
ABSTRACT
Single nucleotide polymorphisms (SNPs) that occur within CpG Islands may lead to increased hypermethylation if a SNP allele has the potential to form a CpG dinucleotide, as well as potentially lead to hypomethylation if a SNP allele eliminates a CpG dinucleotide. We analyzed CpG-related SNP allele frequencies in whole genome sequences (WGS) across 5 TCGA cancer datasets, thereby exploiting a more recent appreciation for signaling pathway degeneracy in cancer. The cancer data sets were analyzed for SNPs in CpG islands associated with the oncogenes, HRAS and MYC, and in the CpG islands associated with the tumor suppressor genes, APC, DCC, and RB1. We determined that one SNP allele (rs3824120) in a CpG island associated with MYC which eliminated a CpG was more common in the cancer datasets than in the 100Genomes databases (p < 0.01). For HRAS, 2 SNP alleles (rs112690925, rs7939028) that created CpG's occurred significantly less frequently in the cancer data sets than in the general SNP databases (e.g., rs7939028, p < 0.0002, in comparison with AllSNPs(142)). Also, one SNP allele (rs4940177) that created a CpG in a CpG island associated with the DCC tumor suppressor gene, was more common in the cancer datasets (p < 0.0007). To understand a broader picture of the potential of SNP alleles to create CpG's in CpG islands of tumor suppressor genes, we developed a scripted algorithm to assess the SNP alleles associated with the CpG islands of 43 tumor suppressor genes. The following tumor suppressor genes have the possibility of significant, percent increases in their CpG counts, depending on which SNP allele(s) is present: VHL, BRCA1, BRCA2, CHEK2, PTEN and RB1.
Introduction
Oncoproteins promote cell division while tumor suppressor proteins inhibit cell division and promote differentiation. Cancer is driven by the imbalance in the activity of these 2 types of proteins. DNA methylation can repress the expression of these genes and the lack of DNA methylation can lead to higher levels of expression. The cytosines in CpG dinucleotides can be methylated to form 5-methyl-cytosine, which leads to formation of a transcriptional repression complex.Citation1 CpG islands are groups of CpG dinucleotides. For a genomic region to be identified as a CpG island, in the hg19 version of the human reference genome, the CpG island size must be at least 200 base pairs and have a CG content of 50% or greater. CpG islands are thus identified algorithmically and identified by the Genome Browser (genome.ucsc.edu) for the hg19 version of the human genome.
Single-nucleotide polymorphisms (SNPs) are single base pair variations that occur commonly (> 1%) in the population, that could facilitate repression if they result in a CpG, or that could facilitate a higher level of expression if a CpG is eliminated. This project assessed SNPs regions for 5 cancer data sets of the cancer genome atlas (TCGA) (http://cancergenome.nih.gov/), for 2 oncogenes and 3 tumor suppressor genes, to determine whether the datasets reflected a skewed distribution of SNP alleles leading to creation or elimination of CpG's in the hg19 designated CpG islands for these oncogenes and tumor suppressor genes.
We also note that, while any SNP located at a CpG dinucleotide designated as a CpG by the reference genome would have alternate alleles that can only eliminate the CpG, it is more complicated to know which SNP alleles create CpG's, an issue potentially relevant to tumor suppressor gene repression. Thus, we generated a scripted algorithm to assess the potential of all the SNP alleles for all the CpG islands associated with 43 tumor suppressor genes to generate a GpG. This algorithm has provided an indication of which tumor suppressor genes are most vulnerable to conversion to a state whereby a CpG island could be more robust depending on the SNP alleles present in an individual.
Methods
Analysis of 5 TCGA cancer data sets
Five TCGA cancer datasets (http://cancergenome.nih.gov/) were chosen where there were at least 50 samples of data available from the UCSC Cancer Genomic Hub browser: Colon adenocarcinoma (COAD), esophageal (ESCA), bladder (BLCA), head and neck (HNSC), and lung cancer (LUAD). Whole genome sequence files (WGS) for primary solid tumors were selected for each cancer data set and their manifest files were downloaded. The data processing is described in . CpG islands for this study were selected by CpG dinucleotide count of 32 or above and by proximity to the transcriptional start site of the respective genes. The CpG islands were confirmed by visual inspection with the genome browser, and an example CpG island is in . The nucleotide positions (hg19 reference genome) for the CpG islands for each gene studied are in . The manifest files indicated above were used in conjunction with the Protocol_v5.sh script for data downloaded and were parsed using the process “mpileup” from “samtools” (Trust Sanger Institute), finding variants in the reads and dumping them into a CSV file that could be further transformed using the GetContext.sh script along with the PHP script cg.php, to determine whether CpG dinucleotides were created or destroyed by SNP allelic variants. The variants were then matched against the All SNPs(142) database to select those CpG alterations that were a result of SNPs, instead of apparent mutations. The rs numbers, designating the SNPs, were then collected in a table along with their minor allele frequencies to allow investigation as to whether SNPs that lead to CpG creation or elimination were associated with the cancers represented by the above datasets (). A record of all rs numbers that can affect the specified CpG Islands from (B) for the 5 cancers is presented in “Samy et al. SOM , RsNumbers and Cancer Counts.” The SNP frequency was calculated in respect to the data sample, seen in for the SNPs among the cancer data sets where the minor allele occurred for at least 12 barcodes in at least one dataset. SNP minor allele frequency was calculated using the Hardy Weinberg equation (1 = p2+2pq+q2) under the assumption that the absence of the SNP allele categorized the sample as homozygous dominant (p2) and that samples with the SNP allele were either heterozygous or homozygous for the recessive allele (2pq+q2). This approach allows for the most conservative estimate of the MAF which means deviations from the expected allele frequency reported by 1000Genomes and the AllSNPs(142) could be even higher if the SNP allele occurred mostly as homozygous instead of heterozygous.
Figure 1. Flow chart for the processing of the BAM files from CGHub for identifying SNPs at CpG positions. (A) An example manifest file used to target a specific cancer patient's tumor DNA sequence to be downloaded from CGHub is provided in the supporting online material (SOM), entitled “Samy et al. SOM , Example Manifest file.” This file is provided as an Excel file, for ease of inspection; and is provided as an XML file for direct use. Note: the Excel version of the manifest file cannot be directly used for downloads. (B) Defined CpG Island regions for each gene are included in . (C) The code used to automate the downloading and recording of variants in the patient files from TCGA is in “Samy et al. SOM , Protocol_v5.” (D) The preliminary Excel files with all variants obtained from the program were combined, labeled with the TCGA cancer abbreviation, and can be found in the “Samy et al. SOM , Preliminary Excel File.” Files are separated by CpG regions in the case of RB1, and the selected regions are in . (E) Adjacent nucleotides were added as a new column to the preliminary Excel files (present in SOM file labeled, “Samy et al. SOM , Preliminary Excel File”) using the code (F) documented as “Samy et al. SOM , GetContext.” (G) The PHP script used to determine how the variants in the DNA changed the CpG island structure is in “Samy et al. SOM , cg.php.” (H) A brief summary of the algorithm guiding the script is in “Samy et al. SOM , CG.PHP Explanation.” (I) The record of all rsNumbers (SNP designations) that can affect the specified CpG Islands from are recorded in the Excel file “Samy et al. SOM , RsNumbers and Cancer Counts.”
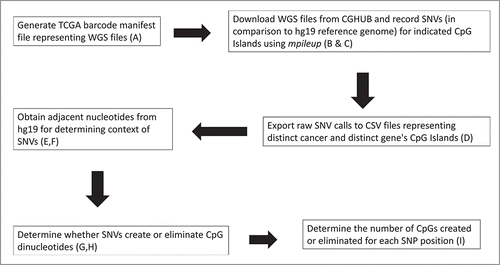
Figure 2. Example CpG Island (CpG count = 237) adjacent to the promoter region of MYC.
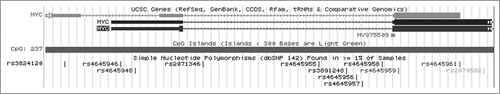
Table 1. CpG islands studied in this report, mapped to the hg19 version of the human genome.
Table 3. Allele frequencies for the non-hg19 SNP alleles that occur most frequently in the indicated data sets and that create or eliminate CpG's. Number of samples in parentheses; MAF = minor allele frequency; bold type represents cases of statistical significance when the frequency of occurrence of the minor allele is different from the frequency of the minor allele in one or both general SNP databases. A comprehensive set of data is in the SOM file labeled, “Samy et al. SOM , RsNumbers and Cancer Counts.”
Analysis of tumor suppressor genes
While SNPs that are a part of a CpG dinucleotide can be easily found by comparing the All SNPs(142) database and the USCS track for CpG dinucleotides, recognizing SNPs that have the potential to create an extra CpG dinucleotide requires testing on a case by case basis. CpG islands within 5000 nucleotides up and downstream that contained SNPs from the AllSNPs(142) database were identified for a sample of 43 tumor suppressor protein genes. SNPs which resulted in a new CpG dinucleotide were programmatically identified and recorded. Further details of the process and the results are in the Results section.
Results
Specific CpG alleles in TCGA cancer data sets
To determine whether any SNPs alleles in the CpG islands of HRAS and MYC eliminated CpG's in their associated CpG islands we performed the processing steps in : WGS files from the CGHub representing 5 distinct cancer datasets were downloaded and all SNVs that did not match the hg19 reference genome were identified and characterized as a mutation or known SNP. We analyzed multiple cancer data sets simultaneously owing to the increasing understanding and appreciation of signaling pathways and oncoprotein function degeneracy across many cancers.
The vast majority of the SNVs were known SNPs and the potential mutations were not further considered. The CpG islands are defined in , an example CpG island designation from the genome browser is shown in , and example processing output is shown in .
Table 2. Example of SNV detection results within the MYC CpG island () for 2 COAD patients after being processed as indicated in . Y, Yes; N, No. For the complete set for this report, see the SOM file entitled, “Samy et al. SOM , Preliminary Excel Files.” rs3824120 is further characterized in rs4645948 and rs4645955 did not meet the standard of the alternate allele appearing at least 12 times in one of the cancer datasets.
We further analyzed 5 SNPs for MYC and HRAS where a SNP allele altered the number of CpG's in the CpG islands for a minimum of 12 barcodes in at least one of the 5 cancer datasets. A summary of the results of the above HRAS and MYC CpG island processing is in . Only one SNP allele, for the rs3824120 SNP, in the MYC CpG island (), eliminated a CpG with a statistically significant, greater frequency of occurrence in the cancer data sets (p < 0.02), i.e., in comparison to the occurrence of this SNP allele in the All SNPs(142) database (but not in the 1000Genomes database). The minor allele for the rs4645958 SNP, also in the MYC CpG island (), eliminates a CpG but occurs less frequently in the cancer datasets in comparison to both the AllSNPs(142) and 1000Genomes databases (p <0.01).
Two minor frequency SNP alleles (rs112690925, rs7939028), within the HRAS CpG island (), created CpGs and occurred less frequently than in the general databases, with p-values ranging from less than .0002 to .02, depending on the SNP and the comparison database, as indicated in .
We next determined whether there were SNP alleles in the CpG islands of the tumor suppressor genes, APC, DCC, and RB1 that would create CpG's, using same processing steps indicated in . Results indicated that only one SNP, rs4940177 in DCC, represented creation of a CpG at a frequency significantly greater than the frequency of this allele in the All SNPs(142) and the 1000G SNP databases (p < .0007).
SNP alleles in tumor suppressor gene CpG islands that have the potential of leading to a greater CpG content
We were interested in the potential of increased CpG content in CpG islands of tumor suppressor genes. This consideration is based on the fact that any oncogene SNP representing a CpG, so designated by the hg19 reference genome, must have an alternate allele that eliminates the CpG, presumably consistent with increased oncogene expression. However, in the case of a tumor suppressor gene, where the interest is in the generation of a CpG, potentially facilitating additional gene repression in cancer development, the creation event is not guaranteed by simply the location of the SNP (at an hg19 indicated CpG island) or by the other alleles of the SNP (i.e., in contrast to the situation of interest with oncogenes). The impact of the alternative SNP alleles, if one of the alternatives were either a C or a G, would depend on the neighboring nucleotides. Thus, we wrote and scripted an algorithm for searching through the CpG islands of 43 tumor suppressor genes to detect SNPs whereby a conversion to a SNP allele different from the hg19 indicated allele (minor frequency allele) would lead to creation of a CpG (Samy et al. SOM, Tumor Suppressor Gene Analysis).
As an example of one tumor suppressor gene, AKAP12 has 4 CpG islands which contain SNPs (). One relevant SNP for the tumor suppressor gene, AKAP12, in the CpG island, CpG_132 (), is indicated in (rs573541357). In this case, the hg19 reference genome allele is a T, whereas the alternative allele for this SNP is a G, thereby creating a CpG (; Samy et al. SOM, Tumor Suppressor Gene Analysis).
Figure 3. Example of a SNP in a CpG Island for AKAP12 that creates a CpG dinucleotide. The T indicated in the figure is the major allele; G is the minor allele, as seen in .
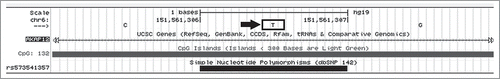
Figure 4. Initial nucleotide sequence of the second CpG Island in AKAP12 (CpG_132). SNPs are underlined, and the (circled) T → G allele change is the first to create a CpG dinucleotide.

Table 4. List of CpG Island regions for the tumor suppressor gene, AKAP12, provided as an example for generating the results of and the results in the SOM file labeled, “Samy et al. SOM, related.”
Table 5. Excerpt of primary processing for assessment of 4 SNPs present in a CpG Island (CpG_132) adjacent to AKAP12. The first SNP indicated is represented by . Further details are present in the SOM file labeled, Samy et al. SOM, Tumor Suppressor Gene Analysis. An example of the results of the primary processing step (for the AKAP12) is present in the SOM, labeled as “Samy et al. SOM, related.”
The effects of several example SNP alleles for AKAP12, in the CpG island, CpG_132 (; ), are indicated in . The results for all of the CpG islands for AKAP12 () are indicated in . Note that 24.7% of the SNPs of the AKAP12 gene CpG islands, collectively, are vulnerable to creating a CpG. And, note that the total CpG count for the AKAP12 CpG islands has the potential of increasing by 9.8%.
The processing results for all 43 of the studied, tumor suppressor genes are in the SOM file labeled, “Samy et al. SOM, Tumor Suppressor Gene Analysis.” The results are summarized in , where the percentage of SNPs that could potentially generate a CpG, for each gene, is indicated, along with the potential percent increase of CpG's, within in each gene's designated CpG islands. The VHL tumor suppressor gene has the greatest potential percent increase in the CpG count, due to alternative SNP alleles. And, some notable tumor suppressor genes with significant, potential percent increases in CpG counts are BRCA1, BRCA2, CHEK2, PTEN and RB1.
While there are many factors that could contribute to differences in the potential for CpG methylation for a given CpG island and tumor suppressor gene, we attempted to determine whether CpG island methylation ranges could be consistent with the range of available CpG's in CpG islands associated with tumor suppressor genes. Thus, we evaluated the range of methylation for AKAP12, which has a wide range of CpG's, due to SNPs (); and TXNIP, which does not vary in CpG percentage (). We obtained the TCGA, methylation β values for these 2 genes, for the 5 cancer data sets indicated in , for 10 randomly selected barcodes each. For every cancer dataset, the AKAP12 gene had a much wider range of methylation values ().
Figure 5. Bar-graph of the tumor suppressor set comparing the vulnerable SNP percentages and the CpG dinucleotide percent changes across all CpG Islands for the indicated genes. Further detail is present in the SOM file labeled, “Samy et al. SOM, Tumor Suppressor Gene Analysis.” Also, the distance of each CpG island containing a SNP, to the start site of transcription for each tumor suppressor gene, is provided in the SOM file labeled, “Samy et al. SOM, Transcription start site, CpG island distances.”
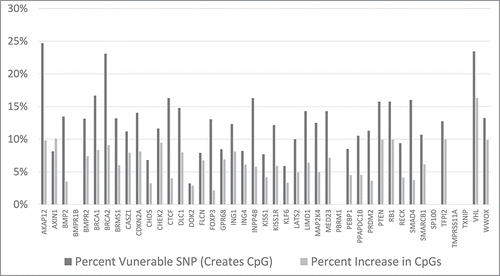
Figure 6. Box and Whisker plots showing the range of methylation β values for 10 randomly selected TCGA barcodes for the 5 cancer datasets studied in , for AKAP12 and TXNIP. Further detail is present in the SOM file labeled, “Samy et al. SOM, Methylation β value analysis.”
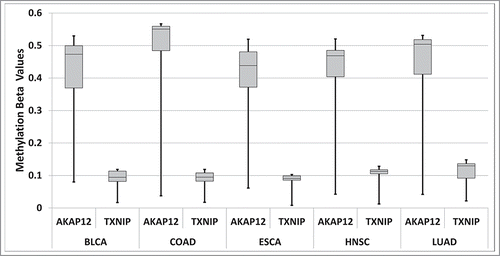
Table 6. CpG island SNP allele effects for AKAP12. Example result from “Samy et al. SOM, Tumor Suppressor Gene Analysis,” which provides all results of minor (non-hg19) SNP allele effects for 43 tumor suppressor genes. Note that “Vulnerable SNP fraction” and “CpG % DNFootnote1 change” are represented for all 43 tumor suppressor genes in .
Discussion
There have been numerous reports of SNPs affecting the hypermethylation of proximal and trans-gene CpG islands,Citation2-6 but there has been very little, if any consideration of SNPs within the CpG islands themselves, particularly with regard to the creation or elimination of a CpG. While the common presumption may be that one or more CpG's is not likely to affect regulation in a significant manner, the work in this report indicates both a decrease in the likelihood of a particular CpG in cancer data sets that is consistent with increased oncoprotein production and a particularly dramatic increase in the occurrence of a CpG that would be consistent with tumor suppressor protein repression. The mechanisms of effect of one CpG in a CpG island remain to be determined, however, it is apparent that the methylation of one to 3 CpG's can represent medically distinct categoriesCitation7-9 in mammals. It is also important to note that signal amplification in cancer, as opposed to the traditional idea of an on and off switch for signaling pathways, is likely important in a cancer phenotype.Citation10 Thus, while one CpG may not have a dramatic impact on expression levels as detected by routine laboratory approaches, the potential for a “shifting of the balance” remains important, particularly in considering such paradigms as feed-forward mechanisms of apoptosis, whereby a shift in transcription factor levels, as opposed to simple presence or absence of a transcription factor, likely determines the difference between cell proliferation and apoptosis.Citation11-14
It has become apparent that cancers traditionally representing different tissue types have extensive molecular overlaps, i.e., have many molecular bases in common for the cancer phenotype.Citation10,15 Thus, by taking a multi-cancer approach, it is possible to enhance statistical power when attempting to learn of potential over-representations of biomarkers in cancer. In this study, the most dramatic associations of CpG island SNPs and cancer were: (i) the lower incidence of CpG forming SNPs in an HRAS CpG island; and (ii) the over-representation of a CpG forming SNP in the CpG island of the DCC tumor suppressor gene. These results raise the question of whether there has been any empirical connection with HRAS and the cancers represented by the TCGA datasets studied here? Thus, for the following cancers, we note the indicated references: ESCA;Citation16 COAD,Citation17 BLCA,Citation18 HNSC,Citation19 and LUAD,Citation20 including indications of elevations of HRAS protein in certain cancers, which would be consistent with a reduced CpG content in the CpG island. A similar question could be raised regarding DCC: ESCA,Citation21 COAD,Citation22 BLCA,Citation23 and HNSC,Citation24 with the latter reference representing lower DCC levels of expression, again consistent with over-representation of CpG's in the CpG island of DCC in carcinogenesis.
The minor allele frequency for the MYC SNP, rs4645958, eliminated a CpG and was significantly under-represented in the cancer data sets, contrary to first expectations and contrary to results with a second MYC SNP and the HRAS SNPs (), discussed above. However, it bears repeating the molecular decision that governs proliferation or apoptosis is a balance in the levels of what are often considered pro-proliferation transcription factors that in fact, at high levels, almost universally lead to apoptosis.Citation13 Thus, it is conceivable that this balance is maintained, in the case of MYC, through higher levels of CpG methylation in some cases, and lower levels in other cases. There are certainly other possible explanations for the over-representation of the rs4645958 CpG in the MYC CpG island, for example a potential lack of relevance of MYC expression levels in certain cancers where an alternative, pro-proliferative signaling pathway could be activated.
Finally, the work above provided an indication of the potential vulnerability of certain tumor suppressor genes to the presence of increased numbers of CpG's in the CpG islands associated with these genes. This work has indicated a number of genes that could represent high priorities for detailed studies on CpG counts in the CpG islands, with the expectation that the most tumor suppressor genes most vulnerable to CpG percentage increases would demonstrate higher CpG counts in patients with certain or several different cancer types.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
1164360_Supplemental_Material.zip
Download Zip (774.5 KB)Related Research Data
References
- Jones PL, Veenstra GJ, Wade PA, Vermaak D, Kass SU, Landsberger N, Strouboulis J, Wolffe AP. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat Genet 1998; 19(2):187-91; PMID:9620779; http://dx.doi.org/10.1038/561
- Voisin S, Almen MS, Zheleznyakova GY, Lundberg L, Zarei S, Castillo S, Eriksson FE, Nilsson EK, Blüher M, Böttcher Y, et al. Many obesity-associated SNPs strongly associate with DNA methylation changes at proximal promoters and enhancers. Genome Med 2015; 7(1):103; PMID:26449484; http://dx.doi.org/10.1186/s13073-015-0225-4.
- Kato N, Loh M, Takeuchi F, Verweij N, Wang X, Zhang W, Kelly TN, Saleheen D, Lehne B, Mateo Leach I, et al. Trans-ancestry genome-wide association study identifies 12 genetic loci influencing blood pressure and implicates a role for DNA methylation. Nat Genet 2015; 47:1282-93; PMID: 26390057; http://dx.doi.org/10.1038/ng.3405
- Lemire M, Zaidi SH, Ban M, Ge B, Aissi D, Germain M, Kassam I, Wang M, Zanke BW, Gagnon F, et al. Long-range epigenetic regulation is conferred by genetic variation located at thousands of independent loci. Nat Commun 2015; 6:6326; PMID: 25716334; http://dx.doi.org/10.1038/ncomms7326.
- Putku M, Kals M, Inno R, Kasela S, Org E, Kozich V, Milani L, Laan M. CDH13 promoter SNPs with pleiotropic effect on cardiometabolic parameters represent methylation QTLs. Hum Genet 2015; 134(3):291-303; PMID:25543204; http://dx.doi.org/10.1007/s00439-014-1521-6
- Barry KH, Moore LE, Sampson J, Yan L, Meyer A, Oler AJ, Chung CC, Wang Z, Yeager M, Amundadottir L, et al. DNA methylation levels at chromosome 8q24 in peripheral blood are associated with 8q24 cancer susceptibility loci. Cancer Prev Res 2014; 7(12):1282-92; PMID:25315430; http://dx.doi.org/10.1158/1940-6207.CAPR-14-0132
- Kojima A, Kobayashi T, Ito S, Murasawa A, Nakazono K, Yoshie H. Tumor necrosis factor-alpha gene promoter methylation in Japanese adults with chronic periodontitis and rheumatoid arthritis. J Periodontal Res 2015; PMID:26247485; http://dx.doi.org/10.1111/jre.12314
- Kalashikam RR, Inagadapa PJ, Thomas AE, Jeyapal S, Giridharan NV, Raghunath M. Leptin gene promoter DNA methylation in WNIN obese mutant rats. Lipids Health Dis 2014; 13:25; PMID: 24495350; http://dx.doi.org/10.1186/1476-511X-13-25
- Tendl KA, Schulz SM, Mechtler TP, Bohn A, Metz T, Greber-Platzer S, Kasper DC, Herkner KR, Item CB. DNA methylation pattern of CALCA in preterm neonates with bacterial sepsis as a putative epigenetic biomarker. Epigenetics 2013; 8(12):1261-7; PMID:24135723; http://dx.doi.org/10.4161/epi.26645
- Ford SA, Blanck G. Signal persistence and amplification in cancer development and possible, related opportunities for novel therapies. Biochim Biophys Acta 2014; 1855(1):18-23; PMID:25450826; http://dx.doi.org/10.1016/j.bbcan.2014.11.001
- Mauro JA, Butler SN, Ramsamooj M, Blanck G. Copy number loss or silencing of apoptosis-effector genes in cancer. Gene 2015; 554(1):50-7; PMID:25307873; http://dx.doi.org/10.1016/j.gene.2014.10.021
- Szekeres K, Koul R, Mauro J, Lloyd M, Johnson J, Blanck G. An Oct-1-based, feed-forward mechanism of apoptosis inhibited by co-culture with Raji B-cells: towards a model of the cancer cell/B-cell microenvironment. Exp Mol Pathol 2014; 97(3):585-9; PMID:25236570; http://dx.doi.org/10.1016/j.yexmp.2014.09.010
- Mauro JA, Blanck G. Functionally distinct gene classes as bigger or smaller transcription factor traps: a possible stochastic component to sequential gene expression programs in cancer. Gene 2014; 536(2):398-406; PMID: 24291030; http://dx.doi.org/10.1016/j.gene.2013.11.013
- Garcia M, Mauro JA, Ramsamooj M, Blanck G. Tumor suppressor genes are larger than apoptosis-effector genes and have more regions of active chromatin: Connection to a stochastic paradigm for sequential gene expression programs. Cell Cycle 2015; 14(15):2494-500; PMID:25945879; http://dx.doi.org/10.1080/15384101.2015.1044179
- Frangione ML, Lockhart JH, Morton DT, Pava LM, Blanck G. Anticipating designer drug-resistant cancer cells. Drug Discov Today 2015; 20:790-3; PMID:25697478; http://dx.doi.org/10.1016/j.drudis.2015.02.005
- Senmaru N, Shichinohe T, Takeuchi M, Miyamoto M, Sazawa A, Ogiso Y, Takahashi T, Okushiba S, Takimoto M, Kato H, et al. Suppression of Erk activation and in vivo growth in esophageal cancer cells by the dominant negative Ras mutant, N116Y. Int J Cancer Journal international du cancer 1998; 78(3):366-71; PMID: 9766573; http://dx.doi.org/10.1002/(SICI)1097-0215(19981029)78:3%3c366::AID-IJC18%3e3.0.CO;2-4
- Gallick GE, Kurzrock R, Kloetzer WS, Arlinghaus RB, Gutterman JU. Expression of p21ras in fresh primary and metastatic human colorectal tumors. Proc Natl Acad Sci U S A 1985; 82(6):1795-9; PMID:3885218; http://dx.doi.org/10.1073/pnas.82.6.1795
- Ulsh LS, Shih TY. Metabolic turnover of human c-rasH p21 protein of EJ bladder carcinoma and its normal cellular and viral homologs. Mol Cell Biol 1984; 4(8):1647-52;PMID: 6092927; http://dx.doi.org/10.1128/MCB.4.8.1647
- Johnson TL, Lloyd RV, Burney RE, Thompson NW. Hurthle cell thyroid tumors. An immunohistochemical study. Cancer 1987; 59(1):107-12; PMID:3539304; http://dx.doi.org/10.1002/1097-0142(19870101)59:1%3c107::AID-CNCR2820590123%3e3.0.CO;2-U
- Yano T, Zissel G, Muller-Qernheim J, Jae Shin S, Satoh H, Ichikawa T. Prostaglandin E2 reinforces the activation of Ras signal pathway in lung adenocarcinoma cells via EP3. FEBS Lett 2002; 518(1–3):154-8; PMID:11997037
- Huang Y, Boynton RF, Blount PL, Silverstein RJ, Yin J, Tong Y, McDaniel TK, Newkirk C, Resau JH, Sridhara R, et al. Loss of heterozygosity involves multiple tumor suppressor genes in human esophageal cancers. Cancer Res 1992; 52(23):6525-30; PMID:1423299
- Fearon ER, Cho KR, Nigro JM, Kern SE, Simons JW, Ruppert JM, Hamilton SR, Preisinger AC, Thomas G, Kinzler KW, et al. Identification of a chromosome 18q gene that is altered in colorectal cancers. Science 1990; 247(4938):49-56; PMID:2294591; http://dx.doi.org/10.1126/science.2294591
- Brewster SF, Gingell JC, Browne S, Brown KW. Loss of heterozygosity on chromosome 18q is associated with muscle-invasive transitional cell carcinoma of the bladder. Br J Cancer 1994; 70(4):697-700; PMID:7917921; http://dx.doi.org/10.1038/bjc.1994.376
- Kim MS, Shin KH, Baek JH, Cherrick HM, Park NH. HPV-16, tobacco-specific N-nitrosamine, and N-methyl-N'-nitro-N-nitrosoguanidine in oral carcinogenesis. Cancer Research 1993; 53(20):4811-6; PMID: 8402666