Wahdan-Alaswad R, Fan Z, Edgerton SM, Liu B, Deng XS, Arnadottir SS, Richer JK, Anderson SM, Thor AD. Glucose promotes breast cancer aggression and reduces metformin efficacy. Cell Cycle 2013; 12(24):3759-69; PMID:24107633; http://dx.doi.org/10.4161/cc.26641
- Wahdan-Alaswad RS, Cochrane DR, Spoelstra NS, Howe EN, Edgerton SM, Anderson SM, Thor AD, Richer JK. Metformin-induced killing of triple-negative breast cancer cells is mediated by reduction in fatty acid synthase via miRNA-193b. Horm Cancer 2014; 5(6):374-89; PMID:25213330; http://dx.doi.org/10.1007/s12672-014-0188-8
- Alimova IN, Liu B, Fan Z, Edgerton SM, Dillon T, Lind SE, Thor AD. Metformin inhibits breast cancer cell growth, colony formation and induces cell cycle arrest in vitro. Cell Cycle 2009; 8(6):909-15; PMID:19221498; http://dx.doi.org/10.4161/cc.8.6.7933
- Liu B, Fan Z, Edgerton SM, Deng XS, Alimova IN, Lind SE, Thor AD. Metformin induces unique biological and molecular responses in triple negative breast cancer cells. Cell Cycle 2009; 8(13):2031-40; PMID:19440038; http://dx.doi.org/10.4161/cc.8.13.8814
- Liu B, Fan Z, Edgerton SM, Yang X, Lind SE, Thor AD. Potent anti-proliferative effects of metformin on trastuzumab-resistant breast cancer cells via inhibition of erbB2/IGF-1 receptor interactions. Cell Cycle 2011; 10(17):2959-66; PMID:21862872; http://dx.doi.org/10.4161/cc.10.17.16359
- Dalal BI, Keown PA, Greenberg AH. Immunocytochemical localization of secreted transforming growth factor-β 1 to the advancing edges of primary tumors and to lymph node metastases of human mammary carcinoma. Am J Pathol 1993; 143(2):381-9.; PMID:8393616
- Parker JS, Mullins M, Cheang MC, Leung S, Voduc D, Vickery T, Davies S, Fauron C, He X, Hu Z, et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol 2009; 27(8):1160-7; PMID:19204204; http://dx.doi.org/10.1200/JCO.2008.18.1370
- Harrell JC, Prat A, Parker JS, Fan C, He X, Carey L, Anders C, Ewend M, Perou CM. Genomic analysis identifies unique signatures predictive of brain, lung, and liver relapse. Breast Cancer Res Treat 2012; 132(2):523-35; PMID:21671017; http://dx.doi.org/10.1007/s10549-011-1619-7
- Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T, Clark L, Bayani N, Coppe JP, Tong F, et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 2006; 10(6):515-27; PMID:17157791; http://dx.doi.org/10.1016/j.ccr.2006.10.008
- Gupta GP, Perk J, Acharyya S, de Candia P, Mittal V, Todorova-Manova K, Gerald WL, Brogi E, Benezra R, Massagué J. ID genes mediate tumor reinitiation during breast cancer lung metastasis. Proc Natl Acad Sci U S A 2007; 104(49):19506-11
- Kang Y, Chen CR, Massague J. A self-enabling TGFbeta response coupled to stress signaling: Smad engages stress response factor ATF3 for Id1 repression in epithelial cells. Mol Cell 2003; 11(4):915-26; http://dx.doi.org/10.1016/S1097-2765(03)00109-6
- Scheel TK, Gottwein JM, Carlsen TH, Li YP, Jensen TB, Spengler U, Weis N, Bukh J. Efficient culture adaptation of hepatitis C virus recombinants with genotype-specific core-NS2 by using previously identified mutations. J Virol 2011; 85(6):2891-906; PMID:21177811; http://dx.doi.org/10.1128/JVI.01605-10
- Gumireddy K, Li A, Kossenkov AV, Cai KQ, Liu Q, Yan J, Xu H, Showe L, Zhang L, Huang Q. ID1 promotes breast cancer metastasis by S100A9 regulation. Mol Cancer Res 2014; 12(9):1334-43; PMID:24948111; http://dx.doi.org/10.1158/1541-7786.MCR-14-0049
- Stankic M, Pavlovic S, Chin Y, Brogi E, Padua D, Norton L, Massagué J, Benezra R. TGF-β-Id1 signaling opposes Twist1 and promotes metastatic colonization via a mesenchymal-to-epithelial transition. Cell Rep 2013; 5(5):1228-42; PMID:24332369; http://dx.doi.org/10.1016/j.celrep.2013.11.014
- Farina AR, Coppa A, Tiberio A, Tacconelli A, Turco A, Colletta G, Gulino A, Mackay AR. Transforming growth factor-beta1 enhances the invasiveness of human MDA-MB-231 breast cancer cells by up-regulating urokinase activity. Int J Cancer 1998; 75(5):721-30; PMID:9495240; http://dx.doi.org/10.1002/(SICI)1097-0215(19980302)75:5<721::AID-IJC10>3.0.CO;2-9
- Kalkhoven E, Kwakkenbos-Isbrücker L, Mummery CL, de Laat SW, van den Eijnden-van Raaij AJ, van der Saag PT, van der Burg B. The role of TGF-β production in growth inhibition of breast-tumor cells by progestins. Int J Cancer 1995; 61(1):80-6; PMID:7705937; http://dx.doi.org/10.1002/ijc.2910610114
- Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res 2010; 70(2):440-6; PMID:20068163; http://dx.doi.org/10.1158/0008-5472.CAN-09-1947
- Pang MF, Georgoudaki AM, Lambut L, Johansson J, Tabor V, Hagikura K, Jin Y, Jansson M, Alexander JS, Nelson CM, et al. TGF-beta1-induced EMT promotes targeted migration of breast cancer cells through the lymphatic system by the activation of CCR7/CCL21-mediated chemotaxis. Oncogene 2015; 35(6):748-60
- Cufi S, Vazquez-Martin A, Oliveras-Ferraros C, Martin-Castillo B, Joven J, Menendez JA. Metformin against TGFbeta-induced epithelial-to-mesenchymal transition (EMT): from cancer stem cells to aging-associated fibrosis. Cell Cycle 2010; 9(22):4461-8; PMID:21088486; http://dx.doi.org/10.4161/cc.9.22.14048
- Qu C, Zhang W, Zheng G, Zhang Z, Yin J, He Z. Metformin reverses multidrug resistance and epithelial-mesenchymal transition (EMT) via activating AMP-activated protein kinase (AMPK) in human breast cancer cells. Mol Cell Biochem 2014; 386(1–2):63-71; http://dx.doi.org/10.1007/s11010-013-1839-8
- Bierie B, Moses HL. Transforming growth factor β (TGF-β) and inflammation in cancer. Cytokine Growth Factor Rev 2010; 21(1):49-59; PMID:20018551; http://dx.doi.org/10.1016/j.cytogfr.2009.11.008
- Pickup M, Novitskiy S, Moses HL. The roles of TGFbeta in the tumour microenvironment. Nat Rev Cancer 2013; 13(11):788-99; PMID:24132110; http://dx.doi.org/10.1038/nrc3603
- Harrell JC, Dye WW, Allred DC, Jedlicka P, Spoelstra NS, Sartorius CA, Horwitz KB. Estrogen receptor positive breast cancer metastasis: altered hormonal sensitivity and tumor aggressiveness in lymphatic vessels and lymph nodes. Cancer Res 2006; 66(18):9308-15; http://dx.doi.org/10.1158/0008-5472.CAN-06-1769
- Harrell JC, Pfefferle AD, Zalles N, Prat A, Fan C, Khramtsov A, Olopade OI, Troester MA, Dudley AC, Perou CM. Endothelial-like properties of claudin-low breast cancer cells promote tumor vascular permeability and metastasis. Clin Exp Metastasis 2014; 31(1):33-45; PMID:23975155; http://dx.doi.org/10.1007/s10585-013-9607-4