
ABSTRACT
To this day, glioblastoma (GBM) remains an incurable brain tumor. Previous research has shown that metformin, an oral anti-diabetic drug, may decrease GBM cell proliferation and migration especially in brain tumor initiating cells (BTICs). As transforming growth factor β 2 (TGF-β2) has been reported to promote high-grade glioma and is inhibited by metformin in other tumors, we explored whether metformin directly interferes with TGF-β2-signaling. Functional investigation of proliferation and migration of primary BTICs after treatment with metformin+/−TGF-β2 revealed that metformin doses as low as 0.01 mM metformin thrice a day were able to inhibit proliferation of susceptible cell lines, whereas migration was impacted only at higher doses. Known cellular mechanisms of metformin, such as increased lactate secretion, reduced oxygen consumption and activated AMPK-signaling, could be confirmed. However, TGF-β2 and metformin did not act as functional antagonists, but both rather inhibited proliferation and/or migration, if significant effects were present. We did not observe a relevant influence of metformin on TGF-β2 mRNA expression (qRT-PCR), TGF-β2 protein expression (ELISA) or SMAD-signaling (Western blot). Therefore, it seems that metformin does not exert its inhibitory effects on GBM BTIC proliferation and migration by altering TGF-β2-signaling. Nonetheless, as low doses of metformin are able to reduce proliferation of certain GBM cells, further exploration of predictors of BTICs' susceptibility to metformin appears justified.
Introduction
Glioblastoma (GBM) is the most common primary malignant brain tumor. It is associated with a devastating prognosis, with median overall survival ranging from 14.6 to 26.3 months in clinical studies.Citation1,2 GBM is characterized by highly invasive growth,Citation3 enhanced angiogenesisCitation4 and severe local immunosuppression.Citation5 The resistance to current therapy is supposedly driven by brain tumor initiating cells (BTIC), a cellular fraction that is able to self-renew and to differentiate into different cell lineages.Citation6
Transforming growth factor beta (TGF-β) is a pivotal immunosuppressive factor in GBM.Citation7 Although both isoforms, TGF-β1 and TGF-β2, are expressed in GBM tissue,Citation8 TGF-β2 seems to be more important for pathogenesis.Citation9 TGF-β2 can both inhibit or activate proliferation and migration of BTICs, with more predominant effects on mesenchymal BTICs.Citation10 TGF-β2 also activates angiogenesisCitation11 and exerts immunosuppressive functions, which promote tumor progressionCitation10 and are likely to dominate in vivo.Citation12 Also, strong expression of TGF-β2 seems to be associated with inferior patient survival.Citation13
Metformin is a widely prescribed antidiabetic drug.Citation14 In addition to its blood glucose-lowering effect, metformin may also exert antineoplastic effects.Citation15 In glioma, metformin has been shown to inhibit proliferationCitation16-20 and invasion,Citation21 to induce apoptosis,Citation16,18,19 autophagyCitation18 and differentiation of BTICs,Citation22 and to increase radio- or chemosensitivity,Citation18,19,23,24 while leaving mature neurons largely unaffected.Citation25 Metformin mainly acts by inhibition of complex-I of the respiratory chain,Citation26 thereby leading to altered AMPK and mTOR-signaling.Citation18,19
The effects of metformin on TGF-β have been investigated in a number of previous studies. Most of the published studies reported inhibitory effects of metformin on mRNACitation27-30 and protein levels of TGF-β,Citation29,31,32 as well as SMAD-signalingCitation29,33,34 mainly in the context of epithelial-to-mesenchymal transition or fibrosisCitation27,28,35-43 in breast,Citation27,28,35 prostate,Citation36 kidney,Citation33,37-41 liver,Citation29 heartCitation42,43 or uterine/ovarian tissue.Citation31 The effects of metformin on TGF-β in glioma cells have not been investigated thus far, which encouraged us to explore whether metformin exerts its effects by modulating TGF-β2-signaling.
Results
Primary cell lines were used as an in vitro model system
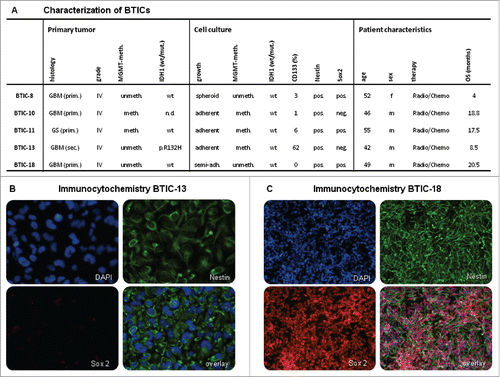
The five different BTIC lines used here were derived from patients who had undergone resection of GBM in the Neurosurgery Department at the University Hospital Regensburg. Primary cell lines were established and used in low passage numbers (P3-P20) to assure maximum resemblance to original tumor cells. For verification of stemness, BTICs were grown in the absence of fetal calf serum (FCS). Tumor take was assessed in 4 out of 5 cell lines with positive results (data not shown). Marker expression was investigated showing Nestin expression in all cell lines indicating stemness, while the stem cell marker CD133 was expressed heterogeneously in 0 to 62% of the cells (). The neural stem cell marker Sox2 was expressed in BTIC-8, −11 and −18, but not in BTIC-10 and −13 (). Clonogenicity and the ability to differentiate after addition of 10% FCS to the cell culture media were confirmed for all BTICs (data not shown). Overall, the ability of BTICs to proliferate in the absence of FCS combined with marker expression, clonogenicity and tumor take in vivo verified that the cell lines used were primary brain tumor initiating cells capable of self-renewal and invasion.
Figure 1. Characterization of BTICs. (A) Cell culture and patient characteristics of the BTICs used in this study. All BTICs were primary BTICs of glioblastoma. MGMT methylation status did not change for most BTIC cell lines after culturing. All BTICs were Nestin positive indicating neurogenesis. (B and C) Immunocytochemistry of BTIC-13 and BTIC-18. These cell lines were chosen exemplarily due to their different endogenous TGF-β2 expression (see ). Both were Nestin positive; however, Sox2, a neural stem cell marker, was expressed only in cell line BTIC-18.
The effects of metformin on proliferation were explored using cell counts and crystal violet assays at a 48-h time point according to relevant literature.Citation16,17 Migration was investigated in spheroid assays at a 20-h time point and in a brain slice culture model. As SD-208, a TGF-β2 receptor blocker, was dissolved in DMSO, we performed DMSO controls. Comparing DMSO controls to medium controls (Fig. S1) shows that DMSO did not significantly influence proliferation and migration.
Metformin inhibits proliferation and migration of BTICs
We investigated the effects of different doses of metformin on proliferation and migration. Proliferation was assessed after 48 h to ensure sufficient proliferation while simultaneously avoiding cell death due to high confluence. Migration was analyzed after 20 h to avoid bias due to excessive proliferation. The reduction of proliferation and migration caused by metformin was dose-dependent and cell line-dependent. High doses of metformin (10 mM) inhibited proliferation in 2 out of 5 examined cell lines ( and Fig. S2) and migration in 3 out of 5 ( and Fig. S2). Metformin in low doses was able to reduce proliferation in 1 out of 5 cell lines assessed by cell counting (), indicating that some GBM cell lines (BTIC-18) are more susceptible to the anti-proliferative effects of metformin than others. Regarding migration, low doses of metformin did not produce significant effects, except for a slight but significant increase in migration of BTIC-18 (). Although in BTIC-13 no anti-migratory effect was observed in spheroid assays after 20 h, long-term effects of 10 mM metformin could be seen in the OBSC, when cells were retreated with 10 mM metformin every other day (). Of note, BTIC-18 stayed close to the initial implantation site, whereas BTIC-13 migrated farther, which made changes after metformin treatment more obvious for BTIC-13.
Figure 2. Effects of metformin on proliferation and migration of BTICs. (A and B) Normalized proliferation of BTIC-13 and BTIC-18 was assessed using cell counts. Absolute proliferation values after 48 h were divided by the 0-h value to obtain a proliferation rate. The proliferation rate of any condition was subsequently normalized to the proliferation of the medium control. (C and D) Spheroid migration of BTIC-13 and BTIC-18 after 20 h. Spheres of BTICs were created 2 d prior to treatment with metformin and photographed after 20 h. The 20-h time point was used to explore migration without proliferation bias. Normalized migration was calculated by dividing the absolute value of the area covered in cells after 20 h by the 0-h area. Afterwards, the migration rates under each condition were normalized to the migration rate of the medium control. (E) Migration of BTIC-13 and BTIC-18 on organotypic brain slice cultures (OBSC) after 14 d of treatment with metformin compared to a medium control. BTIC-13 was lentivirally transduced using a U57 pHR SFFV GFP plasmid (green), while BTIC-18 was transduced with pLenti-H1-(shRNA-Neg-control)-Rsv(RFP-Bsd) (red). Two-day-old spheres of these transduced BTICs were transferred onto the same rat brain slice and monitored at 10× magnification under a fluorescent microscope after 0 and 14 d.
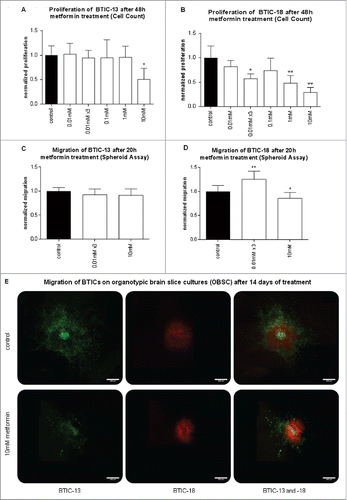
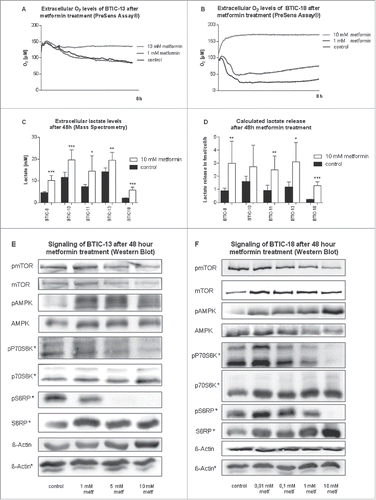
Figure 3. Metabolic effects of metformin on BTICs. (A and B) Extracellular oxygen levels of BTIC-13 and BTIC-18 during 8 h of metformin treatment as determined by a PreSens Assay®. (C and D) Extracellular lactate levels and lactate release per cell per hour corrected for proliferation after 48 h. The average increase in absolute extracellular lactate under 10 mM metformin over the cell lines tested was approximately 5 mM. (E and F) Signaling of BTIC-13 and BTIC-18 after 48 h metformin treatment investigated by Western blot. Confirming previous results, metformin acted as an AMPK-activator and mTOR inhibitor in a dose-dependent manner in both BTIC-13 and -18.
Overall, metformin reduced proliferation significantly in one of 5 cell lines investigated at a dose as low as 0.01 mM thrice a day.
Known mechanisms of metformin's action could be confirmed
Next, we investigated the mechanisms of metformin's action. Using a PreSens Assay®, we confirmed that metformin decreased oxygen consumption in both BTIC-13 and BTIC-18 prior to any obvious changes in proliferation (). Extracellular lactate levels were on average 5 mM higher after 48 h of exposure to 10 mM metformin (). Extracellular lactate did not increase significantly after treatment with 0.01 mM metformin thrice a day (data not shown). After adjustment for proliferation, 4 out of 5 cell lines still secreted significantly more lactate under 10 mM metformin than in the absence of metformin (). Western blots of known signaling pathways of metformin showed a dose-dependent activation of AMPK and inhibition of mTOR with downstream inhibition of P70S6 kinase (P70S6K) and plastid ribosomal protein S6 (S6RP) (). Higher concentrations of metformin were used for the less susceptible BTIC-13.
Metformin does not exert its effects by directly or indirectly altering TGF-β2-signaling
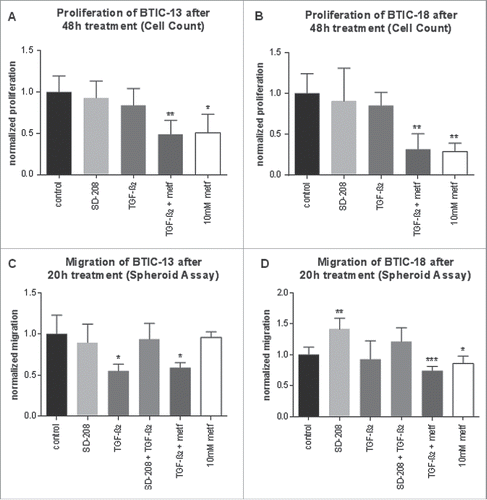
We studied functional, direct and indirect effects of metformin, TGF-β2 and the combination thereof on BTICs. Functionally, there were no differences between the effects of TGF-β1 and TGF-β2. Proliferation measurements in crystal violet assays (Fig. S3A and B) matched the results obtained by cell counting (). There was no significant increase in cytotoxicity after treatment with TGF-β2, but treatment with TGF-β1 led to a slight increase in LDH release (Fig. S3C and D). TGF-β1 and TGF-β2 had no effects on proliferation, but greatly reduced migration in BTIC-13, albeit not in BTIC-18 (). TGF-β2 (10 ng/mL) alone reduced proliferation and migration in 1 out of 5 cell lines. Some cell lines (BTIC-11, -13) were more susceptible to TGF-β2 anti-migratory effects than others ( and Fig. S4). Its antagonist, SD-208 (1 µM), had almost no significant effect on proliferation or migration of BTICs except for an increase in migration in BTIC-18. Hence, the effects of TGF-β2 were either non-significant or, similar to metformin, anti-proliferative and anti-migratory. Interestingly, cell lines strongly affected by metformin (BTIC-8 and -18) were less affected by TGF-β2, while those strongly impaired by TGF-β2 (BTIC-11, -13) reacted less to metformin. Comparing the combination of 10 ng/mL TGF-β2 and 10 mM metformin with their separate administration showed that in those cell lines sensitive to metformin (BTIC-8 and -18), the extent of the anti-proliferative and anti-migratory effects of the combination was very similar to that of metformin alone ( and Fig. S4). In all tested cell lines, a heterogeneous pattern was observed ranging from equal effects of TGF-β2 alone and its combination with metformin (reduction of migration of BTIC-13) to additive effects of the combination (reduction of proliferation of BTIC-10 and -11) ( and Fig. S4). Thus, TGF-β2 was not functionally antagonized by metformin, but rather exerted independent anti-migratory and/or anti-proliferative effects.
Figure 4. Functional effects of metformin and TGF-β2 on proliferation and migration of BTICs. (A and B) Normalized proliferation of BTIC-13 and BTIC-18 was assessed using cell counts. Absolute proliferation values after 48 h were divided by the 0-h value to obtain a proliferation rate. The proliferation rate of any condition was subsequently normalized to the proliferation of the medium control. (C and D) Spheroid migration of BTIC-13 and BTIC-18 after 20 h. Spheres of BTICs were created 2 d prior to treatment with metformin and photographed after 20 and 48 h. The 20-h time point was used to explore migration in order to avoid proliferation bias. Normalized migration was calculated by dividing the absolute value of the area covered in cells after 20 h by the 0-h area. Afterwards, the migration rates under each condition were normalized to the migration rate of the medium control.
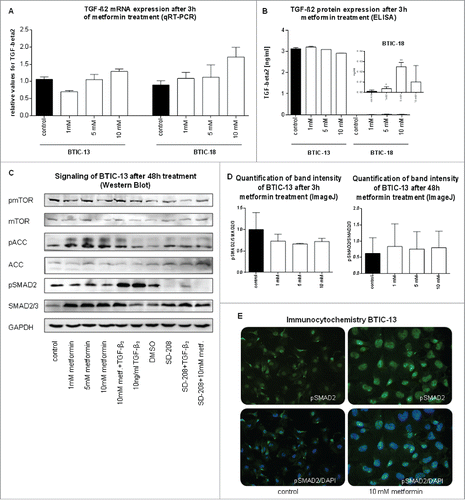
In order to investigate whether metformin directly influenced TGF-β2 mRNA, protein levels, and signaling, we harvested BTIC-13 and -18 after 3 h and 48 h of metformin treatment. Firstly, quantitative real-time PCR showed TGF-β2 mRNA expression to remain stable under treatment with different concentrations of metformin (). Secondly, endogenous TGF-β2 protein levels differed substantially between BTIC-13 and BTIC-18. The former yielded 3,000-fold higher TGF-β2 levels than BTIC-18 regardless of the concentration of metformin (). TGF-β2-signaling in the SMAD2 pathway was not influenced by metformin as demonstrated by Western blot and immunocytochemistry (, Fig. S5). To estimate levels of active AMPK, we investigated the downstream molecule pACC. Here, TGF-β2 served as a positive control increasing SMAD2-signaling without influencing metformin signaling pathways of mTOR or ACC. Metformin, on the other hand, altered mTOR- and ACC-signaling without affecting SMAD-signaling. Effects of metformin on mTOR-signaling were more pronounced in BTIC-18 than BTIC-13. As expected, SD-208 inhibited TGF-β2- signaling while pSMAD2 expression remained stable when metformin was added to SD-208 (, Fig. S5). Hence, it appears that the signaling pathways of metformin and TGF-β2 are independent of each other and that metformin exerts no direct effect on TGF-β2-signaling in GBM BTICs.
Figure 5. Direct effects of metformin on TGF-β2 mRNA levels, extracellular protein expression and SMAD-signaling. (A) TGF-β2 mRNA expression after 3 h of metformin treatment as observed by qRT-PCR. TGF-β2-levels were normalized to those of the medium control. (B) TGF-β2 protein expression after 3 h of metformin treatment as determined by ELISA. Overall, the protein expression of TGF-β2 was much higher in BTIC-13 than BTIC-18. Therefore, the slight increase in TGF-β2 protein level after treatment of BTIC-18 with 5 mM metformin might be due to very low base line levels. (C) Western blot of metformin and TGF-β2 -signaling of BTIC-13 after 48 h of treatment. Pellets from cell counts were employed for Western blotting. (D) Quantification of Western blot band intensity of BTIC-13 after 3 h and 48 h of metformin treatment using ImageJ. (E) Immunocytochemistry of BTIC-13 did not detect a change in pSMAD2 expression despite metformin-induced changes in morphology.
Lastly, we investigated whether indirect effects of metformin might influence TGF-β2-signaling. Metformin administered at millimolar concentrations is known to increase extracellular lactate levels (), thereby causing acidification, which in turn may activate TGF-β2-signaling.Citation44,45 Therefore, we analyzed the effects of lactic acid on TGF-β2-signaling in BTIC-18, as this was the only cell line to exhibit a slight increase in TGF-β2 mRNA and protein expression after metformin treatment (). No significant drop in extracellular pH could be observed upon addition of 3.8 mM lactic acid, which corresponded to the increase in extracellular lactate observed upon treatment of BTIC-18 with 10 mM metformin (see ). Further, after 3 h of incubation, no significant increases in mRNA or protein levels of TGF-β2 as well as SMAD-signaling could be observed (Fig. S6). Results were similar when assays were performed after 48h (data not shown).
STAT3 is known to critically regulate the growth and self-renewal of GBM stem cells beyond the TGFβ2-SMAD pathway.Citation46 We therefore investigated whether metformin inhibits proliferation and/or migration of brain tumor initiating cells by blocking STAT3-signaling. Indeed, treatment with increasing doses of metformin led to an inhibition of STAT3 phosphorylation in BTIC-13 (Fig. S7) and BTIC-18 (data not shown), which aligns with previously published data.Citation47
Discussion
Previous research has shown that metformin is able to influence GBM hallmarks via the inhibition of proliferationCitation16-19 and invasionCitation21 and the induction of apoptosis,Citation16,18,19 autophagyCitation18 and differentiation of BTICs.Citation18 However, the underlying mechanisms of metformin's action are still not fully understood. TGF-β, which is a pivotal growth factor in glioblastoma,Citation7 was shown to be inhibited by metformin in other tumor models,Citation27 but the effects of metformin on TGF-β in glioma cells have not been explored so far. Here, we could demonstrate that metformin did not act as a TGF-β2 antagonist in our in vitro BTIC model. Nevertheless, 10 mM metformin decreased proliferation and migration in some BTICs. These findings are in line with current literature, especially with Würth et al.,Citation17 who reported an IC50 of approximately 9 mM metformin after 48-h treatment. Further, we were able to show for the first time that doses as low as 0.01 mM metformin in triple retreatment were able to significantly reduce proliferation of a susceptible BTIC line, even when investigated under high-glucose conditions, which likely reduces sensitivity to metformin.Citation22 0.01 mM is a concentration that may actually be reached in the brain of a human diabetic on metformin. In a rat model, Łabuzek et al.Citation48 observed that after 3 weeks of oral administration of 300 mg/kg metformin per day, a concentration of 0.044 mM could be measured in the cerebrospinal fluid (CSF). Wilcock and BaileyCitation49 investigated metformin uptake in a diabetic mouse model after oral administration of 50 mg/kg metformin and measured a drug concentration of 0.012 mmol/kg wet weight in the brain. In diabetes treatment, metformin is given at doses as high as 3000 mg/day (around 43 mg/kg for a 70 kg patient).Citation50 Thus, based on mouse data, brain tissue concentrations of 0.01 mM metformin might actually be achieved in human patients. Ongoing clinical trials use 1000 mg metformin twice a day in combination with temozolomide with or without memantine and/or mefloquine (https://clinicaltrials.gov/ct2/show/NCT01430351; NCT02149459). As metformin is well tolerated in patients and selectively affects BTICs,Citation18 higher doses than in diabetic treatment should be explored in clinical trials of GBM.
Importantly, we observed that TGF-β2 alone either exerted no effects or decreased proliferation and migration, which is in line with prior data.Citation10,51 Comparing the effects on proliferation and migration of metformin and TGF-β2 alone and in combination did not suggest an interaction between the 2 agents. Nonetheless, other authors have proposed an antagonistic effect of metformin and TGF-β2 in the context of the epithelial-to-mesenchymal transition.Citation37,41,52-54 While some of these data were only descriptive, other authors proposed mechanisms of a direct interaction between metformin and TGF-β such as AMPK activation through metformin and a subsequent reduction of TGF-β-induced IL-6 transcriptionCitation54 or diminished ROS production.Citation52 In these studies, mainly differentiated fibrocytes, liver, and kidney cells were used and – in contrast to our study - TGF-β1. These studies proposed that metformin activated AMPK, which in turn decreased the phosphorylation of SMAD2/3 and, consequently, lowered expression of TGF-β-inducible genes such as fibronectin and IL-6.Citation53 In contrast to these findings, metformin did not significantly influence TGF-β2 mRNA and protein levels or SMAD-signaling in our BTIC lines. BTIC-18 showed slightly enhanced TGF-β2 mRNA and protein expression after metformin treatment. However, BTIC-18 has very low basal TGF-β2 protein levels, and doses as high as 20 ng/mL TGF-β2 did not result in functionally relevant changes. Thus, the relevance of significant, but very small changes in TGF-β2 expression is questionable, especially when SMAD-signaling is not altered. Yet, changes in alternative signaling pathways cannot be fully excluded. For another BTIC line (BTIC-11), a possible over-additive effect of TGF-β2 and metformin was observed. Pondering these results, we were concerned that increased levels of lactic acid, which readily dissociates into protons and lactate under physiological conditions, might cause activation of TGF-β2 via a decrease in tissue pH. However, even treatment with concentrations of lactic acid observed at the maximum dose of metformin (10 mM), a concentration unlikely to be reached in human brain tissue, did not affect TGF-β2 mRNA or protein expression.
Our in vitro data suggest that metformin and TGF-β2-signaling function independently of each other in GBM BTICs. In vivo, interactions become more complex as TGF-β2 is an important inducer of angiogenesisCitation11 and diminishes immune responses,Citation10 raising the question of how metformin influences them. Adequate in vivo models may help to elucidate the interactions of metformin and TGF-β2 in a more complex environment. In addition, the effects of metformin on STAT3-signaling, another pivotal pathway in glioma,Citation46 should be investigated in more detail.
In summary, our data suggest that metformin's anti-migratory and anti-proliferative effects are not based on direct inhibition of TGF-β2-signaling. Further studies are needed to investigate whether metformin is sufficiently effective in treating glioma at clinically relevant doses and whether there are predictors for sensitivity to metformin. Additionally, it should be explored, whether glioma patients can tolerate higher doses of metformin than currently prescribed for the treatment of type 2 diabetes without causing severe lactic acidosis.
Materials and methods
Tumor specimens and enrichment of BTICs
Upon approval by the ethics committee of the University of Regensburg (No° 11-103-0182) and after receipt of written informed consent, BTICs were established from resected, untreated human malignant gliomas as previously described.Citation55 The local neuropathology department established the diagnosis and WHO grading. Routine histopathology was accompanied by testing for IDH1 mutation (by pyrosequencing or conventional Sanger sequencing) and MGMT promoter methylation status (by methylation specific PCRCitation56). Clinical parameters such as age, gender, type of treatment, and overall survival (according to the RANO criteria) were available for all patients.
For enrichment of BTICs, resected tumor specimens were mechanically (and partly also enzymatically) dissociated, washed with PBS and passed through a cell strainer with 30-µm pore size to obtain a single cell suspension (BD, #352235). BTICs were selected by their ability to proliferate in RHBA-based serum-free culture media (Takara, #Y40001) supplemented with 20 ng/mL of the mitogens EGF (Miltenyi Biotec, #130-097-751) and bFGF (Miltenyi Biotec, #130-093-842), as described.Citation57 Progenitor features of BTIC lines were verified by flow cytometryCitation10 (CD133, CD15, CD44, A2B5), by immunocytochemistry (Nestin, Sox2, GFAP), by clonogenicity assays, and partly by tumor take assays in an immunocompromised mouse model. Cells were incubated at 37°C, 5% CO2, 95% humidity in a standard tissue culture incubator.
Treatment of BTICs with metformin, TGF-β2, TGF-β1 or SD-208
Metformin hydrochloride (Sigma-Aldrich #PHR1084) was dissolved in RHBA medium to yield a 100 mM stock solution. Mammalian TGF-β1 and TGF-β2 (Peprotech #100–21, #100-35B) were dissolved in 10 mM citric acid/0.2% BSA and PBS/0.2% BSA, respectively, to yield stock solutions of 5 µg/mL. TGF-β1 and TGF-β2 were used at a concentration of 10 ng/mL in all assays. SD-208, an inhibitor of the TGFβRII-kinase (Tocris #3269), was dissolved in DMSO to obtain a 10 mM stock solution and used at a concentration of 1 µM.
For all functional assays, cell culture medium was carefully removed 48 h after seeding and then replaced with media containing the respective substance concentrations, or fresh cell culture medium for the controls. For SD-208, we used DMSO as a control in a corresponding concentration. Metformin was also used in triple retreatment, which means addition of metformin to cell culture media thrice a day.
Determination of long-term oxygen consumption under culture conditions
We used the PreSens technology (PreSens technologies, #Oxo DishOD24) to determine long-term oxygen consumption under cell culture conditions. 300,000 cells/well were seeded in 1 mL of cell culture medium on oxygen sensor-equipped 24-well plates. The following day, 500 µL of medium were carefully removed and replaced with new medium containing treatments in double concentration or control. Oxygen consumption rate was determined in µmol/L over 48 h and evaluated using Graph Pad Prism software version 5.0 (GraphPad Software, Inc.).
Determination of extracellular lactate concentrations
Measurement of extracellular lactate was performed using mass spectrometry as described.Citation58 Lactate levels were adjusted for tumor cell proliferation using a formula including the final cell number, doubling time, culture media volume and culture time according to Jain et al.Citation59
Treatment of BTICs with lactic acid
BTICs were treated with lactic acid from Fluka (#69771) at the same concentrations as measured after treatment with 10 mM metformin. Briefly, 400,000 cells were seeded in 6-cm plates and treated for 48 h with lactic acid. After 2 h and before harvest, pH was controlled. Cells and supernatants were harvested to prepare total RNA or protein as described below. Cell lysates and supernatants of untreated cells were used as controls in all assays.
pH measurement
For pH measurement, a pH-electrode (HANNA Instruments, #HI 10530) was calibrated at 37°C, and the pH of the cell supernatant was measured directly in the incubator immediately after opening the incubator and cell culture flasks.
RNA isolation
For RNA isolation, cells were incubated in 6-well plates (2×105 cells/mL). Total RNA was isolated using the Nucleo Spin RNA Plus Kit (Macherey-Nagel, #740 984.25) and reversely transcribed using AMV reverse transcriptase (Promega, #A3500) following the manufacturer's instructions.
Polymerase chain reaction (PCR) and quantitative real time PCR (qRT-PCR)
Primers to detect transcripts of interest were: TGF-β2 (forward: 5′ CAC CAT AAA GAC AGG AAC CTG -3′; reverse: 5´- GGA GGT GCC ATC AAT ACC TGC -3´, Genbank Accession No: NM_003238), and RPLPO (large ribosomal protein) (forward: 5′-CTG TCT GCA GAT TGG CTA CCC-3′, reverse: 5′-GAT GGA TCA GCC AAG AAG GC-3′, GenBank Accession No: NM_001002.3) as a housekeeping gene in all assays. Annealing temperatures were optimized for each primer pair.
Quantification of mRNA expression was performed by real-time PCR (Mx3000P Quantitative PCR [qPCR] System, Stratagene) based on SYBR-Green I fluorescence (Brilliant III Ultra Fast SYBR GREEN QPCR Master Mix, Agilent Technologies, 600883) using the ΔΔCT-method. Three serial fivefold dilutions of cDNA or a mixture of all used cDNA-samples, were amplified in duplicates to construct standard curves for both the target and the reference gene, respectively. cDNA samples of BTICs were diluted 1:10. All samples were tested in triplicate. For each reaction, melting curves were used to verify the identity of the amplification products. The target gene amount was divided by the reference (RPLPO) amount. Each of the experimental normalized values was divided by the normalized control (untreated) sample value to generate relative expression levels in fold changes.
Protein isolation
To investigate protein levels of (p)mTOR, (p)AMPK, (p)SMAD2, and GAPDH, whole-cell lysates were prepared with RIPA buffer and 30 µg each of lysate were subjected to Western blotting on a denaturing 10% SDS-PAGE.
TGF-β2 ELISA
We used the quantitative sandwich enzyme linked immunoassay technique for quantitative determination of activated human TGF-β2 concentrations in cell culture supernatants using the commercially available human TGF-β2 specific immunoassay kit (R&D Systems, #DY302). We used triplicates of each sample according to the manufacturer's instruction.
Western blot
For Western blot analysis, 30 µg of total cell lysate were diluted in Laemmli buffer, separated on a 10% SDS-PAGE gel and transferred to nitrocellulose membranes by semi-dry blotting. The membranes were blocked with 5% milk powder or 5% BSA in 0.02% Tween TBS (TBST) for 1 h. Membranes were incubated with specific monoclonal antibodies for pACC (Ser79, 1:1000, #3661), ACC (1:1000, #3676), pmTOR (Ser 2448, 1:1000, #5536), mTOR (1:1000, #2983), pAMPK (Thr 172, 1:1000, #2535), AMPK (1:1000, #2603), pSMAD (Ser 465/467, 1:500, #3101), SMAD 2/3 (1:500, #3102), pSTAT3 (Tyr 705, 1:1000, #9145), STAT3 (1:1000, #9132), pP70S6K (Thr 389, 1:1000, #9205), P70S6K (1:1000, #9202), pS6RP (Ser 235/236, 1:1000, #4858), S6RP (1:1000, #2217, all from Cell Signaling), ß-Actin (1:2000, Sigma Aldrich, #A 5316) or GAPDH (1:2000, #sc-48167 from Santa-Cruz) overnight at 4°C. Immunocomplexes were visualized using horseradish peroxidase-conjugated antibodies (goat anti-rabbit (Advansta #R-05072-500), donkey anti-goat (Santa-Cruz, #sc-2020)) followed by enhanced chemoluminescence (Western Bright Sirius ECL, Biozym #541–021). Western blots were repeated 3 times and quantified using ImageJ, version 1.49.Citation60
Immunocytochemistry
For staining of Nestin, Sox2, pSMAD2 and DAPI, 15,000 cells/well were seeded in laminin-coated coverslips (Fisher Scientific, #1023–4121). 48 h after treatment, supernatants were discarded and cells were fixed with paraformaldehyde for 10 min. Cells were washed 3 times with PBS and blocked with blocking buffer (PBS, 10% donkey serum, 1% BSA, 0.1% Triton-X) for 2 h. Coverslips were incubated with primary antibodies (Nestin: 1:500, Millipore, # Mab5326; Sox2: 1:500, Santa Cruz, #17320; pSMAD2: 1:50, Ser 465/467, Cell Signaling, #3104; SMAD2 1:50, Cell Signaling, #3102) at 4°C overnight, washed thrice with blocking buffer before incubation with secondary antibodies (anti rabbit (GFP), #A21206; anti mouse (TxRED), #A10037; all from Invitrogen), and DAPI (Sigma, #D9542) for nuclear staining for 1 h. Coverslips were washed successively 3 times with blocking buffer and PBS, respectively, before they were mounted and dried overnight. Fluorescence microscopy was performed using the Zeiss Axio Observer.Z1 microscope (Zeiss Axio Observer.Z1, Visitron Systems GmbH, #3834003816). Quantitative and qualitative evaluation of staining was performed by counting stained and DAPI-positive cells in 4 visual fields using a fixed exposure time. Additionally, we compared optimal exposure times for each protein.
Proliferation assays
Cell count
Proliferation of BTICs was determined by counting viable cells using a Neubauer chamber. 150,000 cells/well were seeded in 6-well plates and allowed to adhere for 48 h prior to treatment. After another 48 h, cells were collected and counted. Non-viable cells were identified by trypan blue staining. Experiments were performed in triplicate.
Crystal violet assay
For crystal violet staining, 2,500 (BTIC-13) or 5,000 (BTIC-18) cells/well were seeded on laminin-coated 96-well plates to reduce cell loss due to cell detachment. 48 h after treatment, medium was exchanged with 0.5% crystal violet (Sigma, #C3886) in 20% aqueous methanol solution and cells were stained for 10 min. After washing and drying, the crystal violet was extracted from live cells by the addition of 0.1 M sodium citrate in 50% ethanol for 30 min on a rotating shaker and measured at 550 nm (VarioSkan Flash Multimode Reader, Thermo Scientific, # 5250030). Assays were performed in triplicate.
Cell death assays
LDH activity measurement
We used the LDH cytotoxicity assay (Promega, #G1780) according to the manufacturer's protocol. Briefly, 2500 cells (BTIC-13) or 5000 cells (BTIC-18) were seeded in 96-well plates and incubated with either the indicated concentrations of TGF-β1 (10 ng/mL), TGF-β2 (10 ng/mL), or both (10 ng/mL) for 48 h. LDH activity was then quantified with a plate reader (VarioSkan Flash Multimode Reader, Thermo Scientific, # 5250030) at 490 nm absorption.
Migration assays
Spheroid migration assay
Tumor spheroids were initiated by seeding 2,500 cells in 200 µL medium into agar-coated wells. Mature spheroids were explanted in 100 µL medium to uncoated round-bottom 96-well plates. 100 µL of treatment in double concentration were added. Spheroids were allowed to migrate for 48 h. The area covered by cells migrating away from the spheroid was photographed after 0 h, 20 h and 48 h. We calculated migration rates after 20 h, when migration started to be visible, but still relatively unaffected by proliferation. The spheroid area was measured using ImageJ. Six replicates per condition were used and assays were performed twice.
Migration of BTICs on organotypic brain slice cultures (OBSC)
Rat pups obtained from an in-house facility (Long-Ewans, Sprague Dawley or Wistar) were killed by cervical dislocation and used to obtain organotypic brain slice cultures (OBSC) at day 7. OBSC preparation was performed adapted to a described procedure.Citation61-63 For storage and cutting, brains were kept in a preparation medium containing 5% (v/v) horse serum (Gibco, #26050–088), 1.6% MEM powder (Invitrogen, #11700–077), 50 U (v/v) penicillin, 0.05% (v/v) streptomycin (Sigma, #P4333), 25 mM (w/v) D-glucose (Merck, #22700), 2 mM (w/v) L-glutamine (Sigma,#G7513) and 1 mM (w/v) Tris base (Sigma, #T1503). After removal, rat brains were embedded in 5% low-melting agarose (Biozym, #840.100) and cut into 350 µm thick axial slices on a Vibratome (VT 1000S, Leica, #047230685; settings: 3 5 speed, frequency of 8). Only those slices with completely preserved hippocampal structures were relocated onto insert membranes (0.4 µm; OMNILAB, #353090). Inserts were transferred into equilibrated 6-well plates (OMNILAB, #353046) containing 1.2 mL culture media consisting of 25% (v/v) HBSS (Gibco), 25% (v/v) horse serum, 0.8% MEM powder, 0.58% (w/v) sodium bicarbonate (Sigma, #S5761), 50 U (v/v) penicillin, 0.05% (v/v) streptomycin, 0.05% (w/v) ITS (Roche, #11074 547001), 25 mM (w/v) D-glucose, 4 mM (w/v) Vitamin C (Sigma, #A4544), 4 mM (w/v) L-glutamine, and 0.5 mM (w/v) Tris base. Brain slices were cultured in an incubator at 37°C and 5% CO2 for 24 h before implantation to let them adjust to incubation conditions.
Forty-eight h prior to implantation, lentivirally transduced BTICs were seeded onto agarose coated 96-well plates (10,000 cells/well) to allow for spheroid formation. BTIC-13 was lentivirally transduced using a U57 pHR SFFV GFP plasmid (green) while BTIC-18 was transduced with pLenti-H1-(shRNA-Neg-control)-Rsv(RFP-Bsd) (red). One spheroid per hemisphere was placed onto the lateral ventricle, facing the hippocampal formation.
In order to monitor spheroid migration, implanted spheroids were visualized at 5-fold magnification under a fluorescent microscope (Zeiss Axio Observer.Z1, Visitron Systems GmbH, #3834003816) once a week. Several pictures of one infiltration site were merged when necessary using Pixelmator software, version 3.3.2.
Statistics
We used Graph Pad Prism version 5.0 to calculate a one-way ANOVA to compare the results (mean values and SDs) of control vs. treated BTICs. We used Dunnett's post-hoc test to control for multiple comparisons. The level of significance was set at *P < 0.05, **P < 0.01, and ***P < 0.001.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
1186316_Supplemental_Material.zip
Download Zip (1.4 MB)Acknowledgments
We thank Judith Proske and Ina Weig-Meckl for excellent technical assistance. We also thank all members of the KFO 262 for critical discussions and valuable ideas regarding our project.
Related Research Data
References
- Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 2005; 352:987-96; PMID:15758009; http://dx.doi.org/10.1056/NEJMoa043330
- Stupp R, Hegi ME, Gorlia T, Erridge SC, Perry J, Hong YK, Aldape KD, Lhermitte B, Pietsch T, Grujicic D, et al. Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma with methylated MGMT promoter (CENTRIC EORTC 26071–22072 study): a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol 2014; 15:1100-8; PMID:25163906; http://dx.doi.org/10.1016/S1470-2045(14)70379-1
- Paw I, Carpenter RC, Watabe K, Debinski W, Lo HW. Mechanisms regulating glioma invasion. Cancer Lett 2015; 362:1-7; PMID:25796440; http://dx.doi.org/10.1016/j.canlet.2015.03.015
- Jhaveri N, Chen TC, Hofman FM. Tumor vasculature and glioma stem cells: Contributions to glioma progression. Cancer Lett 2014: http://dx.doi.org/10.1016/j.canlet.2014.12.028; PMID:25527451
- Avril T, Vauleon E, Tanguy-Royer S, Mosser J, Quillien V. Mechanisms of immunomodulation in human glioblastoma. Immunotherapy 2011; 3(4 Suppl):42-4; PMID:21524170; http://dx.doi.org/10.2217/imt.11.39
- Lathia JD, Mack SC, Mulkearns-Hubert EE, Valentim CL, Rich JN. Cancer stem cells in glioblastoma. Genes Dev 2015; 29:1203-17; PMID:26109046; http://dx.doi.org/10.1101/gad.261982.115
- Wick W, Naumann U, Weller M. Transforming growth factor-β: a molecular target for the future therapy of glioblastoma. Curr Pharm Des 2006; 12:341-9; PMID:16454748; http://dx.doi.org/10.2174/138161206775201901
- Riemenschneider MJ, Hirblinger M, Vollmann-Zwerenz A, Hau P, Proescholdt MA, Jaschinski F, Rothhammer-Hampl T, Wosikowski K, Janicot M, Leo E. TGF-ss isoforms in cancer: Immunohistochemical expression and Smad-pathway-activity-analysis in thirteen major tumor types with a critical appraisal of antibody specificity and immunohistochemistry assay validity. Oncotarget 2015; 6:26770-81; PMID:26450853; http://dx.doi.org/10.18632/oncotarget.5780
- Hau P, Jachimczak P, Schlaier J, Bogdahn U. TGF-beta2 signaling in high-grade gliomas. Curr Pharm Biotechnol 2011; 12:2150-7; PMID:21619538; http://dx.doi.org/10.2174/138920111798808347
- Beier CP, Kumar P, Meyer K, Leukel P, Bruttel V, Aschenbrenner I, Riemenschneider MJ, Fragoulis A, Rummele P, Lamszus K, et al. The cancer stem cell subtype determines immune infiltration of glioblastoma. Stem Cells Dev 2012; 21:2753-61; PMID:22676416; http://dx.doi.org/10.1089/scd.2011.0660
- Kuczynski EA, Patten SG, Coomber BL. VEGFR2 expression and TGF-β signaling in initial and recurrent high-grade human glioma. Oncology 2011; 81:126-34; PMID:21985798; http://dx.doi.org/10.1159/000332849
- Weller M, Fontana A. The failure of current immunotherapy for malignant glioma. Tumor-derived TGF-β, T-cell apoptosis, and the immune privilege of the brain. Brain Res 1995; 21:128-51; PMID:8866671; http://dx.doi.org/10.1016/0165-0173(95)00010-0
- Frei K, Gramatzki D, Tritschler I, Schroeder JJ, Espinoza L, Rushing EJ, Weller M. Transforming growth factor-β pathway activity in glioblastoma. Oncotarget 2015; 6:5963-77; PMID:25849941; http://dx.doi.org/10.18632/oncotarget.3467
- Ferrannini E. The target of metformin in type 2 diabetes. N Engl J Med 2014; 371:1547-8; PMID:25317875; http://dx.doi.org/10.1056/NEJMcibr1409796
- Yin M, Zhou J, Gorak EJ, Quddus F. Metformin is associated with survival benefit in cancer patients with concurrent type 2 diabetes: a systematic review and meta-analysis. Oncologist 2013; 18:1248-55; PMID:24258613; http://dx.doi.org/10.1634/theoncologist.2013-0111
- Isakovic A, Harhaji L, Stevanovic D, Markovic Z, Sumarac-Dumanovic M, Starcevic V, Micic D, Trajkovic V. Dual antiglioma action of metformin: cell cycle arrest and mitochondria-dependent apoptosis. Cell Mol Life Sci 2007; 64:1290-302; PMID:17447005; http://dx.doi.org/10.1007/s00018-007-7080-4
- Wurth R, Pattarozzi A, Gatti M, Bajetto A, Corsaro A, Parodi A, Sirito R, Massollo M, Marini C, Zona G, Fenoglio D, Sambuceti G, Filaci G, Daga A, Barbieri F, Florio T. Metformin selectively affects human glioblastoma tumor-initiating cell viability: A role for metformin-induced inhibition of Akt. Cell Cycle 2012; 12:145-56; PMID:23255107; http://dx.doi.org/10.4161/cc.23050
- Gritti M, Wurth R, Angelini M, Barbieri F, Peretti M, Pizzi E, Pattarozzi A, Carra E, Sirito R, Daga A, Curmi PM, Mazzanti M, Florio T. Metformin repositioning as antitumoral agent: selective antiproliferative effects in human glioblastoma stem cells, via inhibition of CLIC1-mediated ion current. Oncotarget 2014; 5:11252-68; PMID:25361004; http://dx.doi.org/10.18632/oncotarget.2617
- Yu Z, Zhao G, Xie G, Zhao L, Chen Y, Yu H, Zhang Z, Li C, Li Y. Metformin and temozolomide act synergistically to inhibit growth of glioma cells and glioma stem cells in vitro and in vivo. Oncotarget 2015; 6:32930-43; PMID:26431379
- Sesen J, Dahan P, Scotland SJ, Saland E, Dang VT, Lemarie A, Tyler BM, Brem H, Toulas C, Cohen-Jonathan Moyal E, Sarry JE, Skuli N. Metformin inhibits growth of human glioblastoma cells and enhances therapeutic response. PloS One 2015; 10:e0123721; PMID:25867026; http://dx.doi.org/10.1371/journal.pone.0123721
- Gao LB, Tian S, Gao HH, Xu YY. Metformin inhibits glioma cell U251 invasion by downregulation of fibulin-3. Neuroreport 2013; 24:504-8; PMID:23660683; http://dx.doi.org/10.1097/WNR.0b013e32836277fb
- Sato A, Sunayama J, Okada M, Watanabe E, Seino S, Shibuya K, Suzuki K, Narita Y, Shibui S, Kayama T, Kitanaka C. Glioma-initiating cell elimination by metformin activation of FOXO3 via AMPK. Stem Cells Transl Med 2013; 1:811-24; http://dx.doi.org/10.5966/sctm.2012-0058
- Mouhieddine TH, Nokkari A, Itani MM, Chamaa F, Bahmad H, Monzer A, El-Merahbi R, Daoud G, Eid A, Kobeissy FH, Abou-Kheir W. Metformin and Ara-a Effectively Suppress Brain Cancer by Targeting Cancer Stem/Progenitor Cells. Front Neurosci 2015; 9:442; PMID:26635517; http://dx.doi.org/10.3389/fnins.2015.00442
- Yu Z, Zhao G, Li P, Li Y, Zhou G, Chen Y, Xie G. Temozolomide in combination with metformin act synergistically to inhibit proliferation and expansion of glioma stem-like cells. Oncology Lett 2016; 11:2792-800; PMID:27073554
- Wurth R, Pattarozzi A, Gatti M, Bajetto A, Corsaro A, Parodi A, Sirito R, Massollo M, Marini C, Zona G, et al. Metformin selectively affects human glioblastoma tumor-initiating cell viability: A role for metformin-induced inhibition of Akt. Cell Cycle 2013; 12:145-56; PMID:23255107; http://dx.doi.org/10.4161/cc.23050
- Owen MR, Doran E, Halestrap AP. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J 2000; 348 Pt 3:607-14; PMID:10839993; http://dx.doi.org/10.1042/bj3480607
- Vazquez-Martin A, Oliveras-Ferraros C, Cufi S, Del Barco S, Martin-Castillo B, Menendez JA. Metformin regulates breast cancer stem cell ontogeny by transcriptional regulation of the epithelial-mesenchymal transition (EMT) status. Cell Cycle 2010; 9:3807-14; PMID:20890129; http://dx.doi.org/10.4161/cc.9.18.13131
- Cufi S, Vazquez-Martin A, Oliveras-Ferraros C, Martin-Castillo B, Joven J, Menendez JA. Metformin against TGFbeta-induced epithelial-to-mesenchymal transition (EMT): from cancer stem cells to aging-associated fibrosis. Cell Cycle 2010; 9:4461-8; PMID:21088486; http://dx.doi.org/10.4161/cc.9.22.14048
- Jung GS, Jeon JH, Choi YK, Jang SY, Park SY, Kim MK, Shin EC, Jeong WI, Lee IK, Kang YN, Park KG. Small heterodimer partner attenuates profibrogenic features of hepatitis C virus-infected cells. Liver Int 2015; 35:2233-45; PMID:25976932; http://dx.doi.org/10.1111/liv.12871
- Dong Z, Chen CX. Effect of catalpol on diabetic nephropathy in rats. Phytomedicine 2013; 20:1023-9; PMID:23746755; http://dx.doi.org/10.1016/j.phymed.2013.04.007
- Zhang X, Zhang C, Shen S, Xia Y, Yi L, Gao Q, Wang Y. Dehydroepiandrosterone induces ovarian and uterine hyperfibrosis in female rats. Hum Reprod 2013; 28:3074-85; PMID:23980058; http://dx.doi.org/10.1093/humrep/det341
- Xavier DO, Amaral LS, Gomes MA, Rocha MA, Campos PR, Cota BD, Tafuri LS, Paiva AM, Silva JH, Andrade SP, et al. Metformin inhibits inflammatory angiogenesis in a murine sponge model. Biomed Pharmacother 2010; 64:220-5; PMID:20053525; http://dx.doi.org/10.1016/j.biopha.2009.08.004
- Kim BH, Han S, Lee H, Park CH, Chung YM, Shin K, Lee HG, Ye SK. Metformin enhances the anti-adipogenic effects of atorvastatin via modulation of STAT3 and TGF-β/Smad3 signaling. Biochem Biophys Res Commun 2015; 456:173-8; PMID:25462562; http://dx.doi.org/10.1016/j.bbrc.2014.11.054
- Park IH, Um JY, Hong SM, Cho JS, Lee SH, Lee SH, Lee HM. Metformin reduces TGF-beta1-induced extracellular matrix production in nasal polyp-derived fibroblasts. Otolaryngol Head Neck Surg 2014; 150:148-53; PMID:24357397; http://dx.doi.org/10.1177/0194599813513880
- Oliveras-Ferraros C, Cufi S, Vazquez-Martin A, Torres-Garcia VZ, Del Barco S, Martin-Castillo B, Menendez JA. Micro(mi)RNA expression profile of breast cancer epithelial cells treated with the anti-diabetic drug metformin: induction of the tumor suppressor miRNA let-7a and suppression of the TGFbeta-induced oncomiR miRNA-181a. Cell Cycle 2011; 10:1144-51; PMID:21368581; http://dx.doi.org/10.4161/cc.10.7.15210
- Zhang J, Shen C, Wang L, Ma Q, Xia P, Qi M, Yang M, Han B. Metformin inhibits epithelial-mesenchymal transition in prostate cancer cells: involvement of the tumor suppressor miR30a and its target gene SOX4. Biochem Biophys Res Commun 2014; 452:746-52; PMID:25201727; http://dx.doi.org/10.1016/j.bbrc.2014.08.154
- Thakur S, Viswanadhapalli S, Kopp JB, Shi Q, Barnes JL, Block K, Gorin Y, Abboud HE. Activation of AMP-activated protein kinase prevents TGF-beta1-induced epithelial-mesenchymal transition and myofibroblast activation. Am J Pathol 2015; 185:2168-80; PMID:26071397; http://dx.doi.org/10.1016/j.ajpath.2015.04.014
- Lu J, Shi J, Li M, Gui B, Fu R, Yao G, Duan Z, Lv Z, Yang Y, Chen Z, Jia L, Tian L. Activation of AMPK by metformin inhibits TGF-β-induced collagen production in mouse renal fibroblasts. Life Sci 2015; 127:59-65; PMID:25744403; http://dx.doi.org/10.1016/j.lfs.2015.01.042
- Maheshwari RA, Balaraman R, Sen AK, Seth AK. Effect of coenzyme Q10 alone and its combination with metformin on streptozotocin-nicotinamide-induced diabetic nephropathy in rats. Indian J Pharmacol 2014; 46:627-32; PMID:25538335; http://dx.doi.org/10.4103/0253-7613.144924
- Gu J, Ye S, Wang S, Sun W, Hu Y. Metformin inhibits nuclear factor-kappaB activation and inflammatory cytokines expression induced by high glucose via adenosine monophosphate-activated protein kinase activation in rat glomerular mesangial cells in vitro. Chinese Med J 2014; 127:1755-60; PMID:24791887
- Lee JH, Kim JH, Kim JS, Chang JW, Kim SB, Park JS, Lee SK. AMP-activated protein kinase inhibits TGF-β-, angiotensin II-, aldosterone-, high glucose-, and albumin-induced epithelial-mesenchymal transition. Am J Physiol Renal Physiol 2013; 304:F686-97; PMID:23324179
- Cha HN, Choi JH, Kim YW, Kim JY, Ahn MW, Park SY. Metformin Inhibits Isoproterenol-induced Cardiac Hypertrophy in Mice. Korean J Physiol Pharmacol 2010; 14:377-84; PMID:21311678; http://dx.doi.org/10.4196/kjpp.2010.14.6.377
- Wang XF, Zhang JY, Li L, Zhao XY. Beneficial effects of metformin on primary cardiomyocytes via activation of adenosine monophosphate-activated protein kinase. Chin Med J 2011; 124:1876-84; PMID:21740847
- Seliger C, Leukel P, Moeckel S, Jachnik B, Lottaz C, Kreutz M, Brawanski A, Proescholdt M, Bogdahn U, Bosserhoff AK, Vollmann-Zwerenz A, Hau P. Lactate-modulated induction of THBS-1 activates transforming growth factor (TGF)-beta2 and migration of glioma cells in vitro. PloS one 2014 8:e78935; http://dx.doi.org/10.1371/journal.pone.0078935
- Annes JP, Munger JS, Rifkin DB. Making sense of latent TGFbeta activation. J Cell Sci 2003; 116:217-24; PMID:12482908; http://dx.doi.org/10.1242/jcs.00229
- Brantley EC, Benveniste EN. Signal transducer and activator of transcription-3: a molecular hub for signaling pathways in gliomas. Mol Cancer Res 2008; 6:675-84; PMID:18505913; http://dx.doi.org/10.1158/1541-7786.MCR-07-2180
- Carmignani M, Volpe AR, Aldea M, Soritau O, Irimie A, Florian IS, Tomuleasa C, Baritchii A, Petrushev B, Crisan G, Valle G. Glioblastoma stem cells: a new target for metformin and arsenic trioxide. J Biol Regul Homeostat Agents 2014; 28:1-15; PMID:24750786
- Labuzek K, Suchy D, Gabryel B, Bielecka A, Liber S, Okopien B. Quantification of metformin by the HPLC method in brain regions, cerebrospinal fluid and plasma of rats treated with lipopolysaccharide. Pharmacol Rep 2010; 62:956-65; PMID:21098880; http://dx.doi.org/10.1016/S1734-1140(10)70357-1
- Wilcock C, Bailey CJ. Accumulation of metformin by tissues of the normal and diabetic mouse. Xenobiotica 1994; 24:49-57; PMID:8165821; http://dx.doi.org/10.3109/00498259409043220
- http://www.sz-produkte.de/pdf/fi/2014_10_51007278_fi_met_sz_500_850mg_it.pdf.
- Hannigan A, Smith P, Kalna G, Lo Nigro C, Orange C, O'Brien DI, Shah R, Syed N, Spender LC, Herrera B, et al. Epigenetic downregulation of human disabled homolog 2 switches TGF-β from a tumor suppressor to a tumor promoter. J Clin Invest 2010; 120:2842-57; PMID:20592473; http://dx.doi.org/10.1172/JCI36125
- Lim JY, Oh MA, Kim WH, Sohn HY, Park SI. AMP-activated protein kinase inhibits TGF-β-induced fibrogenic responses of hepatic stellate cells by targeting transcriptional coactivator p300. J Cell Physiol 2012; 227:1081-9; PMID:21567395; http://dx.doi.org/10.1002/jcp.22824
- Lin H, Li N, He H, Ying Y, Sunkara S, Luo L, Lv N, Huang D, Luo Z. AMPK Inhibits the Stimulatory Effects of TGF-β on Smad2/3 Activity, Cell Migration, and Epithelial-to-Mesenchymal Transition. Mol Pharmacol 2015; 88:1062-71; PMID:26424816; http://dx.doi.org/10.1124/mol.115.099549
- Zhao Z, Cheng X, Wang Y, Han R, Li L, Xiang T, He L, Long H, Zhu B, He Y. Metformin inhibits the IL-6-induced epithelial-mesenchymal transition and lung adenocarcinoma growth and metastasis. PloS One 2014; 9:e95884; PMID:24789104; http://dx.doi.org/10.1371/journal.pone.0095884
- Moeckel S, Meyer K, Leukel P, Heudorfer F, Seliger C, Stangl C, Bogdahn U, Proescholdt M, Brawanski A, Vollmann-Zwerenz A, et al. Response-predictive gene expression profiling of glioma progenitor cells in vitro. PloS one 2014; 9:e108632; PMID:25268354; http://dx.doi.org/10.1371/journal.pone.0108632
- Esteller M, Hamilton SR, Burger PC, Baylin SB, Herman JG. Inactivation of the DNA repair gene O6-methylguanine-DNA methyltransferase by promoter hypermethylation is a common event in primary human neoplasia. Cancer Res 1999; 59:793-7; PMID:10029064
- Pollard SM, Yoshikawa K, Clarke ID, Danovi D, Stricker S, Russell R, Bayani J, Head R, Lee M, Bernstein M, et al. Glioma stem cell lines expanded in adherent culture have tumor-specific phenotypes and are suitable for chemical and genetic screens. Cell Stem Cell 2009; 4:568-80; PMID:19497285; http://dx.doi.org/10.1016/j.stem.2009.03.014
- Dettmer K, Aronov PA, Hammock BD. Mass spectrometry-based metabolomics. Mass Spectrom Rev 2007; 26:51-78; PMID:16921475; http://dx.doi.org/10.1002/mas.20108
- Jain M, Nilsson R, Sharma S, Madhusudhan N, Kitami T, Souza AL, Kafri R, Kirschner MW, Clish CB, Mootha VK. Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science 2012; 336:1040-4; PMID:22628656; http://dx.doi.org/10.1126/science.1218595
- Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 2012; 9:671-5; PMID:22930834; http://dx.doi.org/10.1038/nmeth.2089
- Gogolla N, Galimberti I, DePaola V, Caroni P. Staining protocol for organotypic hippocampal slice cultures. Nat Protoc 2006; 1:2452-6; PMID:17406491; http://dx.doi.org/10.1038/nprot.2006.180
- Gogolla N, Galimberti I, DePaola V, Caroni P. Preparation of organotypic hippocampal slice cultures for long-term live imaging. Nat Protoc 2006; 1:1165-71; PMID:17406399; http://dx.doi.org/10.1038/nprot.2006.168
- Eyupoglu IY, Hahnen E, Heckel A, Siebzehnrubl FA, Buslei R, Fahlbusch R, Blumcke I. Malignant glioma-induced neuronal cell death in an organotypic glioma invasion model. Technical note. J Neurosurgery 2005; 102:738-44; PMID:15871520; http://dx.doi.org/10.3171/jns.2005.102.4.0738