
ABSTRACT
Due to increased glycolysis and poor local perfusion, solid tumors are usually immersed in an acidic microenvironment. While extracellular acidosis is cytotoxic, cancer cells eventually become acclimated to it. While previous studies have addressed the acute effect of acidosis on cancer cells, little is known about how cancer cells survive chronic acidosis. In this study we exposed colorectal cancer (CRC) cells (HCT15, HCT116 and LoVo) to acidic pH (pH 6.5) continuously for over three months and obtained CRC cells that become acclimated to acidic pH, designated as CRC-acidosis-acclimated or CRC-AA. We unexpectedly found that while acute exposure to low pH resulted in an increase in the level of intracellular reactive oxygen species (ROS), CRC-AA cells exhibited a significantly reduced level of ROS when compared to ancestor cells. CRC-AA cells were found to maintain a higher level of reduced glutathione, via the upregulation of CD44 and glutathione reductase (GSR), among others, than their ancestor cells. Importantly, CRC-AA cells were more sensitive to agents that deplete GSH. Moreover, downregulation of GSR by RNA interference was more deleterious to CRC-AA cells than to control cells. Together, our results demonstrate a critical role of glutathione-dependent antioxidant defense in acclimation of CRC cells to acidic extracellular pH.
Introduction
Cancer cells form a dynamic relationship with their microenvironment. Both the cancer cells and their microenvironment evolve during the course of cancer development and progression. While cancer cells can modify their microenvironment by recruiting immune cells, mesenchymal cells and endothelial cells that comprise the cellular components of tumor microenvironment, they also need to evolve in various features in order to survive in the relatively hostile environment, which is usually hypoxic, poorly circulated and low in nutrients.Citation1-4 Because cancer cells are usually rewired for glycolysis, which produces lactic acid, even under aerobic condition (the Warburg effect), and tumor vasculature is usually poorly functional, tumor microenvironment is characteristic of an acidic pH, or called extracellular acidosis, which is usually measured between 5.5∼7.0.Citation5-7 For most cells, an extracellular acidic microenvironment is harmful and genotoxic.Citation8-10 However, some tumor cells may survive and evolve to become more malignant under such a condition.Citation11-17 Interestingly, cancer cells usually have a higher intracellular pH (pH > 7 .4) than normal differentiated adult cells (pH∼7.2).Citation18 This reversed pH gradient is regarded as one of the adaptive features of most cancers and may facilitate survival, proliferation, metabolic adaptation, metastasis and invasion of cancer cells. Moreover, autophagy and reprogrammed cellular metabolism were found to be critical for the survival of cancer cells in an acidic microenvironment.Citation19-21 However, it remains to be determined whether there are other means upon which cancer cells rely for living in the acidic extracellular milieu.
Living cells are always subjected to the effects of reactive oxygen species (ROS) which include superoxide anion (O2·−), hydroxyl radical (HO·) and hydrogen peroxide (H2O2). While ROS at low to moderate levels are essential for cellular signaling that sustains proliferation and differentiation, when they are produced in excess and overwhelm the cellular antioxidant defensive systems, oxidative stress ensues, which may lead to apoptosis, senescence and increased mutation load.Citation22-25 Intracellular ROS level is usually elevated in response to various types of stress and stimuli.Citation23 It was reported that acute exposure to acidic microenvironment can cause an increase in ROS.Citation26 Cancer cells usually experience high level of ROS and have concomitantly acquired robust antioxidant capacity.Citation27-29
While acidic tumor microenvironment is known to accompany tumor survival, growth, invasion and metastasis, most studies so far addressed acute acidosis, in terms of hours to days, and little is known about how cancer cells respond to chronic acidic environment. If acidosis is a selective factor for cancer cells, it is important to know what features the survivors have when compared to ancestors. In order to understand what happens to tumor cells in chronic acidic microenvironment, we exposed colorectal cancer (CRC) cells to acidic pH continuously for a long period and selected the CRC cells that had become acclimated to acidic pH (CRC-AA). We found that contrary to the elevation of ROS in CRC cells acutely exposed to low pH, the ROS level in CRC-AA cells was lower than in their parental cells and was maintained by a high level of GSH.
Results
Characteristics of colon cancer cells adapted to acidic microenvironment
We subjected colorectal cancer cells, HCT15, HCT116, LoVo and SW480 cells, to acidic medium (pH6.5) continuously to select for cells that became acclimated to the acidic microenvironment. While SW480 cells failed to proliferate shortly after being transferred to acidic medium, HCT15, HCT116 and LoVo cells survived and were able to proliferate after passages in acidic medium for at least three months. We designated them CRC-AA for CRC-acidosis acclimated (HCT15-AA, HCT116-AA and LoVo-AA cells, respectively).
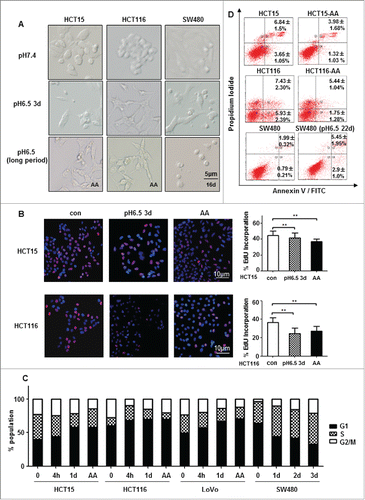
The CRC-AA cells appeared to have lost the cobblestone-like feature that is typical of epithelial cells and is present in their parental cells (). They were more elongated and more scattered. Measurement of proliferation using EdU incorporation assay showed that the CRC-AA cells generally had a reduced rate of proliferation (). Examination of cell cycle distribution showed that CRC-AA cells had significantly higher percentages of cells in G1 phase and reduced percentages of cells in S phase ().Interestingly, SW480 cells, which failed to survive the chronic acidic stress, were not arrested in G1 phase. CRC cells exposed to acidic medium initially showed an increased apoptosis than controls. However, the CRC-AA cells eventually exhibited reduced levels of apoptosis than their parental cells (). High level of apoptosis remained in SW480 cells even after 22 d in acidic medium (), which is probably due to loss of the protection by G1 arrest. These results indicate that CRC-AA cells are distinct from their parental cells in morphology and proliferation. They even have a reduced rate of spontaneous apoptosis.
Figure 1. Chronic acidic microenvironment inhibits proliferation. (A) Cell density and morphology of HCT15 cells (left), HCT116 (middle) and SW480 (right) at pH6.5 were examined under microscopy (×20). HCT15 and HCT116 cells growing in acidic medium were more scattered and elongated. SW480 showed little morphological change except a reduction in cell density. (B) Acidic microenvironment reduced proliferation of CRC cells. Proliferation of HCT15 (top) and HCT116 (bottom) cells cultured at low pH (6.5) for 72 h or for more than 3 months (CRC-AA) were determined by EdU incorporation. Red, EdU; blue, DAPI. (C) Cell cycle distribution of CRC cells growing in acidic medium for various durations. (D) Apoptotic levels of CRC cells adapted to acidic pH (HCT15 and HCT116 cells) and SW480 cells. *, p < 0.05; **, p < 0.01.
ROS level is reduced in colorectal cancer cells acclimated to chronic acidic stress
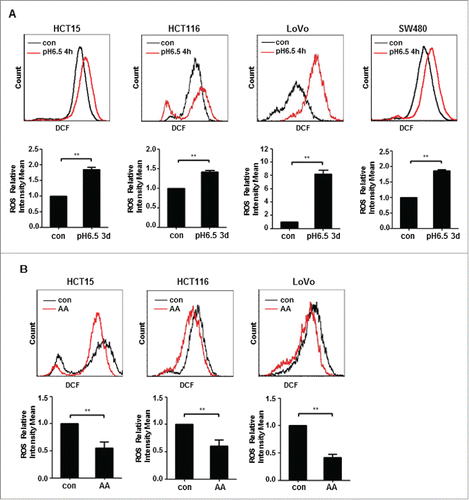
A previous study showed that the level of intracellular ROS in cancer cells exposed to acute acidosis was elevated.Citation26 Measurement of ROS showed that consistent with the previous report, the ROS level was increased when CRC cells were exposed to acidic medium for 4 h (). The elevation of ROS in response to acute acidosis applied to all four cell lines tested. However, all three lines of CRC-AA cells showed a significantly reduced level of ROS when compared to their parental cells (). These results suggest that while CRC cells may respond to acute acidosis by elevating ROS, they generally have a reduced level of ROS when they become acclimated to the low extracellular pH.
Figure 2. ROS is reduced in colon cancer cells acclimated to acidic microenvironment. (A) DCF fluorescent intensity of HCT15, HCT116, LoVo and SW480 cells exposed to acidic medium for 4 h. (B) DCF fluorescent intensity of HCT15 cells, HCT116 cells and LoVo cells growing acidic medium for more than 3 months. *, p < 0.05; **, p < 0.01.
Acclimation of CRC cells to chronic acidosis depends on GSH
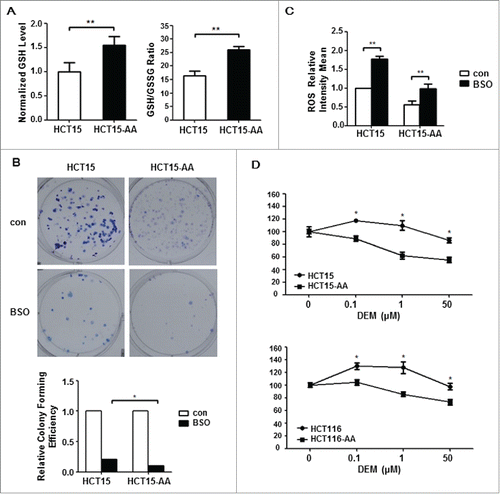
Glutathione (GSH) is regarded to be the most important antioxidant in mammalian cells.Citation30 Reduced GSH can effectively convert ROS into benign molecules. To determine whether GSH is involved in the maintenance of a reduced ROS level in CRC-AA, we measured the level of GSH as well as the ratio of GSH/GSSG in HCT15-AA and their parental cells. As shown in , the level of GSH as well as the ratio of GSH/GSSG was significantly higher in HCT15-AA cells. To test whether the increased level of GSH is responsible for adaptation of CRC cells to acidic pH, we determined colony formation of HCT15-AA and their parental cells in the presence of buthionine sulphoximine (BSO), a GSH-depleting agent. As shown in , BSO inhibited colony formation in both sets of cells, but the reduction was more pronounced for HCT115-AA cells. As expected, the level of ROS was increased in CRC cells treated with BSO (). Furthermore, when HCT15 and HCT116 cells were exposed to diethyl maleate (DEM), another depleting agent of GSH, and scored for viability with MTT assay, both HCT15-AA and HCT116-AA cells showed an increased sensitivity to DEM when compared to their parental cells (). These results suggest that the survival of CRC-AA cells was more dependent on GSH than that of control cells. Together, these results indicate that a low level of ROS, which is probably mediated by increased level of GSH, is critical for CRC cells to live under the acidic microenvironment.
Figure 3. Antioxidant GSH is critical for survival of acidosis-acclimated cancer cells. (A) GSH level as well as GSH/GSSG ratio is higher in HCT15-AA than in their parental cells. The GSH and GSH/GSSG ratio were measured by a GSH and GSSG assay kit. (B) Inhibition of colony formation by BSO in HCT15-AA and their parental cells. Cultured cells were exposed to BSO (10μM) for 12d to allow for colony formation. (C) Induction of ROS by BSO. ROS fluorescence intensity of HCT15 cells treated with BSO (500μM, 12h) was determined by flow cytometry. (D) Inhibition of proliferation of CRC-AA cells and their parental cells by DEM. CRC cells were treated with different concentrations of DEM (0, 0.1, 1, 50μM) for 48 h. Cell viability was measured by MTT assay. *, p < 0.05; **, p < 0.01.
Expression pattern of representative genes involved in GSH metabolism
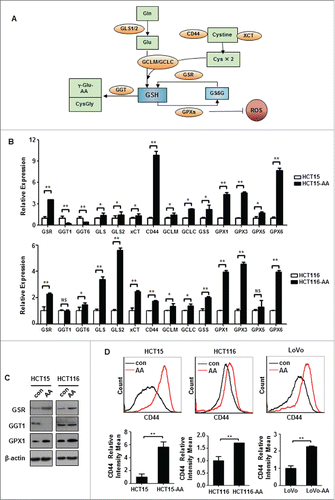
As illustrated in , the level of GSH is affected by its rate of de novo synthesis (via steps catalyzed by GLS1/2 and GCLC/GCLM, and facilitated by CD44 and xCT), by its utilization (catalyzed by GPX or GST) and by its breakdown (catalyzed by GGT).Citation28,30,31 We next determined, by quantitative real-time PCR (qRT-PCR), the expression levels of representative genes involved in GSH metabolism in HCT15-AA and HCT116-AA cells. As shown in , the expression pattern of those genes generally suggests an increased capacity for antioxidant defense in CRC-AA cells when compared to their parental cells. GSR, CD44, GPX1, GPX3 and GPX6 were remarkably upregulated in both HCT15-AA and HCT116-AA cells, though for the other genes the changes in expression were less consistent or the degrees of difference were less pronounced. Western blot analysis confirmed the upregulation of GSR and GPX1 at protein level (). Furthermore, flow cytometry analysis revealed an increase in the expression of CD44 in HCT15-AA, HCT116-AA and LoVo-AA cells over their parental cells (). Taken together, these results suggest that compared to their parental cells, CRC-AA cells may be generally wired to maintain a more robust antioxidant defense that depends on GSH.
Figure 4. Expression levels of genes involved in GSH metabolism. (A) A diagram showing the key factors involved in GSH metabolism. (B) Relative expressions of representative redox genes at mRNA level in CRC-AA and their parental cells, as measured by quantitative real-time PCR. (C) Expression levels of redox proteins in CRC-AA and their parental cells measured by Western blotting. (D) Flow cytometric analysis of CD44 expression levels in CRC-AA and their parental cells. *, p < 0.05; **, p < 0.01.
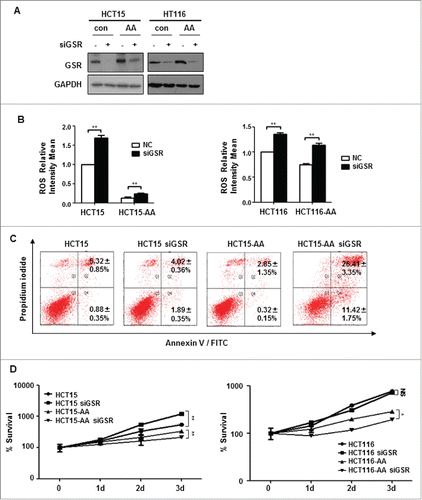
Requirement of GSR for survival of CRC cells in acidic microenvironment
GSR is essential for the conversion of GSSG to reduced GSH. We next determined the effect of GSR depletion on the survival of CRC-AA cells. As shown in , GSR was efficiently knocked down by RNAi in HCT15 and HCT116 cells. As expected, the ROS level was elevated as a result of the GSR depletion (). Strikingly, while the level of apoptosis remained unchanged in response to GSR depletion in parental HCT15 cells, it increased over ten-fold in HCT15-AA cells (), suggesting that GSR is strictly required for the survival of CRC-AA cells. Consistently, HCT15-AA and HCT116-AA cells in which GSR was depleted experienced a significantly reduced proliferation, as determined by MTT assay (). Intriguingly, the parental HCT15 cells in which GSR was depleted even showed an enhanced rate of proliferation. Together, these results provide further support for the notion that the CRC-AA cells are more reliant on GSH than their parental cells.
Figure 5. GSR knockdown compromises the survival of CRC-AA cells. (A) Knockdown of GSR by siRNA in HCT15 and HCT116 cells. RNAi efficiency was determined by Western blotting, GAPDH was used as a loading control. (B) Increase of ROS levels in CRC cells by GSR siRNA. (C) Apoptosis was greatly increased in CRC-AA cells when transfected with GSR siRNA. (D) Viability of CRC-AA cells was reduced when transfected with GSR siRNA, as determined by MTT assay. *, p < 0.05; **, p < 0.01.
Discussion
In this study we subjected colorectal cancer cells to chronic acidosis, selected for and characterized the cells that became acclimated to low pH, designated as CRC-AA. We observed that while acute exposure to low pH resulted in an increase in the level of intracellular ROS in CRC cells, CRC-AA cells exhibited a much reduced level of ROS when compared to their ancestor cells. CRC-AA cells exhibited a higher level of GSH than in their ancestor cells. Consistently, key genes involved in GSH metabolism are found to be expressed in such a way as to maintain a higher level of GSH and to establish a more robust GSH-based antioxidant defense in CRC-AA cells. CD44, glutathione reductase and glutathione peroxidases were generally upreguated in CRC-AA cells. Importantly, CRC-AA cells were found to be more sensitive to depletion of GSH than their ancestor cells. Our results thus provided a new insight into how cancer cells adapt to acidic microenvironment.
We also observed that cancer cells that survived chronic exposure to acidic medium all showed an elevation of cells in G1/G0 phase, indicative of G1 arrest. Interestingly, SW480 cells, which did not exhibit G1 arrest in response to acidic medium and failed to adapt, experienced high level of apoptosis throughout the selection process. These observations suggest that G1 arrest is also important for survival under acidic stress.
It should be noted that antioxidant genes such as CD44, GLS2, GPX1 are generally upregulated in CRC specimens when compared to mucosaCitation32 (GEO dataset accession: GSE20842), thus already providing to CRC a stronger buffer against oxidative stress than to normal tissue. The increased expression of those genes in CRC-AA cells over CRC parental cells would confer further enhanced antioxidant defense under the acidic microenvironment. It should be pointed that while genes involved in GSH metabolism may generally evolve in expression levels to boost the antioxidant defense under acidic condition, individual genes may be regulated differentially in different CRC cell lines or specimens. It is clear that the extent to which the genes were upregulated varied between HCT15-AA and HCT116-AA cells. Furthermore, while GGT1 and GGT6 were downregulated in HCT15-AA cells, they remained unchanged or upregulated in HCT116-AA cells. Therefore, while we may expect an enhanced GSH-based antioxidant defense in CRC-AA cells, the redox pathways and/or genes involved may vary in different CRC cells. Furthermore, because the metabolic pathways of glucose, fatty acids and amino acids are potentially all subjected to reprogramming under stressful conditions,Citation21 changes in those pathways may also contribute to antioxidant defense.
Cancer cells and their microenvironment co-evolve during cancer progression. Thanks to accumulation of acquired mutations and/or epigenetic changes, cancer cells are often reprogramed in their developmental potential and metabolism when compared to normal cells. They can also hijack or harness host cells to establish a microenvironment that is conducive to cancer progression. On the other hand, environmental pressures including physical restraint, hypoxia, shortage of nutrients, inflammatory cytokines and extracellular acidosis also act to select for those that are most adapted to the changing environment. The ability to undergo autophagy appears to confer cancer cells a great advantage under many stressful conditions.Citation33 Autophagy likewise operates to protect cancer cells under acidic stress.Citation19,20 Our results presented in this study indicate that an augmented antioxidant defense is also critical for cell survival under chronic acidic stress.
Materials and methods
Cells and cell culture
Human colon cancer cell lines HCT15, HCT116, SW480 and LoVo were obtained from the Cell Bank of Chinese Academy of Sciences (Shanghai). HCT15 and HCT116 cells were maintained in RPMI-1640, SW480 in Leibovitz's L-15 medium and LoVo in DMEM/F12, all supplemented with 10% FBS, 100U/mL penicillin, and 100 μg/mL streptomycin in a humidified 5% CO2/95% air atmosphere at 37°C. The pH of the growth medium was adjusted to 7.4 or 6.5 with 25 mmol/L each of PIPES and HEPES. HCT15, HCT116 and LoVo cells were continuously cultured and passed in growth medium adjusted to pH 6.5. Cells that survived the chronic exposure to low pH, for at least three months, were designated as HCT15-AA, HCT116-AA and LoVo-AA, respectively. SW480 cells failed to survive the chronic acidic culture condition beyond 21 d.
Chemicals
Diethyl Maleate (DEM) from Sigma Chemical (St Louis, MO). Buthionine sulfoximine (BSO) was from Cayman Chemical (Ann Arbor, MI).
EdU incorporation
EdU (5-Ethynyl-2-deoxyuridine) incorporation was performed using Cell-Light™ EdU Cell Proliferation Detection kit (RiboBio, Guangzhou, China) and was previously described.Citation33 Cells were incubated in the presence of EdU (100 µM) for the final 2 h before being processed for EdU staining.
Analyses of cell cycle distribution and apoptosis by flow cytometry
Cells were harvested using 0.25% Trypsin-EDTA, centrifuged (300 g), and washed once with cold PBS. The pellet was resuspended in ice cold 70% ethanol and stored at −20°C. Once all samples were collected, cells were centrifuged and resuspended in cold PBS. Samples were incubated with 20 μg/mL propidium iodide/0.1% Triton X-100 staining solution with 0.1mg/mL RNase A. Cell cycle distribution was determined using the BD Biosciences FACSCanto II Analyzer. At least 20,000 cells were collected. For analysis of apoptosis, both adherent and supernatant cells were harvested, washed twice in PBS, and resuspended in 1× binding buffer at a density of 1×106 cells/mL. Cells were assayed for apoptosis using an Alexa Fluor 488 annexin V/Dead Cell Apoptosis Kit (Invitrogen) according to the manufacturer's instructions.
Determination of cellular ROS
Changes in intracellular ROS were measured using Reactive Oxygen Species Assay Kit (Beyotime Biotechnology) following the manufacturer's protocols. In brief, cells were labeled with 10μM DCFH-DA (2′,7′-dichlorofluorescin diacetate) for 20 min at 37°C in a humidified atmosphere at 5% CO2. The labeled cells were washed and collected. The fluorescence intensity of DCF was measured by flow cytometry (FACSCanto II, BD Biosciences).
Colorimetric determination of GSH and GSSG
The amounts of total glutathione and GSSG were determined using the GSH and GSSG Assay Kit (Beyotime, China) following the manufacturer's recommendations. The amount of GSH was obtained by subtracting GSSG from the total glutathione.
Cell viability assay
Cells were plated in 96-well cell culture plates at the concentration of 2–5 × 103 cells/well. Twenty-four h later, the medium was removed and replaced with fresh medium with or without DEM. Cell viability was measured at the appropriate time points by using the MTT assay kit (Beyotime, China) following manufacturer's instructions. The absorbance of converted dye is measured at the wavelength of 490 nm and the absorbance is proportional to cell viability.
Clonogenic survival assay
Briefly, the cells were treated, trypsinized, suspended in complete medium, counted and replated in 100-mm tissue culture dishes to allow formation of macroscopic colonies. Plates were incubated at 37°C for 7 to 14 days, fixed with methanol, stained with Giemsa, and colonies containing at least 50 cells in size were counted.
Western blotting analysis
Cells were harvested, rinsed in ice-cold PBS, and lysed in Cell Lysis Buffer (Beyotime, China) for 30 minutes. The lysates were centrifuged at 12,000 rpm to remove any cellular debris. Protein concentrations of the lysates were determined by the BCA protein assay system (Beyotime, China). Equal amounts of protein were separated by 10% SDS-PAGE, transferred to PVDF membrane (Millipore, Billerica, MA), and blocked with 5% nonfat dry milk in TBS-Tween 20 (0.1%, v/v) for 1 h at room temperature. The membrane was incubated with primary antibody overnight. Anti-GGT1 and GPX1 were from Abcam (Cambridge, UK); GPX3 was from Novus (Minneapolis, MN); GSR was from proteintech (Wuhan, China); and β-actin and GAPDH were from Santa Cruz (Dallas, TX). After washing, the membrane was incubated with the appropriate horseradish peroxidase secondary antibody (diluted 1:5,000; Amersham Pharmacia Biotech, Arlington Heights, IL) for 1 h. Following several washes, the blots were developed by enhanced chemiluminescence (Millipore, Billerica, MA).
cDNA synthesisand qPCR
Total RNA was isolated using TRIzol reagent (Invitrogen, Shanghai, China) according to the manufacturer's protocol. For qPCR, cDNA was synthesized by reverse transcription of 1 μg of total RNA with random hexamers. The total volume of reverse transcription reaction was 20 μL. Real-time quantitative PCR was performed using the LightCycler® 480 sequence Detection System (Roche Applied Science, Upper Bavaria, Germany). Human glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was amplified as an internal control. The primer sequences are listed in Supplementary Table 1. The samples were loaded in quadruple, and the results of each sample were normalized to GAPDH.
RNA interference
Cells were transfected with siRNAs (50nM) using Lipofectamine 2000 (Invitrogen, USA) according to the manufacturer's guide. siRNAs corresponding to the cDNA of GSR: 5′-CCAGAAUA CCAACGUCAAATT-3′/5′-UUUGACGUUGGUAUUCUGGTT -3′. RNAi efficiency was determined 48 h after transfection.
Statistical analysis
Student's t test was used to determine the statistical significance between experimental groups. Difference was considered significant if the P value was less than 0.05.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
1158374_Table_S1.docx
Download MS Word (15.5 KB)Funding
This study was supported by National Basic Research Program of China (973 Program) grant (2011CB966200), National Natural Science Foundation Research Grants (81372241,81572785 and 81330050), and State Program of National Natural Science Foundation of China for Innovative Research Group (81321061).
References
- Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med 2013; 19:1423-37; PMID:24202395; http://dx.doi.org/10.1038/nm.3394
- Vaupel P, Kallinowski F, Okunieff P. Blood flow, oxygen and nutrient supply, and metabolic microenvironment of human tumors: a review. Cancer Res 1989; 49:6449-65; PMID:2684393
- Vaupel P. Tumor microenvironmental physiology and its implications for radiation oncology. Seminars Radiation Oncol 2004; 14:198-206; PMID:15254862; http://dx.doi.org/10.1016/j.semradonc.2004.04.008
- Amend SR, Pienta KJ. Ecology meets cancer biology: The cancer swamp promotes the lethal cancer phenotype. Oncotarget 2015; 6:9669-78; PMID:25895024; http://dx.doi.org/10.18632/oncotarget.3430
- Engin K, Leeper DB, Cater JR, Thistlethwaite A J, Tupchong L, McFarlane JD. Extracellular pH distribution in human tumours. Int J Hyperthermia 1995; 11:211-6; PMID:7790735; http://dx.doi.org/10.3109/02656739509022457
- Gerweck LE, Seetharaman K. Cellular pH gradient in tumor versus normal tissue: potential exploitation for the treatment of cancer. Cancer Res 1996; 56:1194-8; PMID:8640796
- Helmlinger G, Yuan F, Dellian M, Jain RK. Interstitial pH and pO2 gradients in solid tumors in vivo: high-resolution measurements reveal a lack of correlation. Nat Med 1997; 3:177-82; PMID:9018236; http://dx.doi.org/10.1038/nm0297-177
- Williams AC, Collard TJ, Paraskeva C. An acidic environment leads to p53 dependent induction of apoptosis in human adenoma and carcinoma cell lines: implications for clonal selection during colorectal carcinogenesis. Oncogene 1999; 18:3199-204; PMID:10359525; http://dx.doi.org/10.1038/sj.onc.1202660
- Morita T, Nagaki T, Fukuda I, Okumura K. Clastogenicity of low pH to various cultured mammalian cells. Mutation Res 1992; 268:297-305; PMID:1379335; http://dx.doi.org/10.1016/0027-5107(92)90235-T
- Leontieva OV, Blagosklonny MV. Yeast-like chronological senescence in mammalian cells: phenomenon, mechanism and pharmacological suppression. Aging (Albany NY) 2011; 3:1078-91; PMID:22156391
- Martinez-Zaguilan R, Seftor EA, Seftor RE, Chu YW, Gillies RJ, Hendrix MJ. Acidic pH enhances the invasive behavior of human melanoma cells. Clin Exp Metastasis 1996; 14:176-86; PMID:8605731; http://dx.doi.org/10.1007/BF00121214
- Rofstad EK, Mathiesen B, Kindem K, Galappathi K. Acidic extracellular pH promotes experimental metastasis of human melanoma cells in athymic nude mice. Cancer Res 2006; 66:6699-707; PMID:16818644; http://dx.doi.org/10.1158/0008-5472.CAN-06-0983
- Robey IF, Baggett BK, Kirkpatrick ND, Roe DJ, Dosescu J, Sloane BF, Hashim AI, Morse DL, Raghunand N, Gatenby RA, Gillies RJ. Bicarbonate increases tumor pH and inhibits spontaneous metastases. Cancer Res 2009; 69:2260-8; PMID:19276390; http://dx.doi.org/10.1158/0008-5472.CAN-07-5575
- Wojtkowiak JW, Verduzco D, Schramm KJ, Gillies RJ. Drug resistance and cellular adaptation to tumor acidic pH microenvironment. Mol Pharmaceutics 2011; 8:2032-8; PMID:21981633; http://dx.doi.org/10.1021/mp200292c
- Estrella V, Chen T, Lloyd M, Wojtkowiak J¸, Cornnell HH, Ibrahim-Hashim A, Bailey K, Balagurunathan Y, Rothberg JM, Sloane BF, et al. Acidity generated by the tumor microenvironment drives local invasion. Cancer Res 2013; 73:1524-35; PMID:23288510; http://dx.doi.org/10.1158/0008-5472.CAN-12-2796
- Gupta SC, Singh R, Pochampally R, Watabe, K, Mo YY. Acidosis promotes invasiveness of breast cancer cells through ROS-AKT-NF-kappaB pathway. Oncotarget 2014; 5:12070-82; PMID:25504433; http://dx.doi.org/10.18632/oncotarget.2514
- Peppicelli S, Bianchini F, Toti A, Laurenzana A, Fibbi G, Calorini L. Extracellular acidity strengthens mesenchymal stem cells to promote melanoma progression. Cell cycle 2015; 14:3088-100; PMID:26496168; http://dx.doi.org/10.1080/15384101.2015.1078032
- Webb BA, Chimenti M, Jacobson MP, Barber DL. Dysregulated pH: a perfect storm for cancer progression. Nat Rev Cancer 2011; 11:671-7; PMID:21833026; http://dx.doi.org/10.1038/nrc3110
- Wojtkowiak JW, Rothberg JM, Kumar V, Schramm KJ, Haller E, Proemsey JB, Lloyd MC, Sloane BF, Gillies RJ. Chronic autophagy is a cellular adaptation to tumor acidic pH microenvironments. Cancer Res 2012; 72:3938-47; PMID:22719070; http://dx.doi.org/10.1158/0008-5472.CAN-11-3881
- Marino ML, Pellegrini P, Di Lernia G, Djavaheri-Mergny M, Brnjic S, Zhang X, Hagg M, Linder S, Fais S, Codogno P, et al. Autophagy is a protective mechanism for human melanoma cells under acidic stress. J Biol Chem 2012; 287:30664-76; PMID:22761435; http://dx.doi.org/10.1074/jbc.M112.339127
- Lamonte G, Tang X, Chen JL, Wu J, Ding CK, Keenan MM, Sangokoya C, Kung HN, Ilkayeva O, Boros LG, et al. Acidosis induces reprogramming of cellular metabolism to mitigate oxidative stress. Cancer Metab 2013; 1:23; PMID:24359630; http://dx.doi.org/10.1186/2049-3002-1-23
- Martindale JL, Holbrook NJ. Cellular response to oxidative stress: signaling for suicide and survival. J Cell Physiol 2002; 192:1-15; PMID:12115731; http://dx.doi.org/10.1002/jcp.10119
- Sena LA, Chandel NS. Physiological roles of mitochondrial reactive oxygen species. Mol Cell 2012; 48:158-67; PMID:23102266; http://dx.doi.org/10.1016/j.molcel.2012.09.025
- Xu B, Wang W, Guo H, Sun Z, Wei Z, Zhang X, Liu Z, Tischfield JA, Gong Y, Shao C. Oxidative stress preferentially induces a subtype of micronuclei and mediates the genomic instability caused by p53 dysfunction. Mutation Res 2014; 770:1-8; PMID:25302047; http://dx.doi.org/10.1016/j.mrfmmm.2014.08.004
- Wei Z, Guo H, Liu Z, Zhang X, Liu Q, Qian Y, Gong Y, Shao C. CUL4B impedes stress-induced cellular senescence by dampening a p53-reactive oxygen species positive feedback loop. Free Radical Biol Med 2015; 79:1-13; PMID:25464270; http://dx.doi.org/10.1016/j.freeradbiomed.2014.11.010
- Seo Y, Kinsella TJ. Essential role of DNA base excision repair on survival in an acidic tumor microenvironment. Cancer Res 2009; 69:7285-93; PMID:19723658; http://dx.doi.org/10.1158/0008-5472.CAN-09-0624
- Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov 2009; 8:579-91; PMID:19478820; http://dx.doi.org/10.1038/nrd2803
- Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov 2013; 12:931-47; PMID:24287781; http://dx.doi.org/10.1038/nrd4002
- Harris IS, Treloar AE, Inoue S, Sasaki M, Gorrini C, Lee KC, Yung KY, Brenner D, Knobbe-Thomsen CB, Cox MA, et al. Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell 2015; 27:211-22; PMID:25620030; http://dx.doi.org/10.1016/j.ccell.2014.11.019
- Meister A. Glutathione-ascorbic acid antioxidant system in animals. J Biol Chem 1994; 269:9397-400; PMID:8144521
- Ishimoto T, Nagano O, Yae T, Tamada M, Motohara T, Oshima H, Oshima M, Ikeda T, Asaba R, Yagi H, et al. CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc(−) and thereby promotes tumor growth. Cancer Cell 2011; 19:387-400; PMID:21397861; http://dx.doi.org/10.1016/j.ccr.2011.01.038
- Gaedcke J, Grade M, Jung K, Camps J, Jo P, Emons G, Gehoff A, Sax U, Schirmer M, Becker H, et al. Mutated KRAS results in overexpression of DUSP4, a MAP-kinase phosphatase, and SMYD3, a histone methyltransferase, in rectal carcinomas. Genes Chromosomes Cancer 2010; 49:1024-34; PMID:20725992; http://dx.doi.org/10.1002/gcc.20811
- White E. Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer 2012; 12:401-10; PMID:22534666; http://dx.doi.org/10.1038/nrc3262
- Liu Q, Xu X, Zhao M, Wei Z, Li X, Zhang X, Liu Z, Gong Y, Shao C. Berberine induces senescence of human glioblastoma cells by downregulating the EGFR-MEK-ERK signaling pathway. Mol Cancer Therapeutics 2015; 14:355-63; PMID:25504754; http://dx.doi.org/10.1158/1535-7163.MCT-14-0634