
ABSTRACT
Akt is a critical mediator of the oncogenic PI3K pathway, and its activation is regulated by kinases and phosphatases acting in opposition. We report here the existence of a novel protein complex that is composed minimally of Akt, PHLPP1, PHLPP2, FANCI, FANCD2, USP1 and UAF1. Our studies show that depletion of FANCI, but not FANCD2 or USP1, results in increased phosphorylation and activation of Akt. This activation is due to a reduction in the interaction between PHLPP1 and Akt in the absence of FANCI. In response to DNA damage or growth factor treatment, the interactions between Akt, PHLPP1 and FANCI are reduced consistent with the known phosphorylation of Akt in response to these stimuli. Furthermore, depletion of FANCI results in reduced apoptosis after DNA damage in accord with its role as a negative regular of Akt. Our findings describe an unexpected function for FANCI in the regulation of Akt and define a previously unrecognized intersection between the PI3K-Akt and FA pathways.
Introduction
The PI3K-Akt signaling network has been shown by cancer genetic studies to be among the most frequently mutated and hyperactivated pathways in human cancers.Citation1-3 PI3K and Akt are two of the most commonly dysregulated oncogenes, and PTEN, a negative regulator of the pathway, is among the most frequently mutated tumor suppressors.Citation4 This pathway controls most aspects of cancer progression including cell proliferation, survival, metabolism, motility, and genomic instability.Citation5,6 The Akt kinase is a central effector of this pathway and its regulation has been a subject of intense investigation. In response to growth factors and other stimuli such as DNA damage, Akt is phosphorylated in its catalytic domain at T308 by PDK1.Citation7,8 For full activation Akt also requires phosphorylation in its hydrophobic motif at S473. In response to growth factors Akt is phosphorylated at this site by mTORC2 Citation9,10; however, in response to DNA damage, phosphorylation at this site occurs via the DNA repair and cell cycle checkpoint kinase DNA-PK.Citation5,11,12 Recently an additional element of Akt activation has been described.Citation13 Akt activation was shown to fluctuate as a function of the mammalian cell cycle, which was due to a requirement for phosphorylation of Akt at S477 and T479 in early S phase by the cyclin-dependent kinase (Cdk2)/cyclin A. Once activated the Akt kinase phosphorylates numerous downstream targets that transduce the multiple pathways regulated by the PI3K-Akt signaling network.Citation5
The discovery of the PHLPP enzymes was first reported in 2005 and they were shown to be novel members of the protein phosphatase 2C grouping of metal-dependent phosphatases.Citation14,15 PHLPP1 is represented by two splicing isoforms referred to as PHLPP1α and PHLPP1β, whereas, PHLPP2 is represented by a single form.Citation16 All three isoforms have a similar domain structure with the exception that PHLPP1α lacks the Ras association domain found in the other two isoforms. PHLPP enzymes were shown to dephosphorylate Akt and several other signaling kinases, such as PKC, S6K1 and Mst1 in their hydrophobic motifs.Citation17-19 The C-terminal PDZ domain of PHLPP is required for its ability to dephosphorylate Akt.Citation14 In addition, four scaffolding proteins have been shown to promote the Akt-PHLPP interaction, namely, FKBP51,Citation20 Scribble,Citation21 NHERF1,Citation22 and SPRK1.Citation23 The PHLPP enzymes have been shown to act as tumor suppressors as demonstrated by mouse models and frequent deletion in many human cancers.Citation22,24-26 For example, PHLPP is mutated as frequently as is PTEN in metastatic prostate cancer.Citation24 Taken together, studies of the PHLPP phosphatases indicate that they are important regulators of the PI3K signaling pathway via their opposition to activation of Akt. In their absence Akt is constitutively hyperactivated leading to the stimulation of proliferation and inhibition of apoptosis, both of which are important contributors to the oncogenic process.
Fanconi anemia (FA) is a rare autosomal recessive disease characterized by congenital abnormalities, bone marrow failure, and an increased incidence of leukemias and squamous cell carcinomas.Citation27-29 FA is genetically complex with 19 genes currently identified as involved in some form of the disease.Citation30 The most established function of the FA pathway is the resolution of DNA interstrand crosslinks (ICLs) induced by such agents as diepoxybutane, although the pathway also participates in the repair of other types of DNA damage such as DNA double strand breaks and UV-induced lesions, and in the regulation of DNA replication forks.Citation31,32,33 The FA core complex, containing eight FA proteins, functions as an E3 ubiquitin ligase that in response to DNA damage monoubiquitinates the FA proteins FANCD2 and FANCI, and is also required for the stability of these two proteins.Citation34-38 These modifications are key regulatory steps in the FA pathway that promote the downstream activities of the pathway such as incision of ICLs, lesion bypass, and homologous recombination.Citation39 Recently it has been shown that monoubiquitinated FANCD2 participates in a cellular function outside of its role in DNA repair, namely, it is involved in the transcriptional regulation of the tumor suppressor gene p63, an activity which suppresses the onset of squamous cell cancers.Citation40,41
Here we report the existence of a novel complex that is composed minimally of Akt, PHLPP1, PHLPP2, FANCI, FANCD2, USP1 and UAF1 (designated as PAF for PHLPP, Akt, and Fanconi). USP1 and UAF1 comprise a deubiquitinating (DUB) enzyme that removes ubiquitin from FANCI and FANCD2 during recovery after DNA repair processing.Citation42,43 Our studies further show that removal of FANCI, but interestingly not FANCD2, results in spontaneous phosphorylation and activation of Akt. This activation is due to a reduction in the interaction between PHLPP1 and Akt in the absence of FANCI, thus elucidating the mechanism by which FANCI regulates Akt phosphorylation. In response to DNA damage or growth factor treatment the interaction between Akt, PHLPP1 and FANCI is reduced in line with the known increase in Akt phosphorylation in response to these stimuli.Citation5 Taken together these findings indicate that there is a convergence between the oncogenic PI3K-Akt pathway and the FA tumor suppressor pathway, and that FANCI is a novel global regulator of AKT activation.
Results
Characterization of the PAF complex
A previous study on deubiquitinating (DUB) enzyme interacting partners reported that the phosphatases PHLPP1 and PHLPP2 interact with the USP1.Citation44 Since FANCI and FANCD2 are substrates of USP1 that are subject to phosphorylation in response to DNA damage,Citation39,43,45 we examined whether they also interact with PHLPP1 since no phosphatase had been described for the FA proteins. We first confirmed that PHLPP1 associates with USP1 (Fig. S1A), and also found that both FANCI and FANCD2 interacted with PHLPP1 ( and S1B). Interestingly, FANCI also interacted with Akt (); however, importantly this interaction did not require PHLPP1 (). In addition, Akt also interacted with FANCD2 and USP1 (). The USP1 partner UAF1 is also a component of PAF, and it enhances the interaction of USP1 with PHLPP1, but is not required for interactions between Akt, PHLPP1 and FANCI (Fig. S1C, D). FANCI also interacted with PHLPP2 (). To determine if FANCI directly interacts with Akt, we purified both proteins by affinity chromatography and carried out an in vitro binding assay. Akt interacted strongly with FANCI with or without a prior stringent high salt wash (). We also performed the same assay with FANCI and PHLPP1, but were unable to demonstrate a direct interaction between these two proteins.
Figure 1. FANCI interacted with PHLPP1 and Akt. (A) Immunoblots of WCL and reciprocal co-immunoprecipitations (co-IPs) in which the indicated tagged PHLPP1 or FANCI interacted with endogenous FANCI or PHLPP1. GAPDH indicates a loading control. Whole cell lysates (WCL) and immunoprecipitations were derived from HEK293 cells transfected with the indicated Flag, HA (hemagglutinin) or Myc tagged constructs. (B) Immunoblots of WCL and reciprocal co-IPs showing FANCI interacted with Akt. (C) Depletion of PHLPP1 indicated it is not required for interaction between FANCI and Akt. Immunoblots of WCL and co-IP of Akt with FANCI and PHLPP1. (D) Akt interacted with FANCD2 and USP1. Immunoblots of WCL and co-IP of HA-Akt with FANCD2 and USP1. (E) Immunoblots of WCL and co-IP showing FANCI interacted with PHLPP2. (F) FANCI interacted with Akt in vitro. Immunoblot of a pull-down assay with affinity purified HA-Akt and Flag-FANCI. Eluate refers to HA-Akt eluted from beads prior to addition to Flag-FANCI beads.
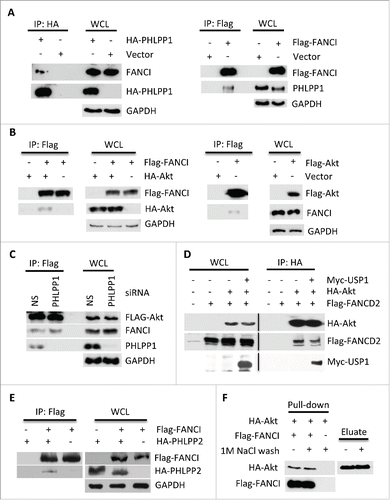
To define the region of FANCI that mediates the interaction with Akt, various truncation mutants of FANCI were co-expressed with HA-Akt. Co-immunoprecipitation (co-IP) experiments indicated that Akt interacted with the N-terminus of FANCI and that residues 1–78 were sufficient for the interaction both in vivo and in vitro (Fig. S2A, B, C). However, deletion of residues 1–60 from the 1–624 aa truncation mutant did not impair the interaction (Fig. S2D). Similar experiments conducted with PHLPP1 indicated that N-terminus of FANCI was also required for co-IP with this protein consistent with the concept that the association between FANCI and PHLPP1 is mediated by Akt (Fig. S2E,F). In addition, in vitro assays indicated that FANCI interacts with the Akt N-terminal and central regions that contain the PH and kinase domains (Fig. S2G,H). Together, the mapping results suggest an extensive region of interaction between FANCI and Akt. Overall, these results indicate the existence of a novel complex (PAF) that is composed minimally of Akt, PHLPP1, PHLPP2, FANCI, FANCD2, USP1 and UAF1.
Depletion of FANCI, but not FANCD2, induces spontaneous phosphorylation of Akt
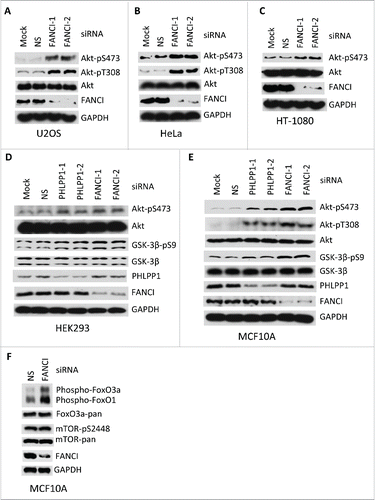
It is well established that the PHLPP enzymes oppose activation of Akt via dephosphorylation of S473,Citation14,15 and also T308.Citation22 To further investigate the function of the PAF complex, particularly with regard to Akt function, we used small interfering RNAs (siRNAs) to deplete other members of the PAF complex. Depletion of FANCI, but not FANCD2 or USP1, resulted in an increased phosphorylation of Akt at both S473 and T308 in both nontransformed and human cancer cell lines ( and S3A-C). Phosphorylation of the Akt downstream targets GSK-3β, oxO3a and FoxO1 were also observed indicating that depletion of FANCI results in activation of Akt (). Interestingly, we did not observe an increase in phosphorylation of mTOR upon FANCI depletion (). Depletion of a member of the FA core complex, FANCE, had no effect on Akt phosphorylation (Fig. S3A). The finding that depletion of FANCD2 or FANCE had no effect on Akt phosphorylation indicates that these results are not due to reduced levels of DNA repair affecting the cell cycle.Citation13 These results therefore indicate that FANCI is a negative regulator of Akt activation presumably in conjunction with PHLPP.
Figure 2. Depletion of FANCI by siRNA induced spontaneous phosphorylation of Akt at S473 and T308. (A-F) Immunoblots of Akt, GSK-3β and FoxO in human nontransformed and cancer cell lines upon FANCI depletion. FANCI-1, FANCI-2, PHLPP1-1 and PHLPP1-2 indicate distinct siRNAs. NS indicates a control siRNA. GAPDH indicates a loading control.
Next we examined whether truncation mutants of FANCI (described above) were able to rescue the phosphorylation defect observed in FANCI-depleted cells. For these experiments we used a somatic cell knockout of FANCI that was generated in HeLa cells (see Fig. S6A). As expected the HeLa FANCI−/− cells exhibited higher levels of Akt pS473 phosphorylation than did the parential HeLa cell line (Fig. S3D). Interestingly, FANCI mutants containing residues 1–78 and 1–328 were not effective at rescuing the Akt phosphorylation defect, whereas, all other FANCI mutants exhibited efficacy in this assay.
FANCI promotes the interaction between Akt and PHLPP1
FANCI has no known catalytic domains suggesting that its possible function within the PAF complex would be to act as an adaptor to maintain the integrity of the complex. In support of this hypothesis, depletion of FANCI by siRNA significantly reduced the association between Akt and PHLPP1 (). Depletion of FANCD2 did not affect the interaction between Akt and PHLPP1, consistent with our finding that this depletion did not induce Akt phosphorylation (Fig. S4A). Interestingly, depletion of FANCI did not affect the interaction between PHLPP1 and the USP1-UAF1 heterodimer indicating that FANCI specifically affects the Akt-PHLPP1 interaction (Fig. S4B). Finally, we also showed that FANCD2 and USP1 are not required for the interaction between Akt and PHLPP1 (Fig. S4C).
Figure 3. DNA damage promoted Akt activation through disruption of the PAF complex. (A) Depletion of FANCI by siRNA decreased the interaction between Akt and PHLPP1. Immunoblots of WCL and reciprocal co-IPs between Flag-Akt and HA-PHLPP1 upon depletion of FANCI. GAPDH indicate loading controls. WCL and immunoprecipitations were derived from HEK293 cells transfected with the indicated Flag or HA tagged constructs. (B-D) UV irradiation disrupted components of the PAF complex. Immunoblots of WCL and co-IPs of Flag-FANCI, HA-Akt, and HA-PHLPP1 as indicated. E, Effects of MMC treatment and recovery on Akt-FANCI interaction and Akt phosphorylation in either HEK293 or U2OS cells transfected with the indicated constructs. Immunoblots of WCL and co-IPs of Flag-Akt and HA-FANCI. “MMC 16 hrs” indicates exposure of cells to the drug for 16 hours; “Six hrs recovery” indicates a subsequent 6 hours of incubation in normal media. GAPDH and CDC5 indicate loading controls.
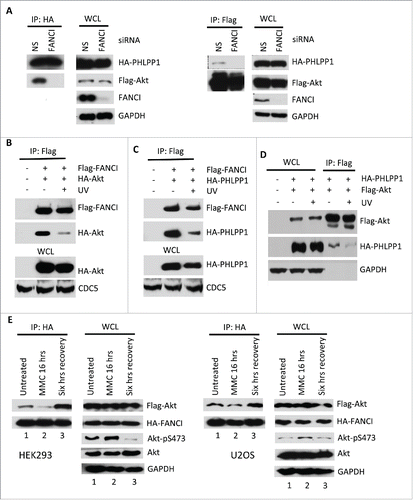
DNA damage has been shown to induce activation of Akt.Citation11,12,46,47 We therefore examined the interactions between Akt, PHLPP1 and FANCI after UV irradiation. UV irradiation reduced the interaction between FANCI and Akt (), between FANCI and PHLPP1 (), and between Akt and PHLPP1 (). Interestingly, UV irradiation did not affect the interaction between Akt and FANCD2 consistent with our finding that FANCD2 does not play a role in the regulation of Akt activation (Fig. S5A). Furthermore, while UV irradiation reduced the interaction between Akt and PHLPP1 and is known to induce USP1 degradation,Citation48 knockdown of USP1 itself had no effect on the Akt-PHLPP1 interaction (Fig. S5B). We also examined these interactions after treatment with the DNA interstrand crosslinking agent mitomycin C (MMC). Treatment of both HEK293 and U2OS cells with MMC resulted in a decrease in the interaction between FANCI and Akt and a corresponding increase in Akt phosphorylation at S473, while a period of recovery in normal media post MMC treatment resulted in a significant increase in the FANCI-Akt interaction and a strong decrease in Akt phosphorylation (). However, if FANCI is depleted by siRNA the phosphorylation of Akt remained high during the recovery period, and the interaction between Akt and PHLPP1 was reduced (Fig. S5C, D). As observed with UV irradiation, MMC treatment also resulted in a reduction in the interaction between FANCI and PHLPP1 (Fig. S5E). These findings indicate that FANCI enhances the interaction between Akt and PHLPP1, and that in response to DNA damage the association between FANCI or PHLPP1 with Akt is decreased resulting in elevated levels of Akt phosphorylation. Other components of the PAF complex such as USP1 and FANCD2 did not appear to regulate Akt activation under these conditions. Interestingly, we did not observe any significant changes in the localization of PAF complex members in response to DNA damage (Fig. S5F).
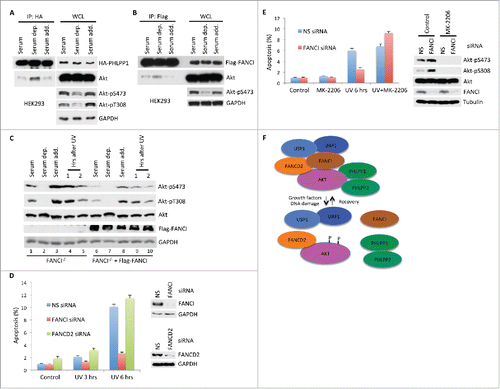
Growth factors are well known as strong inducers of Akt activation.Citation5 Incubation of HEK293 cells in 0.1% serum resulted in a strong increase in the interaction between Akt and PHLPP1 and a corresponding decrease in the phosphorylation of Akt at both S473 and T308 (). Addition of 10% serum resulted in a decrease in the Akt-PHLLP1 interaction and an increase in Akt phosphorylation. Similarly, serum deprivation resulted in an increase in the Akt-FANCI interaction, and a decrease in this interaction ensued upon return of the cells to normal media (). We next examined HeLA FANCI−/− cells, and for comparison a stable derivative HeLa cell line rescued with Flag-FANCI. In response to both serum addition and UV irradiation, the FANCI−/− cell line exhibited higher levels of Akt phosphorylation compared to the FANCI rescued cell line (, compare lanes 1 and 6, etc.), or to the parental HeLa cell line (Fig. S6A). These findings indicate that FANCI is a global regulator of Akt activation in response to both DNA damage and growth factor status.
Figure 4. Serum levels regulated the PAF complex and Akt activation. (A) Immunoblots of WCL and co-IPs between HA-PHLPP1 and Akt after 6 hours of serum deprivation followed by 30 minutes of recovery in normal medium. WCL and immunoprecipitations were derived from either HEK293 or HeLa cells transfected with the indicated Flag or HA tagged constructs. GAPDH indicates a loading control. (B) Immunoblots of WCL and co-IPs between Flag-FANCI and Akt after serum deprivation and recovery in normal medium. (C) Phosphorylation of Akt is differentially affected by serum levels or DNA damage in HeLa FANCI−/− cells compared to cells rescued with Flag-FANCI. Immunoblots of WCL after serum deprivation and recovery in normal medium, or upon treatment with UV irradiation. (D) Depletion of FANCI, but not FANCD2, reduced DNA damage induced apoptosis. Left panel, Annexin V assays showing levels of apoptosis after UV irradiation in MCF10A cells depleted of indicated FANC proteins. “UV 3 hrs” indicates cells were processed for analysis 3 hours after exposure to UV irradiation. Experiments were performed in triplicate; data are shown as mean ± s.d. Right panel, immunoblots showing depletion of FANC proteins by siRNA. (E) Inhibition of Akt restored apoptosis in FANCI depleted cells. Left panel, apoptosis assays were performed as indicated in D with the Akt inhibitor MK-2206. Right panel, immunoblots showing depletion of FANCI and phosphorylation status of Akt. Tubulin indicates a loading control. (F) Schematic indicating the structure of the known components of the PAF complex and its responses to DNA growth factor stimulation or DNA damage, and its response to recovery from these stimuli. “P” indicates phosphorylation sites on Akt at S473 and S308.
FANCI is a regulator of apoptosis via Akt
It has been demonstrated that down regulation of PHLPP prevents apoptosis via increased Akt activity,Citation14,15 and consistent with this result our findings indicate that PHLPP-depleted LN229 glioblastoma cellsCitation22,49 exhibit increased survival when exposed to DNA damage (Fig. S6B). However, FANCI-deficient cells are known to be hypersensitive to DNA damage,Citation37,38,50 thus exhibiting the opposite phenotype to PHLPP-deficient cells. Nevertheless, depletion of FANCI, but not FANCD2, resulted in a substantial reduction in apoptosis upon UV irradiation in MCF10A cells (). This result is consistent with our observation of regulation of Akt activation by FANCI, but not by FANCD2. Furthermore, the reduction in apoptosis by depletion of FANCI was reversed by the addition of the allosteric Akt inhibitor MK-2206Citation51 indicating that this effect was specific to the Akt pathway (). As a further control, we knocked down FANCI expression in MCF10A cells and were able to partially rescue the apoptosis phenotype with a siRNA-refractory expression construct (Fig. S7). These results would suggest that cell death occurring upon DNA damage in FANCI-deficient cells is largely due to cellular necrosis rather than apoptosis.
Discussion
Our results describe a novel function for FANCI as a negative regulator of Akt activation in conjunction with PHLPP phosphatases. This unanticipated finding is the first indication that FANCI acts in a role outside of its well-established function in DNA repair. It also defines a previously unknown link between the FA pathway and the oncogenic PI3K-Akt signaling network. Furthermore, our results indicate that FANCI can act as a tumor suppressor by two independent mechanisms: it prevents oncogenic DNA replication stress via its role in DNA repair, and regulates the activity of the Akt oncogene by promoting the inhibitory function of PHLPP. Thus, for example, in response to DNA damage the efficiency of lesion removal is compromised in FANCI-deficient cells resulting in mutations, while the decrease in apoptosis would hinder the removal of damaged cells. These considerations suggest that FANCI-deficient individuals conceivably might have a greater risk of tumorigenesis than other FA patients. However, this hypothesis is difficult to confirm at present given the rarity of human FANCI patients,Citation52 and the current lack of a mouse model for FANCI. Our result that FANCI is a regulator of Akt activation is the second demonstration of a role for either FANCI or FANCD2 outside of DNA repair. Previously, FANCD2 has been shown to activate the transcription of Tap63.Citation41 It is also interesting to note that although FANCD2 is a component of the PAF complex and is commonly associated with FANCI in a heterodimeric form, it does not exhibit a role in the inhibitory regulation of Akt that we have identified for FANCI. Furthermore, inhibition of FANCD2 is known to sensitize cancer cells to chemotherapeutic agents.Citation53,54
Our findings indicate that the mechanism by which FANCI negatively regulates Akt is via recruitment of the PHLPP1 phosphatase to the PAF complex. Depletion of FANCI reduced the interaction between PHLPP1 and Akt and thereby induced a significant increase in Akt phosphorylation in a number of different human cell lines. Interestingly depletion of PHLPP1 did not affect the interaction between FANCI and Akt. Furthermore, our evidence indicates that the association between FANCI and Akt is direct and interaction mapping studies suggest that the interface between the two proteins may be extensive, although it is unlikely to be constitutive.
Stimuli, such as growth factors or DNA damage, which induce activation of Akt, also cause a disruption of the PAF complex in that the interactions between FANCI, PHLPP1 and Akt are reduced. On the other hand, removal of growth factors or recovery from DNA damage increases the interactions among these proteins, and a striking decrease in activating Akt phosphorylations.Citation5 Thus, there is a strong correspondence between the integrity of the PAF complex and the state of Akt activation. The mechanism by which the PAF complex is regulated in response to these stimuli is at present unknown. One possibility which we investigated, is that monoubiquitination of FANCI might induce its dissociation from Akt. This modification seemed promising since it occurs during S phase or in response to DNA damage, two events which correspond to Akt activation.Citation5,11-13 However, depletion of USP1, which causes a significant increase in FANCI monoubiquitination,Citation37,38 did not result in an increase in Akt phosphorylation, nor did it affect the Akt-PHLPP1 interaction. This result does not completely rule out a role for monoubiquitination of FANCI in the regulation of Akt activation, but it indicates that at least it is not a sufficient event for this process. Another possible model is the recently identified phosphorylations of Akt that occur at S477 and T479 and which are required for Akt activation.Citation13 These phosphorylations occur during S phase by Cdk2/cyclin A, or by mTORC2 or DNA-PK in response to growth factors or DNA damage, respectively. Conceivably, these modifications could promote the disruption of the PAF complex by inducing changes in the conformation of Akt, and we are currently assessing this model.
Taken together our findings describe a novel mechanism for the control of Akt activation and indicate an unanticipated intersection between the PI3K-Akt and the FA pathways. As Akt is one of the most commonly dysregulated oncogenes in human cancers, identification of regulatory mechanisms provide potential avenues for therapeutic intervention.
Materials and methods
Plasmids and constructs
Flag-HA-FANCI and Flag-FANCD2 was obtained from W. Harper and J. Chen, respectively. Various PHLPP constructs were obtained from A. Newton or T. Gao and Akt constructs were obtained from T. Gao. Myc-USP1 was obtained from T Huang.
FANCI truncation mutants and a vector control were created using the primeSTAR GXL DNA polymerase kit (Takara). Primers were synthesized (Sigma) and PCR protocols were followed according to the product manual with an annealing temperature of 60°C. PCR products were recovered from agarose gels and ligated by T4 ligase at room temperature for 30 minutes. All truncation mutants were verified by DNA sequencing. Akt truncation mutants were generously provided by S.-Y. Shieh.Citation55 The FANCI siRNA refractory mutant (FANCI*) was prepared using primers 5′-TCTTTCATTTCTTGGAAGTCATCTGACCCCCCT and 5′-AGGGGGGTCAGATGACTTCCAAGAAATGAAAGA.
siRNAs
FANCI (SASI-Hs02-00325751, SASI-Hs02-00325752), FANCD2 (SASI-Hs01-00137854, SASI-Hs02-00307032), FANCE (SASI-Hs02-00355198, SASI-Hs01-00028355), PHLPP1 (SASI-Hs01-00159083, SASI-Hs01-00159085), and USP1 (SASI-Hs01-00204271, SASI-Hs01-00204272) siRNAs were purchased from Sigma. Non-specific siRNA and the method of transfection have been described previously.Citation56
Antibodies
Rabbit polyclonal anti-Flag (F7425) and anti-tubulin (T5192) antibodies were purchased from Sigma. Peroxidase-conjugated mouse monoclonal anti-HA (sc-7392), anti-FANCI (sc-98532), mouse monoclonal anti-FANCE (sc25303), anti-FANCD2 (sc-20022), and anti-GAPDH (sc-59541) antibodies were purchased from Santa Cruz Biotechnology. Peroxidase-conjugated total anti-Akt (8596), anti-phospho-Ser473-Akt (4060), anti-phospho-Thr308-Akt (13038), anti-pS9-GSK3β (9336), and anti-GSK3β (9315) antibodies were purchased from Cell Signaling. Anti-PHLPP1 (A300-659A) and anti-PHLPP2 (A300-661A) antibodies were purchased from Bethyl Laboratories.
Immunoblots and immunoprecipitation
Whole cell lysate preparation for immunoblots and immunoprecipitations have been described previously.Citation32 Briefly, cells were lysed in EBC (50 mM Tris pH7.5, 100 mM NaCl, 0.5% NP-40) or RIPA (50 mM Tris pH 8, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS) buffer supplemented with protease inhibitors (Pierce, 88266) and phosphatase inhibitors (Roche, 04906845001). One mg of protein lysates were incubated with antibodies and protein A/G plus-agarose (Santa Cruz, sc-2003), anti-Flag affinity gel (Sigma, F2426) or anti-HA affinity gel (Sigma, E6779) at 4°C overnight, and the immunoprecipitated proteins were resuspended in SDS sample buffer for further analysis. For in vitro binding assays, immunoprecipitated Flag- or HA-tagged proteins were washed with 1M NaCl and eluted by 200 μg/ml of HA peptides (Sigma, 12149) or 200 μg/ml of Flag peptide (sigma, F4799) in TBS buffer in the presence of proteinase inhibitors. The eluted proteins were added to the targeted protein's immunoprecipitates in NET-N buffer (100 mM NaCl, 20 mM Tris pH 8.0, 0.5 mM EDTA, 0.5 % (v/v) NP-40) and incubated overnight at 4°C.
Cell culture and annexin V assay
MCF10A cells were cultured in DMEM/F12 medium supplemented with 5% horse serum, 10 μg/ml of insulin, 0.1 μg/ml of cholera toxin, 0.5 μg/ml of hydrocortisone, and 0.02 μg/ml of EGF. HeLa, HEK293, U2OS and HT1080 cells were grown in DMEM medium supplemented with 10% of fetal bovine serum. HeLa FANCI−/− and HeLa FANCD2−/− cell lines were generously provided by J. Chen.Citation57 Transfections with plasmid and siRNA were performed with lipofectamine (Life Technologies) with procedures described in the company's protocols. For assays with DNA damage, cells were subjected to 40 J/m2 UV irradiation or exposed to 250 ng/ml MMC for approximately 16 hrs. Levels of cellular apoptosis were determined by Annexin V assay. Briefly, cells were trypsinized and washed with cold PBS twice before resuspension in Annexin V binding buffer (10 mM HEPES pH 7.4, 140 mM NaCl, 2.5 mM CaCl ). Cells (1 × 105) in 100 ul of Annexin V buffer were incubated with 5 ul of Annexin V-FITC (BD Biosciences, 556420) and 2 ul of propidium iodide (0.1 mg/ml) for 15 minutes and Annexin V positive cells were determined by flow cytometry (Beckman-Coulter Gallios flow cytometer using Gallios acquisition software and Beckman-Coulter Kaluza software for post-acquisition data analysis). The allosteric Akt inhibitor MK-2206 was obtained from ApexBio Technology.
Colony survival assays of LN229 cells were performed as previously described.Citation58 Stable knockdown of PHLPP1 and PHLPP2 in LN229 cells by stable shRNA expression were previously described.Citation22,49
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Author contributions
X.Z., X.L., and S.A. performed the experiments, and X.Z., X.L., M.M.G. and R.L. designed the experiments. R.L. supervised the study and R.L. and M.M.G. wrote the manuscript.
1158375_Supplemental_Material.zip
Download Zip (2.8 MB)Acknowledgments
We thank J. Liu for assistance in preparing plasmid constructs, and J. Chen, L. Li, A. Newton, and W. Harper for providing reagents.
Funding
This work was supported in part by National Institutes of Health grant to R.L. (PO1CA097175). The authors have no conflicts of interest to disclose.
References
- Porta C, Paglino C, Mosca A. Targeting PI3K/Akt/mTOR Signaling in Cancer. Front Oncol 2014; 4:64; PMID:24782981; http://dx.doi.org/10.3389/fonc.2014.00064
- Ocana A, Vera-Badillo F, Al-Mubarak M, Templeton AJ, Corrales-Sanchez V, Diez-Gonzalez L, Cuenca-Lopez MD, Seruga B, Pandiella A, Amir E. Activation of the PI3K/mTOR/AKT pathway and survival in solid tumors: systematic review and meta-analysis. PLoS One 2014; 9:e95219; PMID:24777052; http://dx.doi.org/10.1371/journal.pone.0095219
- Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov 2014; 13:140-56; PMID:24481312; http://dx.doi.org/10.1038/nrd4204
- Stebbing J, Lit LC, Zhang H, Darrington RS, Melaiu O, Rudraraju B, Giamas G. The regulatory roles of phosphatases in cancer. Oncogene 2014; 33:939-53; PMID:23503460; http://dx.doi.org/10.1038/onc.2013.80
- Fayard E, Xue G, Parcellier A, Bozulic L, Hemmings BA. Protein kinase B (PKB/Akt), a key mediator of the PI3K signaling pathway. Curr Topics Microbiol Immunol 2010; 346:31-56; PMID:20517722
- Dummler B, Hemmings BA. Physiological roles of PKB/Akt isoforms in development and disease. Biochem Soc Trans 2007; 35:231-5; PMID:17371246; http://dx.doi.org/10.1042/BST0350231
- Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, Hemmings BA. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J 1996; 15:6541-51; PMID:8978681
- Bellacosa A, Chan TO, Ahmed NN, Datta K, Malstrom S, Stokoe D, McCormick F, Feng J, Tsichlis P. Akt activation by growth factors is a multiple-step process: the role of the PH domain. Oncogene 1998; 17:313-25; PMID:9690513; http://dx.doi.org/10.1038/sj.onc.1201947
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005; 307:1098-101; PMID:15718470; http://dx.doi.org/10.1126/science.1106148
- Bayascas JR, Alessi DR. Regulation of Akt/PKB Ser473 phosphorylation. Mol Cell 2005; 18:143-5; PMID:15837416; http://dx.doi.org/10.1016/j.molcel.2005.03.020
- Bozulic L, Surucu B, Hynx D, Hemmings BA. PKBalpha/Akt1 acts downstream of DNA-PK in the DNA double-strand break response and promotes survival. Mol Cell 2008; 30:203-13; PMID:18439899; http://dx.doi.org/10.1016/j.molcel.2008.02.024
- Boehme KA, Kulikov R, Blattner C. p53 stabilization in response to DNA damage requires Akt/PKB and DNA-PK. Proc Natl Acad Sci U S A 2008; 105:7785-90; PMID:18505846; http://dx.doi.org/10.1073/pnas.0703423105
- Liu P, Begley M, Michowski W, Inuzuka H, Ginzberg M, Gao D, Tsou P, Gan W, Papa A, Kim BM, et al. Cell-cycle-regulated activation of Akt kinase by phosphorylation at its carboxyl terminus. Nature 2014; 508:541-5; PMID:24670654; http://dx.doi.org/10.1038/nature13079
- Gao T, Furnari F, Newton AC. PHLPP: a phosphatase that directly dephosphorylates Akt, promotes apoptosis, and suppresses tumor growth. Mol Cell 2005; 18:13-24; PMID:15808505; http://dx.doi.org/10.1016/j.molcel.2005.03.008
- Brognard J, Sierecki E, Gao T, Newton AC. PHLPP and a second isoform, PHLPP2, differentially attenuate the amplitude of Akt signaling by regulating distinct Akt isoforms. Mol Cell 2007; 25:917-31; PMID:17386267; http://dx.doi.org/10.1016/j.molcel.2007.02.017
- O'Neill AK, Niederst MJ, Newton AC. Suppression of survival signalling pathways by the phosphatase PHLPP. Febs J 2013; 280:572-83; PMID:22340730; http://dx.doi.org/10.1111/j.1742-4658.2012.08537.x
- Gao T, Brognard J, Newton AC. The phosphatase PHLPP controls the cellular levels of protein kinase C. J Biol Chem 2008; 283:6300-11; PMID:18162466; http://dx.doi.org/10.1074/jbc.M707319200
- Liu J, Stevens PD, Li X, Schmidt MD, Gao T. PHLPP-mediated dephosphorylation of S6K1 inhibits protein translation and cell growth. Mol Cell Biol 2011; 31:4917-27; PMID:21986499; http://dx.doi.org/10.1128/MCB.05799-11
- Qiao M, Wang Y, Xu X, Lu J, Dong Y, Tao W, Stein J, Stein GS, Iglehart JD, Shi Q, et al. Mst1 is an interacting protein that mediates PHLPPs' induced apoptosis. Mol Cell 2010; 38:512-23; PMID:20513427; http://dx.doi.org/10.1016/j.molcel.2010.03.017
- Pei H, Li L, Fridley BL, Jenkins GD, Kalari KR, Lingle W, Petersen G, Lou Z, Wang L. FKBP51 affects cancer cell response to chemotherapy by negatively regulating Akt. Cancer Cell 2009; 16:259-66; PMID:19732725; http://dx.doi.org/10.1016/j.ccr.2009.07.016
- Li X, Yang H, Liu J, Schmidt MD, Gao T. Scribble-mediated membrane targeting of PHLPP1 is required for its negative regulation of Akt. EMBO Rep 2011; 12:818-24; PMID:21701506; http://dx.doi.org/10.1038/embor.2011.106
- Molina JR, Agarwal NK, Morales FC, Hayashi Y, Aldape KD, Cote G, Georgescu MM. PTEN, NHERF1 and PHLPP form a tumor suppressor network that is disabled in glioblastoma. Oncogene 2012; 31:1264-74; PMID:21804599; http://dx.doi.org/10.1038/onc.2011.324
- Wang P, Zhou Z, Hu A, Ponte de Albuquerque C, Zhou Y, Hong L, Sierecki E, Ajiro M, Kruhlak M, Harris C, et al. Both decreased and increased SRPK1 levels promote cancer by interfering with PHLPP-mediated dephosphorylation of Akt. Mol Cell 2014; 54:378-91; PMID:24703948; http://dx.doi.org/10.1016/j.molcel.2014.03.007
- Chen M, Pratt CP, Zeeman ME, Schultz N, Taylor BS, O'Neill A, Castillo-Martin M, Nowak DG, Naguib A, Grace DM, et al. Identification of PHLPP1 as a tumor suppressor reveals the role of feedback activation in PTEN-mutant prostate cancer progression. Cancer Cell 2011; 20:173-86; PMID:21840483; http://dx.doi.org/10.1016/j.ccr.2011.07.013
- Hellwinkel OJ, Rogmann JP, Asong LE, Luebke AM, Eichelberg C, Ahyai S, Isbarn H, Graefen M, Huland H, Schlomm T. A comprehensive analysis of transcript signatures of the phosphatidylinositol-3 kinase/protein kinase B signal-transduction pathway in prostate cancer. BJU Int 2008; 101:1454-60; PMID:18336616; http://dx.doi.org/10.1111/j.1464-410X.2008.07540.x
- Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010; 18:11-22; PMID:20579941; http://dx.doi.org/10.1016/j.ccr.2010.05.026
- Kupfer GM. Fanconi anemia: a signal transduction and DNA repair pathway. Yale J Biol Med 2013; 86:491-7; PMID:24348213
- Sakaguchi H, Nakanishi K, Kojima S. Inherited bone marrow failure syndromes in 2012. Int J Hematol 2013; 97:20-9; PMID:23271412; http://dx.doi.org/10.1007/s12185-012-1249-9
- Shukla P, Solanki A, Ghosh K, Vundinti BR. DNA interstrand cross-link repair: understanding role of Fanconi anemia pathway and therapeutic implications. Eur J Haematol 2013; 91:381-93; PMID:23859405; http://dx.doi.org/10.1111/ejh.12169
- Dong H, Nebert DW, Bruford EA, Thompson DC, Joenje H, Vasiliou V. Update of the human and mouse Fanconi anemia genes. Hum Genomics 2015; 9:32; PMID:26596371; http://dx.doi.org/10.1186/s40246-015-0054-y
- Panneerselvam J, Pickering A, Han B, Li L, Zheng J, Zhang J, Zhang Y, Fei P. Basal level of FANCD2 monoubiquitination is required for the maintenance of a sufficient number of licensed-replication origins to fire at a normal rate. Oncotarget 2014; 5:1326-37; PMID:24658369; http://dx.doi.org/10.18632/oncotarget.1796
- Berquist BR, Wilson DM, 3rd. Pathways for repairing and tolerating the spectrum of oxidative DNA lesions. Cancer Lett 2012; 327:61-72; PMID:22353689; http://dx.doi.org/10.1016/j.canlet.2012.02.001
- Crossan GP, Patel KJ. The Fanconi anaemia pathway orchestrates incisions at sites of crosslinked DNA. J Pathol 2012; 226:326-37; PMID:21956823; http://dx.doi.org/10.1002/path.3002
- Clark DW, Tripathi K, Dorsman JC, Palle K. FANCJ protein is important for the stability of FANCD2/FANCI proteins and protects them from proteasome and caspase-3 dependent degradation. Oncotarget 2015; 6:28816-32; PMID:26336824
- Meetei AR, de Winter JP, Medhurst AL, Wallisch M, Waisfisz Q, van de Vrugt HJ, Oostra AB, Yan Z, Ling C, Bishop CE, et al. A novel ubiquitin ligase is deficient in Fanconi anemia. Nat Genet 2003; 35:165-70; PMID:12973351; http://dx.doi.org/10.1038/ng1241
- Gregory RC, Taniguchi T, D'Andrea AD. Regulation of the Fanconi anemia pathway by monoubiquitination. Semin Cancer Biol 2003; 13:77-82; PMID:12507559; http://dx.doi.org/10.1016/S1044-579X(02)00102-5
- Smogorzewska A, Matsuoka S, Vinciguerra P, McDonald ER, 3rd, Hurov KE, Luo J, Ballif BA, Gygi SP, Hofmann K, D'Andrea AD, et al. Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell 2007; 129:289-301; PMID:17412408; http://dx.doi.org/10.1016/j.cell.2007.03.009
- Sims AE, Spiteri E, Sims RJ, 3rd, Arita AG, Lach FP, Landers T, Wurm M, Freund M, Neveling K, Hanenberg H, et al. FANCI is a second monoubiquitinated member of the Fanconi anemia pathway. Nat Struct Mol Biol 2007; 14:564-7; PMID:17460694; http://dx.doi.org/10.1038/nsmb1252
- Kim H, D'Andrea AD. Regulation of DNA cross-link repair by the Fanconi anemia/BRCA pathway. Genes Dev 2012; 26:1393-408; PMID:22751496; http://dx.doi.org/10.1101/gad.195248.112
- Panneerselvam J, Pickering A, Zhang J, Wang H, Tian H, Zheng J, Fei P. A hidden role of the inactivated FANCD2: upregulating DeltaNp63. Oncotarget 2013; 4:1416-26; PMID:23965832; http://dx.doi.org/10.18632/oncotarget.1217
- Park E, Kim H, Kim JM, Primack B, Vidal-Cardenas S, Xu Y, Price BD, Mills AA, D'Andrea AD. FANCD2 activates transcription of TAp63 and suppresses tumorigenesis. Mol Cell 2013; 50:908-18; PMID:23806336; http://dx.doi.org/10.1016/j.molcel.2013.05.017
- Cohn MA, Kowal P, Yang K, Haas W, Huang TT, Gygi SP, D'Andrea AD. A UAF1-containing multisubunit protein complex regulates the Fanconi anemia pathway. Mol Cell 2007; 28:786-97; PMID:18082604; http://dx.doi.org/10.1016/j.molcel.2007.09.031
- Nijman SM, Huang TT, Dirac AM, Brummelkamp TR, Kerkhoven RM, D'Andrea AD, Bernards R. The deubiquitinating enzyme USP1 regulates the Fanconi anemia pathway. Mol Cell 2005; 17:331-9; PMID:15694335; http://dx.doi.org/10.1016/j.molcel.2005.01.008
- Sowa ME, Bennett EJ, Gygi SP, Harper JW. Defining the human deubiquitinating enzyme interaction landscape. Cell 2009; 138:389-403; PMID:19615732; http://dx.doi.org/10.1016/j.cell.2009.04.042
- Ishiai M, Kitao H, Smogorzewska A, Tomida J, Kinomura A, Uchida E, Saberi A, Kinoshita E, Kinoshita-Kikuta E, Koike T, et al. FANCI phosphorylation functions as a molecular switch to turn on the Fanconi anemia pathway. Nat Struct Mol Biol 2008; 15:1138-46; PMID:18931676; http://dx.doi.org/10.1038/nsmb.1504
- Dragoi AM, Fu X, Ivanov S, Zhang P, Sheng L, Wu D, Li GC, Chu WM. DNA-PKcs, but not TLR9, is required for activation of Akt by CpG-DNA. EMBO J 2005; 24:779-89; PMID:15678105; http://dx.doi.org/10.1038/sj.emboj.7600539
- Feng J, Park J, Cron P, Hess D, Hemmings BA. Identification of a PKB/Akt hydrophobic motif Ser-473 kinase as DNA-dependent protein kinase. J Biol Chem 2004; 279:41189-96; PMID:15262962; http://dx.doi.org/10.1074/jbc.M406731200
- Huang TT, Nijman SM, Mirchandani KD, Galardy PJ, Cohn MA, Haas W, Gygi SP, Ploegh HL, Bernards R, D'Andrea AD. Regulation of monoubiquitinated PCNA by DUB autocleavage. Nat Cell Biol 2006; 8:339-47; PMID:16531995
- Agarwal NK, Zhu X, Gagea M, White CL, 3rd, Cote G, Georgescu MM. PHLPP2 suppresses the NF-kappaB pathway by inactivating IKKbeta kinase. Oncotarget 2014; 5:815-23; PMID:24553260; http://dx.doi.org/10.18632/oncotarget.1774
- Dorsman JC, Levitus M, Rockx D, Rooimans MA, Oostra AB, Haitjema A, Bakker ST, Steltenpool J, Schuler D, Mohan S, et al. Identification of the Fanconi anemia complementation group I gene, FANCI. Cell Oncol 2007; 29:211-8; PMID:17452773
- Hirai H, Sootome H, Nakatsuru Y, Miyama K, Taguchi S, Tsujioka K, Ueno Y, Hatch H, Majumder PK, Pan BS, et al. MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol Cancer Ther 2010; 9:1956-67; PMID:20571069; http://dx.doi.org/10.1158/1535-7163.MCT-09-1012
- Ameziane N, Sie D, Dentro S, Ariyurek Y, Kerkhoven L, Joenje H, Dorsman JC, Ylstra B, Gille JJ, Sistermans EA, et al. Diagnosis of fanconi anemia: mutation analysis by next-generation sequencing. Anemia 2012; 2012:132856; PMID:22720145; http://dx.doi.org/10.1155/2012/132856
- Patil AA, Sayal P, Depondt ML, Beveridge RD, Roylance A, Kriplani DH, Myers KN, Cox A, Jellinek D, Fernando M, et al. FANCD2 re-expression is associated with glioma grade and chemical inhibition of the Fanconi Anaemia pathway sensitises gliomas to chemotherapeutic agents. Oncotarget 2014; 5:6414-24; PMID:25071006; http://dx.doi.org/10.18632/oncotarget.2225
- Shen C, Houghton PJ. Targeting FANCD2 for therapy sensitization. Oncotarget 2014; 5:3426-7; PMID:24913333; http://dx.doi.org/10.18632/oncotarget.2070
- Cheng YC, Chen PH, Chiang HY, Suen CS, Hwang MJ, Lin TY, Yang HC, Lin WC, Lai PL, Shieh SY. Candidate tumor suppressor B-cell translocation gene 3 impedes neoplastic progression by suppression of AKT. Cell Death Dis 2015; 6:e1584; http://dx.doi.org/10.1038/cddis.2014.550
- Geng L, Zhang X, Zheng S, Legerski RJ. Artemis links ATM to G2/M checkpoint recovery via regulation of Cdk1-cyclin B. Mol Cell Biol 2007; 27:2625-35; PMID:17242184; http://dx.doi.org/10.1128/MCB.02072-06
- Huang Y, Leung JW, Lowery M, Matsushita N, Wang Y, Shen X, Huong D, Takata M, Chen J, Li L. Modularized functions of the Fanconi anemia core complex. Cell Rep 2014; 7:1849-57; PMID:24910428; http://dx.doi.org/10.1016/j.celrep.2014.04.029
- Ahkter S, Richie CT, Zhang N, Behringer RR, Zhu C, Legerski RJ. Snm1-deficient mice exhibit accelerated tumorigenesis and susceptibility to infection. Mol Cell Biol 2005; 25:10071-8; PMID:16260620; http://dx.doi.org/10.1128/MCB.25.22.10071-10078.2005