
ABSTRACT
Most solid tumors are aneuploid, carrying an abnormal number of chromosomes, and they frequently missegregate whole chromosomes in a phenomenon termed chromosome instability (CIN). While CIN can be provoked through disruption of numerous mitotic pathways, it is not clear which of these mechanisms are most critical, or whether alternative mechanisms could also contribute significantly in vivo. One difficulty in determining the relative importance of candidate CIN regulators has been the lack of a straightforward, quantitative assay for CIN in live human cells: While gross mitotic abnormalities can be detected visually, moderate levels of CIN may not be obvious, and are thus problematic to measure. To address this issue, we have developed the first Human Artificial Chromosome (HAC)-based quantitative live-cell assay for mitotic chromosome segregation in human cells. We have produced U2OS-Phoenix cells carrying the alphoidtetO-HAC encoding copies of eGFP fused to the destruction box (DB) of anaphase promoting complex/cyclosome (APC/C) substrate hSecurin and sequences encoding the tetracycline repressor fused to mCherry (TetR-mCherry). Upon HAC missegregation, daughter cells that do not obtain a copy of the HAC are GFP negative in the subsequent interphase. The HAC can also be monitored live following the TetR-mCherry signal. U2OS-Phoenix cells show low inherent levels of CIN, which can be enhanced by agents that target mitotic progression through distinct mechanisms. This assay allows direct detection of CIN induced by clinically important agents without conspicuous mitotic defects, allowing us to score increased levels of CIN that fall below the threshold required for discernable morphological disruption.
Introduction
Chromosome Instability (CIN)Citation1-3 is defined as the frequent missegregation of whole chromosomes. CIN can be both advantageous and problematic for tumor cellsCitation4-6: On one hand, moderate CIN levels in tumors are associated with poor prognosis because they generate phenotypic diversity, driving evolutionary adaptation and promoting the acquisition of metastatic potential and drug resistance.Citation5,7,8 Most solid tumors and about 50% of haematopoietic cancers are aneuploid,Citation3,9,10 reflecting chromosome gain or loss. On the other hand, levels of CIN above a critical threshold can kill cancer cellsCitation11,12 and inhibit tumor growth.Citation6 Because many tumors may already display higher CIN levels than normal cells, further enhancement of CIN has been proposed as a strategy to differentially kill cancer cells using anti-cancer therapeutics.Citation11-13
A number of mechanisms have been suggested to drive CIN by undermining major pathways required for accurate chromosome segregation.Citation4,5,13,14 It is not clear which of these mechanisms are most critical in vivo, and it remains possible that other mechanisms could also contribute to CIN within tumors. One difficulty in determining the relative importance of candidate CIN regulators has been the lack of a straightforward, quantitative assay for CIN in live human cells: Gross mitotic abnormalities, such as the presence of multiple lagging chromosomes during anaphase, can be detected visually in cultured cells. However, moderate levels of CIN (≤1 chromosome missegregation per division) may not be obvious, and are thus challenging and laborious to detect and quantify in live cells. Rather, chromosome missegregation rates have been quantified by laborious techniques such as coupling clonal cell analysis with karyotype analysis, fluorescence in situ hybridization (FISH) or fixed cell analysis of cancer cells expressing LacI-GFP with LacO arrays integrated in single chromosomes.Citation1,15-17 A rapid and quantitative assay to measure CIN induced in mammalian cells by potential chemotherapeutic agents would thus be extremely useful.
Human artificial chromosomes (HACs) have been extensively developed as potential vectors for gene therapy, and it has previously been shown that their segregation relies on the same machinery that mediates endogenous chromosome segregation.Citation18,19 The regional centromere of the AlphoidtetO-HAC was engineered using a 40-kb synthetic alphoid DNA array that contains the 42-bp tetracycline operator (tetO) sequences incorporated into every other alphoid DNA monomer.Citation20 Two approaches have been previously utilized to adopt this HAC for CIN studies: In the first approach, a constitutively expressed eGFP transgene was inserted into the AlphoidtetO HACCitation21 in human fibrosarcoma HT1080 cells, so that cells inheriting the HAC expressed eGFP and cells lacking the HAC did not. This system was recently used to study the effect of 62 different anticancer drugs on chromosome segregation using flow cytometry.Citation22 In the second approach, the HAC carries a constitutively expressed short hairpin RNA (shRNA) against a eGFP transgene that is integrated into the genome of HT1080 cells, so that HAC loss is required before eGFP accumulates within the cells.Citation23 In these approaches, GFP protein or shRNA respectively persist in daughter cells for a considerable time after HAC loss, so that HAC loss is scored is 14 d after drug treatment. Due to the extended delay for scoring, calculations of HAC loss rates are relatively indirect and imprecise.
To overcome these problems, we have designed a live cell assay for the fidelity of HAC segregation allowing immediate visualization of faithful chromosome segregation: We reengineered the AlphoidtetO HAC to express a fluorescent marker that is cyclically degraded during each mitosis, enhanced green fluorescent protein (eGFP) fused to the destruction box (DB) domain of the anaphase promoting complex/cyclosome (APC/C) substrate hSecurin. Missegregation of the HAC during any mitosis results in the production of daughter cells that lack the HAC and that therefore remain non-fluorescent during the subsequent cell cycle. The reengineered HAC also expresses the tetracycline repressor protein fused to monomeric cherry fluorescent protein (tetR-mCherry), which binds to tetO arrays within the HAC itself, giving us an independent marker for assessment of HAC segregation. This assay provides an excellent, quantitative measurement of CIN, with HAC missegregration in ≤ 0.5% of divisions within a human U2OS-based cell line (U2OS-Phoenix). Moreover, we show that this assay provides the capacity for direct detection of CIN induced by well-studied and clinically important agents without the necessity to score for conspicuous morphological defects.
Results
Reengineering of the alphoidtetO HAC and isolation of the U2OS-Phoenix cell line
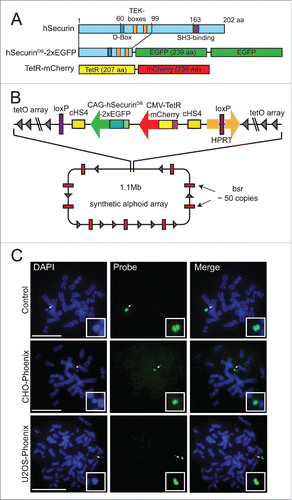
To develop a more rapid assay for HAC loss, we reengineered the AlphoidtetO HAC to encode tandem repeats of the enhanced green fluorescent protein (eGFP) fused to the 1–99 aa N-terminal domain of anaphase promoting complex/cyclosome (APC/C) substrate hSecurin containing its destruction box (DB) and TEK-boxes.Citation24,25 We also introduced sequences encoding the tetracycline repressor protein fused to monomeric cherry fluorescent protein (tetR-mCherry) (). The HAC was reengineered by a targeting construct carrying these fusions into the HAC in hamster CHO cells using Cre-LoxP mediated recombination (Fig. S1a). We then transferred the HAC via MMCT (for details see Methods) into a number of human cell lines, and screened for cells that both faithfully maintained the HAC and strongly expressed both fluorescent markers. We found that human osteosarcoma-derived U2OS cells containing the HAC (U2OS-Phoenix) (Fig. S1a) showed these properties, and we confirmed by FISH that the HAC was indeed maintained in our cells in a stable and autonomous fashion through cell divisions ().
Figure 1. Isolation of the U2OS-Phoenix cell line. (a) Cartoon depicting fusion constructs that were introduced into the LoxP site of the AlphoidtetO HAC backbone as markers of CIN within one cell division. N terminal aa 1–99 of hSecurin was fused with tandem copies of enhanced Green Fluorescent Protein (eGFP). Also shown is E. coli tet repressor TetR fused with mCherry (For more details see Materials and Methods). (b) Schematic representation of the HAC containing the constructs with their position and orientation within the HAC (bsr: gene conferring Blasticidin resistance, for more details about HAC construction refer to papers 20 and 21, for more details about loading of the targeting construct into the HAC see Materials and methods). (c) FISH using the FITC-tetO PNA probe detecting the newly constructed HAC in the CHO and U2OS cells. CHO cells containing AlphoidtetO HAC was used as a control. Chromosomes are counterstained with DAPI (blue). Arrows indicate HACs. In the insert, HACs are shown in higher magnification. Size bars = 15 μm.
U2OS-phoenix cells can quantify HAC missegregation within a single cell cycle
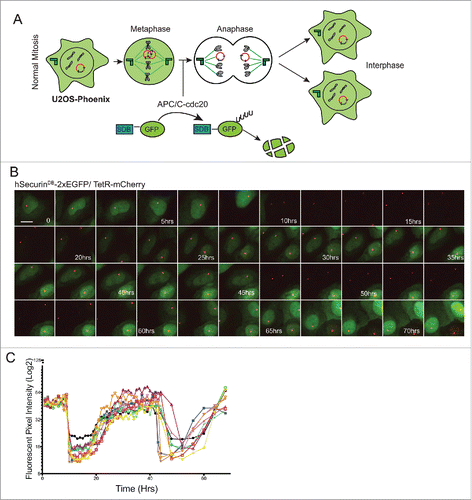
We monitored HAC segregation in two ways: The APC/C recognizes DB-containing proteins and promotes their degradation upon mitotic exit.Citation24,26 Thus DB-eGFP fusions expressed from HACs are rapidly degraded at anaphase onset, and re-accumulate in the two daughter cells after G1 phase (). Daughter cells that do not obtain a copy of the HAC are GFP negative in the subsequent interphase (Fig. S3a). In addition, because tetR-mCherry binds to the tetO arrays, we could follow the HAC itself by following the mCherry signal (). Live cell imaging of U2OS-Phoenix cells for two subsequent cycles showed a clear cyclic degradation pattern of the eGFP coupled to the cell cycle (). We also quantified the mean fluorescent intensity of the GFP signal in 10 cells over two cell cycles each (). The cyclic pattern of the GFP degradation could also be observed by flow cytometry using asynchronous population of U2OS-Phoenix cells (Fig. S2). These data show that we can detect HAC missegregation within a single cell cycle. This is a substantial improvement beyond previous HAC-based assays, wherein HAC loss was measured in HT1080 cells after a period of approximately two weeks.Citation22,23
Figure 2. GFP expression in U2OS-Phoenix cells is coupled to the cell cycle. (a) Cartoon depicting U2OS-Phoenix cells undergoing one round of error free mitosis. At the onset of anaphase, active APC/C-cdc20 recognizes and ubiquitinates DB-containing proteins promoting their degradation. Thus, hSecurinDB-2xeGFP fusions expressed from HACs are rapidly degraded at the onset of anaphase, and re-accumulate in the 2 daughter cells after G1 phase. (b) Still images from a live cell imaging experiment following hSecurinDB-2xGFP and TetR-mCherry signals in U2OS-Phoenix cells using a spinning disc confocal microscope. The images follow one cell in 2 rounds of consecutive mitosis. The GFP levels in the imaged cell and its daughter are coupled to the cell cycle. Size bars = 30 μm (c) The mean fluorescent intensity of the GFP signal was quantified in 10 cells from movies such as in (b) and plotted against time.
HAC missegregates in a rate comparable to endogenous chromosomes in U2OS-phoenix cells
To assess the level of missegregation of endogenous chromosomes in the U2OS-Phoenix cells, asynchronous population of U2OS-Phoenix cells were fixed and analyzed (). In this analysis, 5.52% of U2OS-Phoenix cells showed lagging chromosomes and bridges (). The modal number of chromosomes in the U2OS cell line is in the hypertriploid range (U2OS-Phoenix cells were counted to have about 62 chromosomes), and we extrapolated the average rate of segregation defects for individual chromosomes to be at least 0.089%. In this analysis, 0.48% of U2OS-Phoenix cells showed multipolar spindles and about 6% displayed micronuclei (). For comparison, we counted events of HAC missegregation in unsynchronized, fixed cells. Only one HAC missegregation event was observed among 341 anaphases (0.29%), thus, the observed fidelity of HAC segregation was comparable to the segregation of endogenous chromosomes (). To further monitor the events of HAC missegregation in the U2OS-Phoenix cells, we followed the HAC by live cell imaging on a spinning disk confocal microscope in 195 cells. HAC missegregation was observed only in one instance (0.51%) (; S3b). This number deviated slightly from the one calculated in , likely because of the low number of observed events. Taken together, these findings indicate that HAC missegregrates in U2OS-Phoenix cells in ≤ 0.5% of cell divisions.
Figure 3. The rate of HAC missegregation in U2OS-Phoenix is comparable to endogenous chromosomes. (a) Images of U2OS-Phoenix cells at anaphase showing events of lagging chromosomes, chromosome bridges, and HAC missegregation. Unsynchronized cells were fixed and DAPI stained and data was collected using a fluorescent microscope. Size bars = 30 μm. (b) Pie graphs showing percentage of U2OS-Phoenix cells with either lagging chromosomes and bridges, HAC missegregation, multipolar spindles or micronuclei. (1 23 events in 417 anaphases;2 1 event in 341 anaphases;3 2 events in 417 anaphases;4 69 (1) and 14 (≥ 2) in 1395 interphases counted). No HACS were observed in micronuclei.
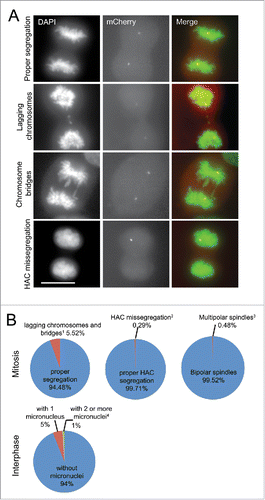
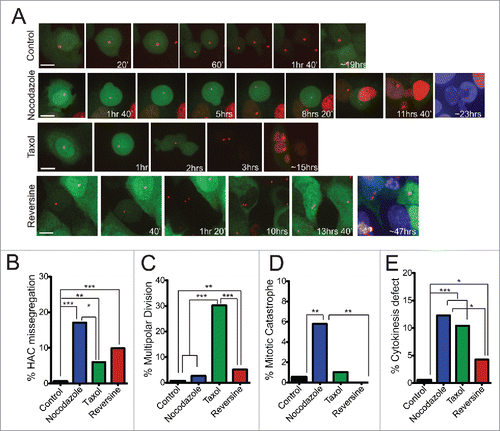
Figure 4. Increased levels of HAC mis-segregation upon treatment with chemotherapeutic agents. (a) Still images from live cell imaging experiments of U2OS-Phoenix cells showing examples of proper HAC segregation in control and examples of HAC missegregation in various treatments. TetR-mCherry and hSecurinDB-2xGFP signals were followed using a spinning disk confocal microscope. The daughter cells of a mother cell that undergoes a HAC missegregation event have either 2 HACs (GFP positive) or no HAC (GFP negative) and survive for over 20hrs in case of Nocodazole and Reversine treatments. The daughter cells were DAPI stained at the end of the movie to visualize the ones that do not carry a HAC. Size bars = 30 μm. (b-e) Bar graphs depicting percentage of U2OS-Phoenix cells that missegregate the HAC (ncontrol=195; nNocodazole=111; nTaxol=67; nReversine=111) (b), undergo multipolar division (ncontrol=196; nNocodazole=114; nTaxol=96; nReversine=117) (c), undergo mitotic catastrophe (ncontrol=197; nNocodazole=121; nTaxol=97; nReversine=117) (d), and have problems in cytokinesis (ncontrol=196; nNocodazole=114; nTaxol=96; nReversine=117) (e) quantified from live cell imaging experiments (***p<0 .001, **p≤0 .01, *p<0 .05). Multipolar divisions were omitted from HAC missegregation counts.
HAC segregation in U2OS-phoenix cells is responsive to chemicals that disrupt mitotic progression
A number of mitotic pathways are targeted by existing chemotherapeutic agents or drugs that are under development in clinical trials.Citation27 The protein targets of these drugs include microtubules (MTs), MT motors and kinases, especially those involved in the spindle assembly checkpoint (SAC). The SAC is a cell cycle regulatory pathway that prevents anaphase onset prior to the point when all chromosomes have achieved correct kinetochore attachment to spindle MTs and alignment on the metaphase plate. We sought to measure the levels of CIN observed after targeting MT dynamics or the SAC at drug concentrations that did not cause mitotic arrest.
We followed HAC segregation by live cell imaging in the presence of microtubule poisons Taxol (2–6 nM, generic name Paclitaxel) and Nocodazole (25 nM). Taxol is a widely used anti-mitotic drugCitation28 that at low, clinically relevant doses, induces aneuploidy without a severe mitotic delay.Citation29,30 Mitotic recovery from Nocodazole-induced MT depolymerization increases the incidence of merotelic kinetochore attachments.Citation15 In the presence of either drug, we observed a significant increase in HAC missegregation (). Taxol-treated cells showed elevated levels of multipolar divisions followed by cell deathCitation31 ( and S5a) but did not show a significant mitotic delay (Fig. S4). Nocodazole-treated cells showed substantial delay in mitosis (Fig. S4) accompanied by mitotic catastrophe (). Both Nocodazole- and Taxol-treated U2OS-Phoenix cells displayed elevated levels of cytokinesis defects (). 75% of HAC missegregation events in Taxol and 48% in Nocodazole were followed by cell death (Fig. S5b), suggesting either the occurrence of endogenous chromosome missegregation in parallel with the HAC or secondary effects of these drugs on other pathways. Our results indicate that microtubule poisons, even at sub-lethal doses, induce defects that either activate the SAC (mitotic delay) and/or cause substantial chromosome loss. These defects then are ultimately incompatible with long-term cell survival.
We also assessed how disruption of the SAC impacts HAC segregation. Emerging evidence suggest that reducing the levels of SAC components causes an increase in chromosome missegregation but not lethality. The latter is only achieved when cells are subjected to a combinatorial action of drugs that target both the SAC and the mitotic spindle.Citation11,12 In particular, partial knockdown of the Monopolar spindle 1 (Mps1) kinase, an essential component of the SAC, weakens the SAC but does not affect U2OS cell viability.Citation11 Doses of Reversine (200–300 nM) that specifically inhibit the Mps1 kinaseCitation32 significantly increased HAC missegregation, but did not similarly increase the levels of other mitotic defects such as multipolar divisions, cytokinesis failure, and mitotic catastrophe (). As expected, most (75%) of the cells that missegregated the HAC survived in the presence of Reversine (Fig. S5b), suggesting that the levels of endogenous chromosome missegregation induced by this drug was not sufficient to drive lethal events in the majority of cells. Taken together, our data indicate that HAC segregation in U2OS-Phoenix cells is responsive to drugs that increase CIN levels.
Discussion
We have devised a live cell assay for direct visualization of faithful HAC segregation. We expressed a fusion of eGFP to the Securin destruction box (DB) domain from the AlphoidtetO HAC. This fusion is cyclically degraded during each mitosis. Mitotic missegregation results in the production of daughter cells that lack the HAC, and that therefore remain non-fluorescent in the subsequent cell cycle. The HAC also encodes the tetR protein fused to monomeric cherry fluorescent protein (tetR-mCherry), which binds to tetO arrays within the HAC itself, giving a second marker for assessment of HAC segregation. This system allows quantitative measurement of CIN within a human U2OS-based cell line (U2OS-Phoenix), and we found that HAC missegregration rates were only marginally higher than the estimated missegregation rates of endogenous chromosomes. In addition, HAC segregation in U2OS-Phoenix cells was responsive to well-studied agents that disrupt mitotic progression. Interestingly, Reversine, an inhibitor of the SAC kinase Mps1, was more effective than Taxol, an anti-MT agent, in promoting HAC missegregation at the concentrations tested (), despite the fact that it was less disruptive to spindle structures or cytokinesis. Thus, our assay detected a significant increase in HAC loss induced by Reversine without obvious mitotic abnormalities or visual evidence of lagging chromosomes.
Our assay provides at least three key advantages over earlier approaches: First, it is much easier to score CIN in this assay than through labor-intensive, chromosome-based approaches. Chromosome-based assays to measure CIN include coupling clonal cell analysis with karyotype analysis, fluorescence in situ hybridization (FISH) or fixed cell analysis of cancer cells expressing LacI-GFP with LacO arrays integrated in single chromosomes.Citation1,15-17 By contrast, our live cell assay requires minimal manipulation of cells, and it is easily visualized and quantitative. Moreover, it can be adapted to high-throughput screens by following the GFP signal. Notably, our assay allows us to score CIN within one cell cycle, so it is also more rapid and direct that previously reported HAC-based assays: HAC-based screening for CIN has been developed using either constitutive AlphoidtetO HAC-based expression of eGFP,Citation22 or AlphoidtetO HAC-based expression of an shRNA against a genomically integrated eGFP transgene.Citation23 Because GFP protein or shRNA respectively persist in daughter cells for a considerable time after HAC loss, CIN is scored roughly two weeks after drug treatment in these assays. As a result, HAC loss rates are estimated indirectly, and are thus less precise that those obtained through our direct observations.
Second, screens to discover novel mitotic modulators as potential chemotherapeutic agents have frequently assessed overt defects such as mitotic arrest, spindle abnormalities or the presence of lagging chromosomes.Citation33 These screens thus preferentially identify compounds that produce visually obvious phenotypes, as can be seen with MT poisons. As a result, these screens may overlook other compounds that do not cause conspicuous distortions in cellular morphology during mitosis, even if they substantially elevate levels of chromosome missegregation. By contrast, we are able to score CIN without relying upon gross morphological defects (), such as the presence of obvious lagging chromosomes or chromosome bridges. We can therefore detect and quantify modulations of CIN that fall below the detection of live cell screens that score for phenotypic defects.
Third, the properties of the HAC make it highly useful to examine mitotic modulators of CIN. The HAC is non-essential and missegregates at a slightly higher rate than endogenous chromosomes (), providing a sensitive platform for CIN detection that does not interfere with cell viability. Since loss of endogenous essential chromosomes can cause cell death, the selective loss of cells after missegregation may complicate the scoring of CIN assays that rely on those chromosomes. Some causes of CIN, such as structural chromosome abnormalities or DNA replication stress,Citation34,35 do not involve the mitotic machinery per se but rather processes that are generally required for genome maintenance. Because the HAC used in our assay is a small circular DNA (1.1 Mb),Citation20,36 we anticipate that our assay will relatively favor the detection of agents that disrupt centromere or kinetochore functionCitation13 over processes that are operative throughout the length of chromosomes. It is possible that chemicals that target centromere or kinetochore function may be relatively desirable as therapeutic agents, as they do not promote mutations elsewhere in the genome as a secondary consequence of treatment. This bias may be useful, to the extent that it can focus subsequent analysis regarding the mechanism of action for compounds under study.
In summary, we have developed a quantitative live-cell assay for mitotic chromosome segregation in human cells, which utilizes U2OS-Phoenix cells to assess the rates of CIN within one cell division, and we have demonstrated that this assay can be utilized to quantitatively assess the levels of CIN induced by agents that target mitotic progression through distinct mechanisms. In the future, this assay will be adapted for high-throughput chemical screens to identify novel drugs and genes that modulate CIN levels, and it is well suited for synthetic lethal screens in which combinatorial effects of disrupting two or more pathways can be studied.
Materials and methods
Cell culture and reagents
U2OS and U2OS-Phoenix cells were grown in DMEM (Life Technologies, Grand Island, NY), containing 10% FBS (Atlanta Biologicals, Flowery Branch, GA), 2 mM GlutaMAX Supplement (Life Technologies, Grand Island, NY), 100 IU/ml penicillin, and 100 μg/ml streptomycin. HPRT-minus Chinese hamster ovary (CHO) cells carrying the alphoidtetO-HAC were grown in Ham's F-12 (Life Technologies, Grand Island, NY) containing 10% FBS with 8g/ml BS (Life Technologies, Grand Island, NY), 2 mM GlutaMAX Supplement, 100 IU/ml penicillin, and 100 μg/ml streptomycin. All cells were maintained at 37° C with 5% CO2. Drugs were added in constant presence in the media unless otherwise indicated. Nocodazole (Sigma-Aldrich, St. Louis, MO) was used at 25nM, Taxol (Sigma-Aldrich, St. Louis, MO) at 2–6 nM and Reversine (Sigma-Aldrich, St. Louis, MO) at 200–300 nM concentrations.
Construction of the targeting vector pSM1 carrying the CIN markers
The open reading frames (ORFs) of two eGFPs in tandem were PCR amplified from a pEGFP-N1 vector (Clontech laboratories, Inc., Mountain View, CA) containing tandem GFPs (provided by Malte Renz) and cloned into the p#264-GFP knot linker CAGpr vector (provided by Artem Kononenko) between the BstBI and AvrII restriction sites. The first 297 bps of the ORF of hSecurin was then PCR amplified from a human cDNA library and cloned between the PacI and BstBI restriction sites of the p#264-GFP knot linker CAGpr vector containing the 2 tandem eGFPs. This created the hSecurinDB-2xEGFP chimeric ORF under the CAG promoter. In addition, the TetR ORF was amplified from TetR-f-GFP_HyTK vector (provided by Alexander Samoshkin) and cloned between the sacI and AgeI restriction sites at the MCS of the pmCherry-N1 vector (Clontech laboratories, Inc., Mountain View, CA). CMV-TetR-mCherry-polyA was then PCR amplified from the pmCherry-N1 vector and introduced into the p#264-GFP knot linker CAGpr at the KpnI restriction site. The final targeting vector was named pSM1. The integrated fusion constructs were spanned by cHS4 insulators. pSM1 also contained a 3′HPRT-loxP cassette that allowed its insertion into the single loxP loading site of the alphoidtetO-HAC propagated in HPRT-minus hamster CHO cellsCitation36 (Fig. S1a). Insertion of pSM1 into the loxp site of alphoidtetO-HAC reconstituted the HPRT gene.
Loading of the pSM1 targeting vector into the LoxP site of the alphoid tetO-HAC in CHO cells
A total of 1.5 μg of the pSM1 targeting vector (described earlier) and 0.5 μg of the Cre expression pCpG-iCre vectorCitation37 DNA were co-transformed using X-tremeGENE 9 DNA transfection reagent (Roche, Indianapolis, IN) at the 3:1 ratio of reagent to DNA into HPRT-deficient CHO cells containing the alphoidtetO-HACCitation36 grown on a 6 well plate. HPRT-positive clones were selected after 2 to 3 weeks growth in medium containing sodium Hypoxanthine (0.1 mM), Aminopterin (0.4 µM) and Thymidine (16 µM) (HAT) (Life Technologies, Grand Island, NY). 6 to 10 clones were usually selected for each transfection. Diagnostic PCR for reconstitution of the HPRT gene was performed to insure correct loading of the transgene cassette into the HAC as described in.Citation38 The HPRT gene was reconstituted in almost 100% of analyzed clones (Fig. S1b), indicating a high efficiency of accurate gene loading. After loading of the transgene cassette into the alphoidtetO-HAC, the CHO cells were maintained in culture in medium containing the HAT supplement.
Microcell-mediated chromosome transfer
The alphoidtetO-HAC containing the hSecurinDB-2xEGFP and TetR-mcherry cassette was transferred from CHO-Phoenix cells into U2OS cells using a standard microcell-mediated chromosome transfer (MMCT) protocol as described earlier inCitation39 with minor changes (Fig. S1a). Briefly, HVJ Envelope Cell Fusion Kit, GenomONE-CF EX (Cosmo Bio Co., Ltd., Tokyo, Japan) was used instead of PEG to fuse the purified microcells from CHO-Phoenix into the U2OS cells. In addition, Blasticidin S HCL (Life Technologies, Grand Island, NY) at a concentration of 4 μg/ml was used to select clones containing the HAC. 3–20 Blasticidin resistant clones were usually obtained in one MMCT experiment and were analyzed by FISH for the presence of the autonomous form of the HAC (). These clones were also analyzed for the presence of any co-transferred CHO chromosomes using a diagnostic PCR test for rodent-specific SINE elements as described below (Fig. S1c).
Genomic DNA preparation and diagnostic PCR
Genomic DNA for PCR analysis was prepared using a QIAmp DNA Mini Kit (QIAGEN Inc., Valencia, CA, USA). Reconstitution of the HPRT gene after Cre/lox-mediated recombination was determined by specific primers, Lox137-R 5′-agccttctgtacacatttcttctc-3′ and Rev #6 5′-gctctactaagcagatggccacagaactag-3′. Cross contamination by hamster chromosomes was determined by specific primers detecting hamster short interspersed elements (SINEs): Furin F: 5′-actcagagatccactgcaccaggatccaagggagg-3′ and Furin R: 5′-ccgctcgagcggctacaccacagacaccattgttgg ctactgctgcc-3′; and primers amplifying human specific SPANX locus: PrimSX-RR 5′-ctacctcttcccttcccttc-3′ and PrimSX-F5 5′ tgggacactgcctgtatgat-3′.
FISH analysis
The presence of the HAC in CHO-Phoenix and U2OS-Phoenix cells in an autonomous form was confirmed by FISH analysis as previously describedCitation21 with minor changes. PNA (peptide nucleic acid) labeled probe used was against the tetO-alphoid array (FITC-OO-ACCACTCCCTATCAG) (Panagene, South Korea) and images were examined as described below in Preparation and Imaging of fixed samples.
Preparation and imaging of fixed samples
U2OS-Phoenix cells were grown on coverslips, washed with Phosphate-Buffered Saline (PBS) pH 7.4 and immediately fixed with 4% paraformaldehyde (PFA) followed by an acetone:methanol (1:1) fixation at −20°C. The samples were then DAPI stained and examined using Zeiss Axioskop microscope (Zeiss, Thornwood, NY), with DAPI, fluorescein isothiocyanate, or Texas Red filters (Omega Optical, Brattleboro, VT) utilizing Zeiss 100x 1.4 NA Plan Apo objective lens. Images were captured using an Orca ER CCD camera (Hamamatsu Photonics, Japan) and analyzed with OpenLAB software suite (Agilent Technologies, Santa Clara, CA). Pie chart were made using Excel (Microsoft Corporation, Redmond, WA).
Live cell imaging
U2OS-Phoenix cells were grown in 35-mm glass-bottom tissue culture dishes (MatTek Cultureware, Ashland, MA) and imaged on an Olympus IX71 inverted microscope (Olympus America Inc., Center Valley, PA) configured with an Ultraview spinning disk confocal system (Ultraview ERS Rapid Confocal Imager; PerkinElmer, Waltham, MA) and controlled by Volocity software (PerkinElmer, Waltham, MA) utilizing a Zeiss 40 Å/ 1.4 NA oil objective. The microscope was enclosed within a temperature- and CO2-controlled environment that maintained an atmosphere of 37°C and 3–5% humidified CO2. GFP was excited with a 488-nm laser line and mCherry was excited with a 568-nm laser line. No more than 20% per cent power in the 488-nm line was applied. A series of 2 µm optical sections were acquired every 10 min for 3–5 days. Image analyses were carried out using either Volocity (PerkinElmer, Waltham, MA) or ImageJ (National Institutes of Health, Bethesda, MD) softwares. Images presented in figures are maximum intensity projections of entire z-stacks unless otherwise indicated.
Analysis of live cell imaging data and biostatistics
Graphs in , S4 and S5 were made using Prism 5 (GraphPad Software, Inc., La Jolla, CA). Unpaired t test with Two-tailed P values and a confidence interval of 95% was performed using Prism 5 (GraphPad Software, Inc., La Jolla, CA) to analyze the time in mitosis data in Figure S4. The Pearson Chi square test was performed for the data in using Stata statistical software (StataCorp, College Station, TX, USA) to examine the likelihood of differences between groups. When the expectant cell count was less than 5, the Fisher's exact test was used instead. In , “mitotic catastrophe” was determined by live cell imaging experiments of cells undergoing mitosis. Cells that entered mitosis and subsequently died within 10 hours without chromosome segregation and with a round cell shape were counted as cells undergoing “mitotic catastrophe.” Cell death was characterized by the extensive blebbing and fragmentation of the plasma membrane followed by the detachment of the cell from the plate. These events were easily detected following the bright mitotic hSecurinDB-2xGFP signal as well as DIC microscopy.
Flow cytometry
U2OS-Phoenix cells were blocked in mitosis and harvested by mitotic shake-off. Mitotic cells were then washed twice with cold PBS, 1X (Corning Inc., Corning, NY) and then immediately fixed for 30 minutes at room temperature with 2% PFA solution containing 100 mM NaCl, 300 mM sucrose, 3 mM MgCl2, 1 mM EGTA, and 10 mM PIPES with a pH of 6.8. The fixed cells were then washed 2–3 times with PBS and stored overnight in 1–5 mls of 70% cold Ethanol at −20°C. Cells were then washed 2–3 times with cold PBS and re-suspended in 1ml of PBS containing 50μg/ml Propidium Iodide and 100μg/ml RNAse and incubated in the dark for 30 mins at RT. Samples were then run on BD FACSCalibur (BD Biosciences, San Jose, CA) and analyzed with FLOWJO, LLC data analysis software (Ashland, OR).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Author contributions
S.M., A.A., and M.D. designed and analyzed the experiments. V.L. provided the CHO cells carrying the alphoid tetO-HAC and had some input in experimental analysis. S.M. and N.S. executed and analyzed the flow cytometry experiment. S.M. executed all remaining experiments. S.M. and MD wrote the manuscript with input from all authors.
Supplemental Files
Download MS Word (756.9 KB)Acknowledgments
We thank all members of the Dasso and Larionov laboratories for helpful discussions. We thank Artem Kononenko for providing the p#264-GFP knot linker CAGpr vector and help with HAC reengineering techniques, Alexander Samoshkin for providing the TetR-f-GFP_HyTK vector and technical help, and Hee-Sheung Lee for help with MMCT. We thank Dr. Malte Renz (Albert Einstein College of Medicine) for providing the tandem GFP plasmids. We thank Dr. Talar Markossian (Loyola University Medical Center) for helpful discussions regarding statistical analysis and for assistance in running Stata analysis. This work was supported through NICHD Intramural Project HD008954.
Related Research Data
References
- Lengauer C, Kinzler KW, Vogelstein B. Genetic instability in colorectal cancers. Nature 1997; 386:623-7; PMID:9121588; http://dx.doi.org/10.1038/386623a0
- Lengauer C, Kinzler KW, Vogelstein B. Genetic instabilities in human cancers. Nature 1998; 396:643-9; PMID:9872311; http://dx.doi.org/10.1038/25292
- Pfau SJ, Amon A. Chromosomal instability and aneuploidy in cancer: from yeast to man. EMBO Reports 2012; 13:515-27; PMID:22614003; http://dx.doi.org/10.1038/embor.2012.65
- Holland AJ, Cleveland DW. Losing balance: the origin and impact of aneuploidy in cancer. EMBO Reports 2012; 13:501-14; PMID:22565320; http://dx.doi.org/10.1038/embor.2012.55
- McGranahan N, Burrell RA, Endesfelder D, Novelli MR, Swanton C. Cancer chromosomal instability: therapeutic and diagnostic challenges. EMBO Reports 2012; 13:528-38; PMID:22595889; http://dx.doi.org/10.1038/embor.2012.61
- Weaver BA, Silk AD, Montagna C, Verdier-Pinard P, Cleveland DW. Aneuploidy acts both oncogenically and as a tumor suppressor. Cancer Cell 2007; 11:25-36; PMID:17189716; http://dx.doi.org/10.1016/j.ccr.2006.12.003
- Swanton C, Nicke B, Schuett M, Eklund AC, Ng C, Li Q, Hardcastle T, Lee A, Roy R, East P, et al. Chromosomal instability determines taxane response. Proc Natl Acad Sci U S A 2009; 106:8671-6; PMID:19458043; http://dx.doi.org/10.1073/pnas.0811835106
- Pavelka N, Rancati G, Zhu J, Bradford WD, Saraf A, Florens L, Sanderson BW, Hattem GL, Li R. Aneuploidy confers quantitative proteome changes and phenotypic variation in budding yeast. Nature 2010; 468:321-5; PMID:20962780; http://dx.doi.org/10.1038/nature09529
- Pariente N. A balancing act: focus on aneuploidy. EMBO Reports 2012; 13:472; PMID:22653484; http://dx.doi.org/10.1038/embor.2012.66
- Heim S, Mitelman F. Cancer cytogenetics New York: Wiley-Liss, 1995.
- Janssen A, Kops GJ, Medema RH. Elevating the frequency of chromosome mis-segregation as a strategy to kill tumor cells. Proc Natl Acad Sci U S A 2009; 106:19108-13; PMID:19855003; http://dx.doi.org/10.1073/pnas.0904343106
- Janssen A, Kops GJ, Medema RH. Targeting the mitotic checkpoint to kill tumor cells. Hormones Cancer 2011; 2:113-6; PMID:21475725; http://dx.doi.org/10.1007/s12672-010-0059-x
- Thompson SL, Bakhoum SF, Compton DA. Mechanisms of chromosomal instability. Curr Biol 2010; 20:R285-95; PMID:20334839; http://dx.doi.org/10.1016/j.cub.2010.01.034
- Gordon DJ, Resio B, Pellman D. Causes and consequences of aneuploidy in cancer. Nat Rev Genetics 2012; 13:189-203; PMID:22269907.
- Thompson SL, Compton DA. Examining the link between chromosomal instability and aneuploidy in human cells. J Cell Biol 2008; 180:665-72; PMID:18283116; http://dx.doi.org/10.1083/jcb.200712029
- Bakker B, van den Bos H, Lansdorp PM, Foijer F. How to count chromosomes in a cell: An overview of current and novel technologies. Bio Essays 2015; 37(5):570-7.
- Thompson SL, Compton DA. Chromosome missegregation in human cells arises through specific types of kinetochore-microtubule attachment errors. Proc Natl Acad Sci U S A 2011; 108:17974-8; PMID:21997207; http://dx.doi.org/10.1073/pnas.1109720108
- Bergmann JH, Martins NM, Larionov V, Masumoto H, Earnshaw WC. HACking the centromere chromatin code: insights from human artificial chromosomes. Chromosome Res 2012; 20:505-19; PMID:22825423; http://dx.doi.org/10.1007/s10577-012-9293-0
- Kouprina N, Earnshaw WC, Masumoto H, Larionov V. A new generation of human artificial chromosomes for functional genomics and gene therapy. Cell Mol Life Sci 2013; 70:1135-48; PMID:22907415; http://dx.doi.org/10.1007/s00018-012-1113-3
- Nakano M, Cardinale S, Noskov VN, Gassmann R, Vagnarelli P, Kandels-Lewis S, Larionov V, Earnshaw WC, Masumoto H. Inactivation of a human kinetochore by specific targeting of chromatin modifiers. Dev Cell 2008; 14:507-22; PMID:18410728; http://dx.doi.org/10.1016/j.devcel.2008.02.001
- Lee HS, Lee NC, Grimes BR, Samoshkin A, Kononenko AV, Bansal R, Masumoto H, Earnshaw WC, Kouprina N, Larionov V. A new assay for measuring chromosome instability (CIN) and identification of drugs that elevate CIN in cancer cells. BMC Cancer 2013; 13:252; PMID:23694679; http://dx.doi.org/10.1186/1471-2407-13-252
- Lee HS, Lee NC, Kouprina N, Kim JH, Kagansky A, Bates S, Trepel JB, Pommier Y, Sackett D, Larionov V. Effects of Anticancer Drugs on Chromosome Instability and New Clinical Implications for Tumor-Suppressing Therapies. Cancer Res 2016; 76:902-11; PMID:26837770; http://dx.doi.org/10.1158/0008-5472.CAN-15-1617
- Kim JH, Lee HS, Lee NC, Goncharov NV, Kumeiko V, Masumoto H, Earnshaw WC, Kouprina N, Larionov V. Development of a novel HAC-based “gain of signal” quantitative assay for measuring chromosome instability (CIN) in cancer cells. Oncotarget 2016; 7(12):14841-14856.
- Barford D. Structure, function and mechanism of the anaphase promoting complex (APC/C). Quarterly Rev Biophys 2011; 44:153-90; http://dx.doi.org/10.1017/S0033583510000259
- Zou H, McGarry TJ, Bernal T, Kirschner MW. Identification of a vertebrate sister-chromatid separation inhibitor involved in transformation and tumorigenesis. Science 1999; 285:418-22; PMID:10411507; http://dx.doi.org/10.1126/science.285.5426.418
- Musacchio A, Salmon ED. The spindle-assembly checkpoint in space and time. Nat Rev Mol Cell Biol 2007; 8:379-93; PMID:17426725; http://dx.doi.org/10.1038/nrm2163
- Janssen A, Medema RH. Mitosis as an anti-cancer target. Oncogene 2011; 30:2799-809; PMID:21339734; http://dx.doi.org/10.1038/onc.2011.30
- Weaver BA. How Taxol/paclitaxel kills cancer cells. Mol Biol Cell 2014; 25:2677-81; PMID:25213191; http://dx.doi.org/10.1091/mbc.E14-04-0916
- Brito DA, Rieder CL. The ability to survive mitosis in the presence of microtubule poisons differs significantly between human nontransformed (RPE-1) and cancer (U2OS, HeLa) cells. Cell Motility Cytoskeleton 2009; 66:437-47; http://dx.doi.org/10.1002/cm.20316
- Ikui AE, Yang CP, Matsumoto T, Horwitz SB. Low concentrations of taxol cause mitotic delay followed by premature dissociation of p55CDC from Mad2 and BubR1 and abrogation of the spindle checkpoint, leading to aneuploidy. Cell Cycle 2005; 4:1385-8; PMID:16138009; http://dx.doi.org/10.4161/cc.4.10.2061
- Zasadil LM, Andersen KA, Yeum D, Rocque GB, Wilke LG, Tevaarwerk AJ, Raines RT, Burkard ME, Weaver BA. Cytotoxicity of paclitaxel in breast cancer is due to chromosome missegregation on multipolar spindles. Sci Translational Med 2014; 6:229ra43; http://dx.doi.org/10.1126/scitranslmed.3007965
- Santaguida S, Tighe A, D'Alise AM, Taylor SS, Musacchio A. Dissecting the role of MPS1 in chromosome biorientation and the spindle checkpoint through the small molecule inhibitor reversine. J Cell Biol 2010; 190:73-87; PMID:20624901; http://dx.doi.org/10.1083/jcb.201001036
- Lampson MA, Kapoor TM. Unraveling cell division mechanisms with small-molecule inhibitors. Nat Chem Biol 2006; 2:19-27; PMID:16408087; http://dx.doi.org/10.1038/nchembio757
- Burrell RA, McClelland SE, Endesfelder D, Groth P, Weller MC, Shaikh N, Domingo E, Kanu N, Dewhurst SM, Gronroos E, et al. Replication stress links structural and numerical cancer chromosomal instability. Nature 2013; 494:492-6; PMID:23446422; http://dx.doi.org/10.1038/nature11935
- Bakhoum SF, Kabeche L, Murnane JP, Zaki BI, Compton DA. DNA-damage response during mitosis induces whole-chromosome missegregation. Cancer Discov 2014; 4:1281-9; PMID:25107667; http://dx.doi.org/10.1158/2159-8290.CD-14-0403
- Iida Y, Kim JH, Kazuki Y, Hoshiya H, Takiguchi M, Hayashi M, Erliandri I, Lee HS, Samoshkin A, Masumoto H, et al. Human artificial chromosome with a conditional centromere for gene delivery and gene expression. DNA Res 2010; 17:293-301; PMID:20798231; http://dx.doi.org/10.1093/dnares/dsq020
- Kim JH, Ebersole T, Kouprina N, Noskov VN, Ohzeki J, Masumoto H, Mravinac B, Sullivan BA, Pavlicek A, Dovat S, et al. Human gamma-satellite DNA maintains open chromatin structure and protects a transgene from epigenetic silencing. Genome Res 2009; 19:533-44; PMID:19141594; http://dx.doi.org/10.1101/gr.086496.108
- Kim JH, Kononenko A, Erliandri I, Kim TA, Nakano M, Iida Y, Barrett JC, Oshimura M, Masumoto H, Earnshaw WC, et al. Human artificial chromosome (HAC) vector with a conditional centromere for correction of genetic deficiencies in human cells. Proc Natl Acad Sci U S A 2011; 108:20048-53; PMID:22123967; http://dx.doi.org/10.1073/pnas.1114483108
- Tomizuka K, Yoshida H, Uejima H, Kugoh H, Sato K, Ohguma A, Hayasaka M, Hanaoka K, Oshimura M, Ishida I. Functional expression and germline transmission of a human chromosome fragment in chimaeric mice. Nat Genetics 1997; 16:133-43; PMID:9171824; http://dx.doi.org/10.1038/ng0697-133