
ABSTRACT
CSL is a key transcriptional repressor and mediator of Notch signaling. Despite wide interest in CSL, mechanisms responsible for its own regulation are little studied. CSL down-modulation in human dermal fibroblasts (HDFs) leads to conversion into cancer associated fibroblasts (CAF), promoting keratinocyte tumors. We show here that CSL transcript levels differ among HDF strains from different individuals, with negative correlation with genes involved in DNA damage/repair. CSL expression is negatively regulated by stress/DNA damage caused by UVA, Reactive Oxygen Species (ROS), smoke extract, and doxorubicin treatment. P53, a key effector of the DNA damage response, negatively controls CSL gene transcription, through suppression of CSL promoter activity and, indirectly, by increased p21 expression. CSL was previously shown to bind p53 suppressing its activity. The present findings indicate that p53, in turn, decreases CSL expression, which can serve to enhance p53 activity in acute DNA damage response of cells.
Introduction
CSL (CBF1, SUH, Lag1, also known as RBPJκ) is a highly conserved transcription factor with both Notch dependent and independent roles.Citation1 This DNA binding protein is endowed with intrisic transcription repressive function, and is converted into an activator of transcription by association with the proteolytically cleaved activated forms of Notch receptors. CSL binds to regulatory regions of target genes in a dynamic and cell-type specific manner, and genes to which it binds with high affinity can be similarly induced by CSL down-modulation and Notch activation.Citation1 We have recently showed that CSL expression in dermal fibroblasts is key for skin homeostasis, and that its down-modulation results in a cancer-inducing stromal environment.Citation2 Loss of CSL gene function leads to the conversion of dermal fibroblasts into cells with cancer associated fibroblast (CAF) properties, with subsequent recruitment of inflammatory cells and establishment of “cancer fields”, with expanding premalignant and malignant keratinocytic lesions.Citation2,3 The in vivo significance of these findings is supported by the fact that CSL is less expressed in stromal fibroblasts of premalignant and malignant skin SCC lesions relative to surrounding normal skin.Citation4 However, the mechanisms involved in CSL down-regulation remain to be investigated.
Surprisingly, little is known on control of CSL gene expression. The Notch pathway is built on a series of positive and negative feedback loops often operating in a cell type-specific manner.Citation5 This complex mode of regulation results from convergent and reciprocal control of expression of the canonical components: Notch ligands, Notch receptors, CSL, and transcriptional repressors of the Hes/Hey family, as well as co-factors like MAML proteins.Citation5 The Drosophila melanogaster and Caenorhabditis elegans homologs of CSL, Suppressor of Hairless (SuH) and LAG-1 respectively, can either activate or repress their own expression, determining cell fate.Citation6,7 Little else is known of control mechanisms of CSL gene transcription. At the protein level, other pathways converge on control of its function. For instance, in mammalian cells Wnt signaling can inhibit CSL activity by direct binding of Dishevelled, and CSL stability is regulated by Presenilin-2 and p38 MAP-kinase, while its cellular localization can be regulated in Xenopus laevis or mammalian cells by RITA or SMRT protein complexes.Citation8-11
Recent advances in human genomic and transcriptomic analyses provide insights into individual differences in susceptibility to disease, with tissue-specific control of gene expression as key determinant.Citation12 For novel insights into control of CSL gene expression, we started from the study of individual variations in gene expression in human dermal fibroblasts (HDFs) derived from many different individuals. Gene ontology analysis of co-regulated genes pointed to the DNA damage/stress response as a possible negative regulator of CSL expression, which we experimentally assessed in HDFs upon various treatments. Importantly, we found that p53, a key element in the DNA damage/stress response, is involved in negative control of CSL transcription by direct binding to the CSL gene and/or through its effector p21. The Notch and p53 pathways interact at multiple levels, in a context dependent manner.Citation13 We recently uncovered that, in HDFs, CSL functions as a direct negative regulator of p53 activity and that induction of p53-dependent cellular senescence provides a fail-safe mechanism against the consequences of compromised CSL activity.Citation4 The present findings indicate that p53, in turn, represses CSL transcription, which can serve to enhance p53 activity in the acute response of cells to DNA damaging cancer-threatening conditions.
Results
Individual variations in Notch/CSL signaling genes
Substantial differences can exist in levels of gene expression among cells from different individuals that are key to understand variations in susceptibility to disease.Citation12 As mentioned, CSL plays an important role in dermal fibroblasts as negative regulator of senescence and CAF activation.Citation2,4 To probe into individual variations in expression of CSL and related genes, we undertook RNA sequencing analysis of second passage HDFs derived from 46 healthy individuals (GSE77371 and Table S1). RNA sequencing reads showed that, besides CSL (), several components of the Notch family are expressed at significant levels in HDFs, with NOTCH2 being more highly expressed than NOTCH1 as we previously reportedCitation2 (). Among Notch ligands, JAG1 expression was greater than DLL1 (paired t-test p < 0.0001), while JAG2, DLL2 and DLL4 expression was not detectable (). Among canonical Notch/CSL targets, HES4 was more expressed than HES1 or HES6 (paired t-test p < 0.0001), with HES1 being expressed only in some strains (38/46) (). Comparative RT-qPCR and RNA-seq analysis of lowly expressed genes like DLL1 in a selected set of samples showed comparable individual variations (). CSL expression was modulated around a RPKM average of 9.8, with higher levels being found in some HDF strains and lower in others (CV, coefficient of variation of 0.26) (). Two other genes, NOTCH3 and JAG1, showed high heterogeneity of expression among the various strains (CV > 0.6), while NOTCH2 was highly expressed in all, with very little variation (CV = 0.15) ().
Figure 1. Individual variations in gene expression in HDFs. (A-D) Second passage HDFs from 46 different individuals were analyzed by RNA sequencing. Shown are RPKM (reads per kilobase transcriptome per million mapped reads) values for the indicated genes (See also Supplementary Table 1). (E) Comparison of DLL1 RNA expression levels obtained by RNA sequencing vs. RT-qPCR analysis. (F) Graphic illustrations of Pearson's R correlation analysis carried out on log2 RPKM values from RNAseq data used in A-D, for indicated Notch signaling pathway components against each other or against CSL. Blue and red squares correspond to positive and negative values respectively. Significant correlation (|Pearson score| > 0.5) is indicated in the colorimetric scale as green stars. (G) Comparative correlation analysis as performed in F between CSL and Notch signaling genes was determined from either RNA-seq or RT-qPCR data. (H) Same analysis as in F was performed between CAF markers genes (selected from a previously published listCitation4) against each other and against CSL. (I) The global set of RNA-seq data used in A-D was analyzed for positive (in blue) or negative (in red) correlation to CSL (|Pearson score| > 0.5, p < 0.0005). The list of genes in each group is given in Supplementary Table 2. Gene ontology (Metacore Networks) analysis with statistically significant enrichment (p < 0.05) was performed on genes whose expression is positively (left panel) or negatively (right panel) correlated to CSL levels. (J) Shown are genes encoding proteins with transcription factor activity positively (blue) or negatively (red) correlated to CSL selected from Supplementary Table 2.
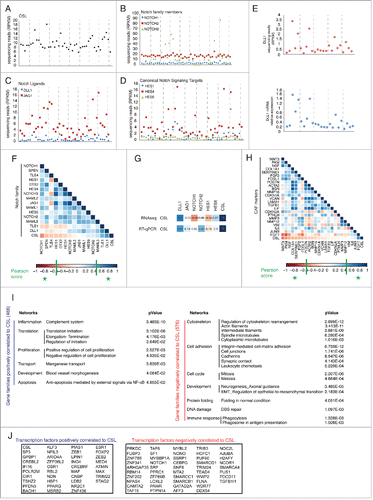
Notch signaling components have the potential of controlling each other's expression in either a positive or negative fashion.Citation5,14 Correlation analysis of RNA-seq data showed that CSL levels among various cell strains were inversely regulated with those of NOTCH1, consistent with CSL functioning as a repressor of this gene,Citation5 while CSL expression was positively correlated with DLL1 and TLE1, suggesting common control mechanisms (). Interestingly, a trend of positive and negative correlation between expression of CSL and other Notch signaling components was also observed (). Although not statistically significant, this was confirmed by RT-qPCR (). Among the inversely related genes we note HES1, which we previously showed to be up-regulated in HDFs by CSL silencing.Citation5
Identification of CSL-coregulated genes
We recently showed that down-modulation of CSL expression leads to fibroblast senescence, with the concomitant induction of a CAF phenotype.Citation4 Consistent with these findings, CSL expression was overall inversely correlated to that of CAF effector genes with WNT3, PKM and NGF showing the best Pearson scores (p < 0.005) ().
For more general insights into CSL regulation and function, we broadened our analysis of the RNA-seq profiles for identification of all genes significantly correlated with CSL in either a positive or negative manner. Expression of 466 genes was positively correlated with that of CSL across the 46 strains (Pearson score > 0.5, p < 0.0005), while 576 genes were negatively correlated (Pearson score < −0.5, p < 0.0005) ( and Table S2). Gene ontology and pathway analysis showed that genes positively correlated with CSL expression were enriched for families related to complement system, proliferation, blood vessel morphogenesis and suppression of apoptosis (via NF-κB). By contrast, oppositely correlated genes were enriched for families related to cytoskeleton and cell adhesion, mitosis and meiosis, epithelial-to-mesenchymal transition and development, protein folding and DNA damage. Among CSL positively or negatively correlated genes many were transcription factors, as shown in . For further studies on a possible connection between CSL expression and these various cellular functions, we focused on DNA damage response.
Stress/DNA damaging conditions as negative regulators of CSL expression
Intrigued by the possibility that DNA damage as a consequence of stress could regulate CSL levels, we assessed levels of CSL expression in various HDF strains upon exposure to UVA, the form of UV light with higher penetration power, capable of reaching the dermal compartment of the skin and directly affecting dermal fibroblasts.Citation15 RT-qPCR analysis showed that CSL expression was down-modulated in all tested HDF strains at various times after UVA exposure, with CDKN1A being oppositely regulated ().
Figure 2. Genotoxic and oxidative stresses negatively regulate CSL expression in HDFs. (A and B) Indicated strains of HDFs were irradiated with 1J/cm2 UVA, collected at the indicated time points and CSL (A) and CDKN1A (B) mRNA levels were measured by RT-qPCR using 36β4 for normalization. N = 5 different experiments for Fb79 and 2 for all other strains. (C) Indicated strains of HDFs were treated with 20 mU/ml glucose oxidase for 4 h, and CSL and CDKN1A mRNA levels were measured by RT-qPCR as in A. N = 2 different experiments for each strain. (D) HDFs (strain Fb79) were treated with 50 μg/ml of Cigarette Smoke Extract for 48 h and assessed as in A for CSL and CDKN1A mRNA levels. N = 2 different experiments. (E) HDFs (strain Fb79) were treated with doxorubicin (0.4 μM) for 24 h and assessed as in A for CSL and CDKN1A mRNA levels. N = 2 different experiments. * p < 0.0001, ** p = 0.0008.
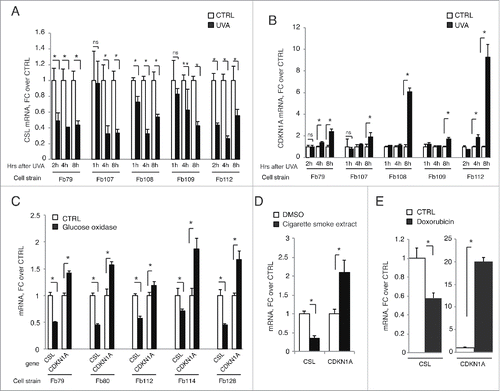
UVA is a major cause of skin photoaging and cancer, with production of reactive oxygen species (ROS) as main mediator of its effects, including those on the DNA.Citation16 As with UVA, CSL down-modulation and CDKN1A up-regulation were observed upon treatment of several HDF strains with glucose oxidase, which results in continuous production of ROS via oxidation of glucose in gluconolactone and hydrogen peroxideCitation17 (). Similar results were observed after treating cells with extracts of cigarette smoke, another potent inducer of oxidative stress – as well as DNA damageCitation18 (). Chemotherapeutic DNA-damaging agents have also been reported to cause stromal fibroblasts senescence and CAF activation.Citation19 As in response to the other insults, treatment with doxorubicin resulted in downregulation of CSL and opposite CDKN1A induction ().
P53 as a negative regulator of CSL expression
The opposite regulation of CSL and CDKN1A expression in response to several stress/DNA damaging conditions suggested that p53 may be involved. To directly assess whether p53 functions as a negative regulator of CSL expression, HDFs were stably infected with a lentivirus for inducible p53 expression. As shown in , induction of wild-type p53 resulted in consistent down-regulation of CSL, at both RNA and protein levels, in all tested HDF strains. No such effects were observed after similar expression of a DNA binding domain p53 mutant protein (R248W) ().
Figure 3. p53 is a negative regulator of CSL expression. (A) HDFs stably infected with lentiviruses for doxycycline-inducible expression of either wild- type p53 (WTp53) or a R248W mutant p53 (mutp53) versus empty vector (CTRL) were treated with doxycycline (Dox) for 48 h. CSL mRNA levels were measured by RT-qPCR using 36β4 for normalization. N = 2 different experiments for each strain. (B) Indicated HDF strains were treated as in A, and CSL and p53 protein levels were assessed by immunoblot analysis using γ-tubulin as equal loading control. P53, CSL and γ-tubulin blots were performed by sequential blotting of the same membrane without stripping (left panel). Relative protein levels were quantified using the ImageStudio program, with γ-tubulin for normalization (right panel). N = 2 different experiments for each strain. (C and D) Indicated HDF strains were treated with Nutlin-3a (10 μM) or DMSO vehicle alone for 48 h. CSL mRNA (C) and CSL and p53 protein (D) levels were assessed as in A and B, respectively. N = 2 different experiments for each strain. (E and F) HDFs were treated with Nutlin-3a (10 μM) or DMSO vehicle for the indicated time points. CSL and p53 protein (E) and p21 mRNA (F) levels were assessed as in B and A, respectively. (G and H) Indicated strains of HDFs infected with control (CTRL) vs. p53 silencing (shp53) retroviruses were analyzed for CSL mRNA and protein levels as in A and B respectively. N = 2 for Fb79 and Fb80. * p< 0.0001.
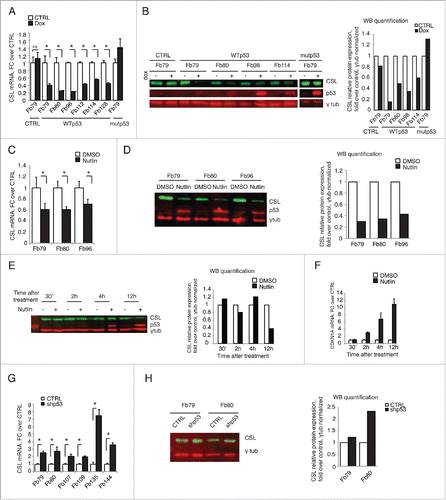
As an alternative approach, HDFs were treated with Nutlin-3a, a MDM2 inhibitor and inducer of endogenous p53 activity through p53 protein stabilization. Even in this case, CSL expression was consistently down-regulated in all tested strains (). A detailed time course after Nutlin-3a treatment showed p53 accumulation and concomitant induction of p21 (as a read-out for p53 activation) preceding CSL down-modulation ().
Mirroring these results, p53 silencing in several strains of HDFs led to a significant induction of CSL expression both at RNA and protein levels ().
Multiple mechanisms of negative control of CSL expression by p53
One mechanism whereby p53 can negatively control expression of target genes is through direct weak binding to their regulatory regions.Citation20 The CSL locus encompasses more than 150 kb. Bioinformatic analysis (with MatInspector, Genomatix) of the enhancer and promoter regions (identified on the basis of the human primary fibroblasts data in the ENCODE database) () revealed the presence of 144 putative binding sites for p53. To assess whether p53 binds to any of these, we performed ChIP-seq (ChIP combined with massive parallel DNA sequencing) analysis of HDFs under basal conditions and after p53 stabilization by Nutlin-3a treatment () (GSE77371). As a positive p53 target, we analyzed the CDKN1A locus and found, as expected, 3 regions of p53 strong binding under basal conditions, with 2 additional binding peaks in Nutlin-3a treated cells ( and Table S3). By contrast, at the CSL locus, we found a single p53 binding peak, only in the Nutlin-3a treated cells ( and Table S3). This was located in intron 4 of the CSL gene, a position outside of predicted regulatory regions and consistent with the one found in a previous survey of p53 binding sites in IMR90 lung fibroblastsCitation21 (Data accessible at NCBI's GEO database,Citation22 accession GSE31558). The binding peak enrichment for p53 at the CSL locus was lower than those at the CDKN1A locus but comparable to the one we found at the PTK2 (Fak kinase) gene ( and Table S3), a previously reported gene under direct negative p53 control.Citation23,24
Figure 4. p53 controls CSL expression via multiple mechanisms. (A) Map of the entire CSL locus showing the transcribed region (green bar), transcription start sites (TSS1 and 2) and exons (E1-E11) and, below, localization of insulators (blue), promoters (red) and enhancers (yellow) as predicted for human primary fibroblasts on the basis of the ENCODE data base (as detailed in Materials and Methods). (B) Graphic illustrations of ChIP-seq analysis of p53-binding peaks in the CDKN1A, CSL, and PTK2 loci in HDFs treated with Nutlin-3a (10 μM for 48 h) or DMSO control (magenta and blue colors, respectively). P53 binding peaks induced by Nutlin-3a treatment are highlighted (red boxes). A full list of p53-binding peaks is provided in Supplementary Table 2. (C) Top: Map of the 1.94 kb CSL promoter region with predicted p53 recognition sequences (blue boxes) and corresponding primers used for the ChIP experiments (magenta arrows) and adjacent exons (green boxes). Bottom: ChIP assay of p53 binding to the CSL promoter and the CSL exon 4 region in HDFs treated for 48 h with Nutlin-3a (10 μM) or DMSO control. Binding enrichment to a upstream CSL region devoid of p53 recognition sequences was used as negative control (negative CTRL), while binding to a p53 binding site in the CDKN1A promoter (chr6: 36,644,111–36,644,216) was used as positive control. (D) HDFs were co-transfected in triplicate wells with a luciferase reporter construct with the CSL promoter (CSLpr-1.94kb) region shown in the map in (C), with either the G/G or the A/A alleles for the rs7657866 SNP, together with increasing amounts of a plasmid over-expressing p53. Luciferase activity was measured 30 h later, using Renilla internal control for normalization. Shown are results of 2 independent experiments (Exp. 1 and 2). (E) HDFs (Fb79) stably infected with lentiviruses for doxycycline-inducible p21 expression were treated or not with doxycycline for 48 h and CSL mRNA levels measured by RT-qPCR using 36β4 for normalization. N=3 independent experiments. (F) Same samples as in (E) were used for immunoblotting with CSL and p53 antibodies using γ-tubulin as equal loading control. CSL and γ-tubulin blots were performed by sequential blotting of the same membrane without stripping (left panel). Relative protein levels were quantified using the ImageStudio program, with γ-tubulin for normalization (right panel). Representative of 2 independent experiments. (G) Summary diagram of the reciprocal negative regulation of CSL and p53 expression and activity, respectively. As discussed in the text, in response to acute genotoxic/oxidative stress, such as UVA or ROS, p53 is activated and represses CSL, which is in turn not longer able to lessen p53 activity. Thus, CSL down-modulation allows for full p53 activation. * p < 0.0001, *** p=0.0006, # p = 0.0089, ## p = 0.0228.
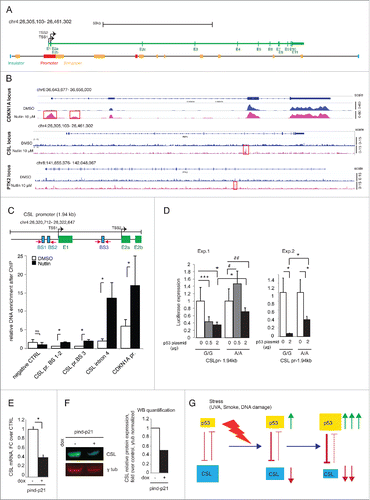
Despite the lack of ChIP-seq p53 binding peaks in regulatory regions of the CSL locus, weaker p53 binding cannot be ruled out due to the relatively low sensitivity of the method. Sequence analysis of the CSL promoter region (chr4: 26320712–26322647) revealed the presence of 3 predicted p53 binding sites, 2 clustered upstream of the transcription start site (BS1 and BS2) and the other in the first intronic region (BS3) (). These sites have various mismatches relative to the consensus p53 recognition sequence, which may result in a low binding affinity.Citation25, 26 In fact, direct chromatin immunoprecipitation (ChIP) assays showed only weak p53 binding to these sites, in Nutlin-3a treated HDFs, in contrast to the more significant binding to the intron 4 region of the CSL gene or the CDKN1A promoter ().
Bioinformatic analysis of genomic variations in the human population showed the presence of a single nucleotide polymorphism (SNP; rs7657866) that abrogates the predicted p53 recognition sequence of the BS3 site in the CSL promoter (as determined by MatInspector, Genomatix). The minor nucleotide allele (A) is present in a homozygous form in 8.42% of the human population and in 33,2% in heterozygous form with the major allele (G). In our collection of HDF strains, by direct nucleotide sequencing we found 3 strains with the A/A genotype. Modulation of endogenous CSL expression in these strains in response to Nutlin-3a treatment was not consistently different from that of strains harboring the major allele. To directly assess whether p53 can negatively control CSL promoter activity and whether in this context the identified SNP is of functional significance, we cloned the CSL promoter spanning this region from cells homozygous for both the G/G and A/A alleles. As shown in , activity of the CSL promoter with the major G/G allele (CSLpr-1.94kb G/G) was effectively suppressed by p53 in a dose-dependent manner. Activity of the CSL promoter with the minor A/A allele was differentially suppressed by p53, requiring higher concentrations ().
Besides direct binding to negative target genes, another complementary mechanism by which p53 can negatively control gene expression is through one of its effectors, CDKN1A (p21).Citation27 To assess whether increased p21 levels reproduce the effects of p53 activation on CSL gene transcription, HDFs were infected with a lentivirus expressing p21. As shown in , increased p21 levels resulted in suppression of CSL expression, similarly to what we observed after induction of p53 expression.
Discussion
Gene expression can vary significantly between individuals, possibly accounting for different susceptibility to complex trait diseases like cancer.Citation28 CSL functions in stromal fibroblasts as direct repressor of senescence and CAF-determinant genes, and down-modulation of CSL expression is a key step for CAF conversion.Citation4 An important question is therefore how this gene is controlled. CSL is best known for its role as key effector of canonical Notch signaling. However, the negative correlation between CSL and Notch1 expression in primary fibroblasts derived from a large number of individuals suggests that the 2 genes play a separate and possibly opposite function in these cells. Gene ontology analysis revealed that among gene families negatively correlated with CSL expression several were involved in cytoskeleton rearrangements, known to be a feature of myofibroblasts and CAF activation.29,30 On the other hand, the top positively correlated genes were involved in the complement system, with a predominance of RCA/CCP (regulators of complement activation/complement control proteins) genes, such as Factor H, C1 inhibitor and MCP. The role of the complement system in cancer has long been debatedCitation31: on one hand, complement is thought to be supportive of the immune-mediated killing of tumor cells. On the other hand, complement activation promotes inflammation, which is correlated with tumor onset and progression.Citation32 The “complement” genes that we found to be positively correlated with CSL are negative regulators of complement activation. This arises the exciting possibility that an important element of CAF conversion resulting from compromised CSL is complement activation, which provides an inflammatory tumor-inducing environment.Citation33 Variations in CSL expression levels correlated significantly also with a large number of genes that may be both downstream CSL targets and/or upstream regulators. Among the first category are likely genes involved in CAF determination, coding for a number of growth factors/cytokines as well as determinants of cytoskeleton organization and cell motility. Among the second category of genes are likely those involved in the DNA damage/stress response.
A number of exogenous insults leading to direct or indirect DNA damage, such as UVA irradiation, exposure to Reactive Oxygen Species (ROS), smoke extracts and doxorubicin, caused down-modulation of CSL expression in the tested fibroblast strains. UVA is thought be an important etiological factor of skin photo-aging and cancerCitation15,34 and, due to its great penetration power, it can directly affect the dermal compartment of the skin. Its effects are mostly mediated by the enhanced production of Reactive Oxygen Species (ROS), which cause secondary DNA damage as well as changes in many other aspects of cell physiology.Citation35 Cigarette smoke extract, another stress inducer, is composed by several toxic compounds, such as nicotine and hydrogen monoxide, which target directly or indirectly the DNA and induce fibroblast senescence and CAF activation.Citation18 Doxorubicin, a chemotherapeutic DNA damaging agent, also causes stromal fibroblast senescence and CAF activation.Citation19 All these stress conditions resulted in significant down-modulation of CSL expression with concomitant induction of CDKN1A levels, a common indicator of p53 activity. While we focused on p53 for further studies, UVA exposure represses CSL expression also in HDFs with p53 gene silencing (Fig. S1A, B), indicating that other determinants of the DNA damage/stress response converge on negative regulation of this gene. A possible mechanism is promoter DNA methylation, as CSL expression is down-modulated with the same kinetics of Notch2 (Fig. S1C), whose down-modulation upon UVA treatment is due to DNA methylation.Citation2
The transcriptional repressing function of p53 is relatively poorly understood.Citation36 It can be mediated, in part, by direct p53 binding to target genes. It has been proposed that the p53 consensus recognition sequences of induced and repressed genes are different, with validated repressor sites having a weaker affinity with longer spacers between quarter-sites and/or changed orientation of these sites.Citation26 We identified a specific site within the CSL locus in intron 4, to which p53, at enhanced levels, binds. However, this site has a complete p53 responsive element with a theoretical high transactivation score and lies outside any predicted regulatory regions of the CSL gene, making it unlikely to be involved in negative control of its expression. More functionally relevant is likely to be the ability of p53 to negatively control CSL promoter activity. Indeed we show that CSL promoter activity is suppressed in exogenous assays by increased p53 in a dose dependent manner, with lesser inhibition resulting from a nucleotide polymorphism in the CSL promoter abrogating a p53 recognition sequence.
Another mechanism by which p53 can negatively control gene transcription is through one or more of its effectors, such as p21 (CDKN1A). p21 can function as a negative regulator of gene expression, independently of its effects on the cell cycle, through a variety of mechanisms including binding and negative regulation of E2F1 activity,Citation27,37–39 recruitment of E2F4 repression complexes,Citation37 and binding and modulation of Estrogen Receptor α –dependent transcription.Citation40 The impact of increased or decreased p21 activity in stromal fibroblasts remains to be conclusively established, as it has been variously reported that p21 overexpression induces CAF conversionCitation41 or only cellular senescence without induction of the senescence-associated CAF-related secretory phenotype.Citation42 In our own work, we found that silencing of the p21 gene, like that of p53, can synergize with CSL knockdown in induction of CAF marker expression,Citation4 pointing to an overlap of p53 and p21 transcription repressive function beyond control of the CSL gene.
Overall, an emerging scenario exists of a reciprocal negative feedback loop between p53 and CSL activity, which could be responsible for overriding individual differences in gene expression, allowing a proper response to stress factors and thus maintaining stromal tissue homeostasis. We recently showed that CSL directly binds to p53 suppressing its activity and, as part of the chronic process leading to cancer development, p53-induced senescence can function as fail-safe mechanism against cancer/stromal cell expansion resulting from compromised CSL function.Citation4 In turn, the present findings indicate that p53 negatively controls CSL expression, thus providing a positive feedback mechanism for enforcement of p53 activity in the acute response of cells to DNA damaging cancer-threatening conditions ().
Materials and methods
Human samples and cell culture
Discarded human foreskin tissue samples for primary cell preparation were obtained from the Department of Pediatrics, Lausanne University Hospital, with institutional approvals and informed consent as part of institutional requirements.
Conditions for culturing of cells, viral infection, siRNA-mediated gene silencing, RT–qPCR and ChIP were as previously reported.Citation2,43,44 For derivation of HDFs, surgically excised foreskins were fragmented into pieces after removal of the hypodermis, followed by incubation in 1% collagenase I solution at 37°C under shaking. The dissociated tissue was diluted, filtered and washed by centrifugation and subsequently cultured in 10% FBS DMEM without Phenol Red (Gibco), in order to avoid any absorbance of UV light by the medium. For stress inducing experiments, cells were treated when they reached 60% confluence. UVA irradiation (1J/cm2) was performed in a UVA irradiator (Vilber) monitored with a dosimeter (InternationaLight, Inc.). Chemicals used for the study were as follows: 20 mU/ml of glucose oxidase (Sigma), 50 μg/ml of Cigarette Smoke Extract (Murty Pharmaceuticals), 0.4 μM of doxorubicin hydrochloride (Sigma), 10 μM of Nutlin-3a (Sigma) or suitable controls. Doxycycline hyclate (Sigma) was used at a dose of 750 ng/ml at the indicated time in experiments with cells transduced with lentiviruses for doxycline-inducible expression.
RT-qPCR and immunoblotting
RT–qPCR were carried out as previously describedCitation4 in triplicate using the following primers: 36β4 Fwd: 5′-GCAATGTTGCCAGTGTCTGT-3′, Rev: 5′-GCCTTGACCTTTTCAGCAAG-3′; CSL Fwd: 5′-GGCTGGAATACAAGTTGAACA-3′, Rev: 5′-AGAGCAAAAGCTCAAAGGTG-3′; p21 Fwd: 5′-AGCAGAGGAAGACCATGTGGACCT-3′, Rev: 5′-GAAGATGTAGAGCGGGCCTTTGAGG-3′. Immunoblotting experiments were carried out as previously describedCitation4 using the following antibodies: for CSL, D10A4 (Cell Signaling), for p53, sc-126 (Santa Cruz Biotechnology) and for γ-tubulin, clone GTU-88 (Sigma). Twenty μg of proteins were loaded on 10% acrylamide gels, and detection was performed using the infrared fluorescent IRDye® secondary antibodies (LI-COR®) and the Odissey CLx (LI-COR®) infrared imaging system. Image analysis were performed using the Image Studio software (LICOR®).
Plasmids and siRNAs
To make the pIND20-p53WT and pIND20-p53R248W plasmids, PCR products using the primers Fwd: 5′-CACCATGGAGGAGCCGCAGTCAGATCCTAGCGTCGA-3′ and Rev: 5′-TCAGTCTGAGTCAGGCCCTTCTGTCTTGAA-3′ and pBABE-puro-p53WT and pBABE-puro-p53R248W vectors as respective templates (obtained from Dr A. Rustgi, University of Pennsylvania, PACitation45), were cloned into the pENTR-D-TOPO entry vector (Life Technologies) and transferred through the Gateway LR Clonase system (Life Technologies) into the pINDUCER20 destination vectorCitation46 using the pENTRTM directional TOPO cloning kit. The same strategy was used to produce pIND20-CSL, using the following PCR primers Fwd: 5′-CACCATGGACCACACGGAGGGCTCGCCCGCGGAGGAGCC-3′ and Rev: 5′-TTAGGATACCACTGTGGCTGTAGATGATGTGAC-3′ with pBMNRBPj plasmidCitation47 as template. The pIND20-p21 was obtained from Dr SJ Elledge (Brigham and Women's Hospital, Boston, MA).Citation46 The pRS-p53 (shp53) and control pRS vectors were obtained from Dr. R. Agami (The Netherlands Cancer Institute, Amsterdam, The Netherlands).
RNAseq analysis
Total RNAs from HDFs were extracted using the directZol RNA miniprep kit (Zymo Research) with on-column DNase treatment. RNA quality was verified on the Bioanalyzer (Agilent) and 1 μg of total RNA with RIN> 8 was used for library preparation using the NEBNext® Ultra kit (NEB) according to manufacturer instructions. Single read was done on the Illumina HiSeq 2000 sequencer at the Genomic Technologies Facility (University of Lausanne). Sequencing quality check was performed using FastQC (v 0.10.1 http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). PolyA tails, adapter sequences and low quality reads were cut using CutadaptCitation48 (v 1.4.1), using a base quality cutoff of 5. The alignment was performed using Tophat2Citation49 (v 2.0.9) and GRCh37 as reference genome. Reads were sorted by position in the reference genome using Picard (http://broadinstitute.github.io/picard/v1.96). Indexing was performed using sambamba (http://lomereiter.github.io/sambamba/v0.4.7) and alignment statistics using bamtool stats (http://sourceforge.net/projects/bamtools/v2.3.0). RNAseq quality was checked using the R package NOISeqCitation50 (v 2.6.0), analyzing coverage, percentage of reads mapping to different genes and fraction of rRNA. Reads were counted and gene expression evaluated using HTSeq (v 0.6.1).
Data have been deposited in NCBI's Gene Expression OmnibusCitation22 and are accessible through GEO series accession number GSE77370 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE77370).
Bioinformatic analyses
The graphic illustrations showing the correlation analysis between various genes and CSL were produced using the R package corrplots (v 0.73), starting from the RNAseq RPKM reads. Correlation analyses were performed on the matrix of log2 RPKM values for the 46 foreskin samples after combat algorithm to adjust data for batch effect.Citation51 Genes with |Pearson coefficient| > 0.5 and p < 0.0005 were considered as significantly correlated to CSL. Correlation matrices were generated in R using the cor() function. The corrplot package was then used to graphically display correlation matrices. The functional annotation tool available within DAVID Website (http://david.abcc.ncifcrf.gov/) was used to look for any over-represented biological process level 5 (BP5) of the Gene Ontology (GO). Process networks and pathway maps analysis was performed with MetaCoreTM version 6.2 (Thomson Reuters).
Locus analysis, ChIP assay and ChIP-seq
For human CSL locus analysis, we used ENCODE data (http://genome.ucsc.edu/ENCODE) giving information on chromatin organization. Based on the Chromatin in State Segmentation by HMM data, we defined promoters and enhancers in the CSL locus. Exons were defined taking in account all exons described for the 4 main CSL transcript variants described in the NCBI database. The variants analyzed are: NM_005349.3, NM_203283.2, NM_015874.4, NM_203284.2.
ChIP assays with antibodies against p53 versus non-immune IgG control were carried out as previously describedCitation4 followed by determination of binding enrichment for the indicated sites; primer sequences were as follows:
CDKN1Apr_BSF: 5′-TGGACTGGGCACTCTTGTC-3′, CDKN1Apr_BSR: 5′-AGAAAGCCAATCAGAGCCAC-3′, negativeCTRL_BSF: 5′-CCCTAATGGTCTAAAAAGGG-3′, negativeCTRL_BSR: 5′-ACTACCACTTACAGTGCCTG-3′, CSLintron4_BSF: 5′-TTTCCAAATAAGGAAGGGGC-3′, CSLintron4_BSR: 5′-CTCTAAAGCCTCCTATCAAC-3′. CSLpr_BS1.2F: 5′-GGCGCCCGGAGCTGGT-3′ CSLpr_BS1.2R: 5′- TGGGAAATTTCCTAAACCTCG-3′ CSLpr_BS3F: 5′- AAGGGCCTCCTCATTAGCAT-3′ CSLpr_BS3R: 5′- TAATCTCGGCGTTTGGTGCA-3′. CDKN1Apr_BS primers were used as a positive control for binding of p53 to the CDKN1A promoter. negativeCTRL_BS were primers designed for a putative p53 binding site in the enhancer of CSL, which was not confirmed, and were used as a negative control for p53 binding.
For ChIP-seq analysis, immunoprecipitated DNA from HDFs was processed as for ChIP assaysCitation4 using 5 μg of antibody against p53 per 106 cells. The ChIPed DNA was quantified by fluorimetry on a Qubit system (Invitrogen). A total of 10 ng of DNA was used for library preparation using a NEBNext ChIP-Seq Library Prep reagent set (New England Biolabs Inc.), as recommended by the manufacturer. The sequencing was performed by the Genomic Technologies Facility at Lausanne University. Sequencing quality control was performed using FASTqcCitation52; immunoprecipitation quality control was performed using PhantomPeakCitation53 quality tools and CHANCE.Citation54 Burrows–Wheeler AlignerCitation55 was used for FASTQ file alignments and, for peak detection, MACS softwareCitation56 with default parameters was used. Each peak was associated to the closest transcription starting sites with ChIPpeakAnno,Citation57 a Bioconductor R package. The Integrative Genomics Viewer (IGV) (http://www.broadinstitute.org/igv) was used for graphic illustration of ChIP-Seq peaks, and ENCODE data (http://genome.ucsc.edu/ENCODE) for information on chromatin organization. ChIP-Seq data have been deposited in NCBI's Gene Expression OmnibusCitation22 and are accessible through GEO series accession number GSE77225 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE77225).
For SNP analysis, the 1000 genome database was used in the SPSmart program (http://spsmart.cesga.es).
Promoter activity assay
The CSL promoter-1.94 kb vector (CSLpr-1.94kb) (chr4: 26,320,712–26,322,647) was obtained by amplification of the PCR product using the following primers Fwd- 5′AGACTAGGTGACTCAAGGCA-3′ and Rev- 5′-CCACAAACTCTCGCCAAAAC-3′ and human genomic DNA as template. The PCR fragment was inserted into the pGL3-enhancer vector (Promega, Firefly luciferase) in the BglII/XhoI site and sequence was verified by DNA sequencing. HDFs were co-transfected in triplicate with the CSLpr-1.94kb (0.5 μg/well) together with the indicated amounts of pBABE-p53WT plasmid (obtained from Dr A. Rustgi (University of Pennsylvania, PA)Citation45) as well as 0.05 ug of the phRL-TK plasmid (Promega, Renilla luciferase for internal control). Luciferase activity was determined with the Dual Luciferase assay kit (Promega) 30 h later, using internal Renilla control for normalization.
Statistical analysis
Two-tailed Student's t-tests were performed where appropriate unless specified. Error bars represent SD. P values are as indicated in figure legends.
Abbreviations
| CAFs | = | cancer associated fibroblasts |
| HDFs | = | human dermal fibroblasts |
| RCA/CCP | = | regulators of complement activation/complement control proteins |
| ROS | = | reactive oxygen species |
| UVA | = | ultra violet A |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
1186317_Supplemental_Material.zip
Download Zip (6.8 MB)Acknowledgments
We thank R. Bernards, R. Agami, A. Rustgi and SJ Elledge for the gift of various viral vectors, K. Harshman, P. Angelino and V. Ioannidis for RNA-seq, ChIP-seq DNA sequencing analysis and bioinformatic support. We are grateful to Corentin Pasche and Tatiana Proust for technical help.
Funding
This work was supported by grants from the Swiss National Science Foundation (310030_156191/1), the National Institute of Arthritis and Musculoskeletal and Skin Diseases (R01AR039190; R01AR064786; the content not necessarily representing the official views of NIH), European Research Council (26075083) and Swiss Cancer League (OCS-2922-02-2012 and KFS-3301-08-2013). E. Menietti was supported by a grant from ISREC and P. Ostano by a grant from Lauretana S.P.A.
Related Research Data
References
- Johnson JE, Macdonald RJ. Notch-independent functions of CSL. Curr Top Dev Biol 2011; 97:55-74; PMID:22074602; http://dx.doi.org/10.1016/B978-0-12-385975-4.00009-7
- Hu B, Castillo E, Harewood L, Ostano P, Reymond A, Dummer R, Raffoul W, Hoetzenecker W, Hofbauer GF, Dotto GP. Multifocal epithelial tumors and field cancerization from loss of mesenchymal CSL signaling. Cell 2012; 149:1207-20; PMID:22682244; http://dx.doi.org/10.1016/j.cell.2012.03.048
- Dotto GP. Multifocal epithelial tumors and field cancerization: stroma as a primary determinant. J Clin Invest 2014; 124:1446-53; PMID:24691479; http://dx.doi.org/10.1172/JCI72589
- Procopio MG, Laszlo C, Al Labban D, Kim DE, Bordignon P, Jo SH, Goruppi S, Menietti E, Ostano P, Ala U, et al. Combined CSL and p53 downregulation promotes cancer-associated fibroblast activation. Nat Cell Biol 2015; 17:1193-204; PMID:26302407; http://dx.doi.org/10.1038/ncb3228
- Bray SJ. Notch signalling: a simple pathway becomes complex. Nat Rev Mol Cell Biol 2006; 7:678-89; PMID:16921404; http://dx.doi.org/10.1038/nrm2009
- Liu F, Posakony JW. An enhancer composed of interlocking submodules controls transcriptional autoregulation of suppressor of hairless. Dev Cell 2014; 29:88-101; PMID:24735880; http://dx.doi.org/10.1016/j.devcel.2014.02.005
- Choi VN, Park SK, Hwang BJ. Clustered LAG-1 binding sites in lag-1/CSL are involved in regulating lag-1 expression during lin-12/Notch-dependent cell-fate specification. BMB Rep 2013; 46:219-24; PMID:23615264; http://dx.doi.org/10.5483/BMBRep.2013.46.4.269
- Collu GM, Hidalgo-Sastre A, Acar A, Bayston L, Gildea C, Leverentz MK, Mills CG, Owens TW, Meurette O, Dorey K, et al. Dishevelled limits Notch signalling through inhibition of CSL. Development 2012; 139:4405-15; PMID:23132247; http://dx.doi.org/10.1242/dev.081885
- Kim SM, Kim MY, Ann EJ, Mo JS, Yoon JH, Park HS. Presenilin-2 regulates the degradation of RBP-Jk protein through p38 mitogen-activated protein kinase. J Cell Sci 2012; 125:1296-308; PMID:22302987; http://dx.doi.org/10.1242/jcs.095984
- Wacker SA, Alvarado C, von Wichert G, Knippschild U, Wiedenmann J, Clauss K, Nienhaus GU, Hameister H, Baumann B, Borggrefe T, et al. RITA, a novel modulator of Notch signalling, acts via nuclear export of RBP-J. EMBO J 2011; 30:43-56; PMID:21102556; http://dx.doi.org/10.1038/emboj.2010.289
- Zhou S, Hayward SD. Nuclear localization of CBF1 is regulated by interactions with the SMRT corepressor complex. Mol Cell Biol 2001; 21:6222-32; PMID:11509665; http://dx.doi.org/10.1128/MCB.21.18.6222-6232.2001
- Consortium GT. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 2015; 348:648-60; PMID:25954001; http://dx.doi.org/10.1126/science.1262110
- Dotto GP. Crosstalk of Notch with p53 and p63 in cancer growth control. Nat Rev Cancer 2009; 9:587-95; PMID:19609265; http://dx.doi.org/10.1038/nrc2675
- Kopan R. Notch signaling. Cold Spring Harb Perspect Biol 2012; 1:4(10); PMID:23028119; http://dx.doi.org/10.1101/cshperspect.a011213.
- Halliday GM. Inflammation, gene mutation and photoimmunosuppression in response to UVR-induced oxidative damage contributes to photocarcinogenesis. Mutat Res 2005; 571:107-20; PMID:15748642; http://dx.doi.org/10.1016/j.mrfmmm.2004.09.013
- Bachelor MA, Bowden GT. UVA-mediated activation of signaling pathways involved in skin tumor promotion and progression. Semin Cancer Biol 2004; 14:131-8; PMID:15018897; http://dx.doi.org/10.1016/j.semcancer.2003.09.017
- Wong CM, Wong KH, Chen XD. Glucose oxidase: natural occurrence, function, properties and industrial applications. Appl Microbiol Biotechnol 2008; 78:927-38; PMID:18330562; http://dx.doi.org/10.1007/s00253-008-1407-4
- Laytragoon-Lewin N, Bahram F, Rutqvist LE, Turesson I, Lewin F. Direct effects of pure nicotine, cigarette smoke extract, Swedish-type smokeless tobacco (Snus) extract and ethanol on human normal endothelial cells and fibroblasts. Anticancer Res 2011; 31:1527-34; PMID:21617206
- Soon PS, Kim E, Pon CK, Gill AJ, Moore K, Spillane AJ, Benn DE, Baxter RC. Breast cancer-associated fibroblasts induce epithelial-to-mesenchymal transition in breast cancer cells. Endocr Related Cancer 2013; 20:1-12; PMID:23111755; http://dx.doi.org/10.1530/ERC-12-0227
- Chen X, Wang J, Shen H, Lu J, Li C, Hu DN, Dong XD, Yan D, Tu L. Epigenetics, microRNAs, and carcinogenesis: functional role of microRNA-137 in uveal melanoma. Invest Ophthalmol Vis Sci 2011; 52:1193-9; PMID:21051724; http://dx.doi.org/10.1167/iovs.10-5272
- Botcheva K, McCorkle SR, McCombie WR, Dunn JJ, Anderson CW. Distinct p53 genomic binding patterns in normal and cancer-derived human cells. Cell Cycle 2011; 10:4237-49; PMID:22127205; http://dx.doi.org/10.4161/cc.10.24.18383
- Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 2002; 30:207-10; PMID:11752295; http://dx.doi.org/10.1093/nar/30.1.207
- Golubovskaya VM, Finch R, Kweh F, Massoll NA, Campbell-Thompson M, Wallace MR, Cance WG. p53 regulates FAK expression in human tumor cells. Mol Carcinog 2008; 47:373-82; PMID:17999388; http://dx.doi.org/10.1002/mc.20395
- Golubovskaya V, Kaur A, Cance W. Cloning and characterization of the promoter region of human focal adhesion kinase gene: nuclear factor kappa B and p53 binding sites. Biochim Biophys Acta 2004; 1678:111-25; PMID:15157737; http://dx.doi.org/10.1016/j.bbaexp.2004.03.002
- Menendez D, Inga A, Resnick MA. The expanding universe of p53 targets. Nat Rev Cancer 2009; 9:724-37; PMID:19776742; http://dx.doi.org/10.1038/nrc2730
- Riley T, Sontag E, Chen P, Levine A. Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol 2008; 9:402-12.
- Lohr K, Moritz C, Contente A, Dobbelstein M. p21/CDKN1A mediates negative regulation of transcription by p53. J Biol Chem 2003; 278:32507-16; PMID:12748190; http://dx.doi.org/10.1074/jbc.M212517200
- Barbujani G, Colonna V. Human genome diversity: frequently asked questions. Trends Genet 2010; 26:285-95; PMID:20471132; http://dx.doi.org/10.1016/j.tig.2010.04.002
- Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer 2006; 6:392-401; PMID:16572188; http://dx.doi.org/10.1038/nrc1877
- Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol 2002; 3:349-63; PMID:11988769; http://dx.doi.org/10.1038/nrm809
- Markiewski MM, Lambris JD. Is complement good or bad for cancer patients? A new perspective on an old dilemma. Trends Immunol 2009; 30:286-92; PMID:19428302; http://dx.doi.org/10.1016/j.it.2009.04.002
- Pio R, Ajona D, Lambris JD. Complement inhibition in cancer therapy. Semin Immunol 2013; 25:54-64; PMID:23706991; http://dx.doi.org/10.1016/j.smim.2013.04.001
- Merle NS, Noe R, Halbwachs-Mecarelli L, Fremeaux-Bacchi V, Roumenina LT. Complement System Part II: Role in Immunity. Front Immunol 2015; 6:257; PMID:26074922
- Rittie L, Fisher GJ. UV-light-induced signal cascades and skin aging. Ageing Res Rev 2002; 1:705-20; PMID:12208239; http://dx.doi.org/10.1016/S1568-1637(02)00024-7
- Kawanishi S, Hiraku Y, Oikawa S. Mechanism of guanine-specific DNA damage by oxidative stress and its role in carcinogenesis and aging. Mutat Res 2001; 488:65-76; PMID:11223405; http://dx.doi.org/10.1016/S1383-5742(00)00059-4
- Wang B, Xiao Z, Ko HL, Ren EC. The p53 response element and transcriptional repression. Cell Cycle 2010; 9:870-9; PMID:20160511; http://dx.doi.org/10.4161/cc.9.5.10825
- Benson EK, Mungamuri SK, Attie O, Kracikova M, Sachidanandam R, Manfredi JJ, Aaronson SA. p53-dependent gene repression through p21 is mediated by recruitment of E2F4 repression complexes. Oncogene 2014; 33:3959-69; PMID:24096481; http://dx.doi.org/10.1038/onc.2013.378
- Devgan V, Mammucari C, Millar SE, Brisken C, Dotto GP. p21WAF1/Cip1 is a negative transcriptional regulator of Wnt4 expression downstream of Notch1 activation. Genes Dev 2005; 19:1485-95; PMID:15964998; http://dx.doi.org/10.1101/gad.341405
- Delavaine L, La Thangue NB. Control of E2F activity by p21Waf1/Cip1. Oncogene 1999; 18:5381-92; PMID:10498892; http://dx.doi.org/10.1038/sj.onc.1202923
- Fritah A, Saucier C, Mester J, Redeuilh G, Sabbah M. p21WAF1/CIP1 selectively controls the transcriptional activity of estrogen receptor α. Mol Cell Biol 2005; 25:2419-30; PMID:15743834; http://dx.doi.org/10.1128/MCB.25.6.2419-2430.2005
- Capparelli C, Chiavarina B, Whitaker-Menezes D, Pestell TG, Pestell RG, Hulit J, Ando S, Howell A, Martinez-Outschoorn UE, Sotgia F, et al. CDK inhibitors (p16/p19/p21) induce senescence and autophagy in cancer-associated fibroblasts, “fueling” tumor growth via paracrine interactions, without an increase in neo-angiogenesis. Cell Cycle 2012; 11:3599-610; http://dx.doi.org/10.4161/cc.21884
- Freund A, Patil CK, Campisi J. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J 2011; 30:1536-48; http://dx.doi.org/10.1038/emboj.2011.69
- Hu B, Lefort K, Qiu W, Nguyen BC, Rajaram RD, Castillo E, He F, Chen Y, Angel P, Brisken C, et al. Control of hair follicle cell fate by underlying mesenchyme through a CSL-Wnt5a-FoxN1 regulatory axis. Genes Dev 2010; 24:1519-32; PMID:20634318; http://dx.doi.org/10.1101/gad.1886910
- Lefort K, Mandinova A, Ostano P, Kolev V, Calpini V, Kolfschoten I, Devgan V, Lieb J, Raffoul W, Hohl D, et al. Notch1 is a p53 target gene involved in human keratinocyte tumor suppression through negative regulation of ROCK1/2 and MRCKalpha kinases. Genes Dev 2007; 21:562-77; PMID:17344417; http://dx.doi.org/10.1101/gad.1484707
- Okawa T, Michaylira CZ, Kalabis J, Stairs DB, Nakagawa H, Andl CD, Johnstone CN, Klein-Szanto AJ, El-Deiry WS, Cukierman E, et al. The functional interplay between EGFR overexpression, hTERT activation, and p53 mutation in esophageal epithelial cells with activation of stromal fibroblasts induces tumor development, invasion, and differentiation. Genes Dev 2007; 21:2788-803; PMID:17974918; http://dx.doi.org/10.1101/gad.1544507
- Meerbrey KL, Hu GA, Kessler JD, Roarty K, Li MZ, Fang JE, Herschkowitz JI, Burrows AE, Ciccia A, Sun TT, et al. The pINDUCER lentiviral toolkit for inducible RNA interference in vitro and in vivo. P Natl Acad Sci USA 2011; 108:3665-70; http://dx.doi.org/10.1073/pnas.1019736108
- Liang Y, Ganem D. Lytic but not latent infection by Kaposi's sarcoma-associated herpesvirus requires host CSL protein, the mediator of Notch signaling. Proc Natl Acad Sci U S A 2003; 100:8490-5; PMID:12832621; http://dx.doi.org/10.1073/pnas.1432843100
- Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnetjournal 2011; 17:10-2.
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 2009; 10:R25; PMID:19261174; http://dx.doi.org/10.1186/gb-2009-10-3-r25
- Tarazona S, Garcia-Alcalde F, Dopazo J, Ferrer A, Conesa A. Differential expression in RNA-seq: a matter of depth. Genome Res 2011; 21:2213-23; PMID:21903743; http://dx.doi.org/10.1101/gr.124321.111
- Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 2007; 8:118-27; PMID:16632515; http://dx.doi.org/10.1093/biostatistics/kxj037
- Patel RK, Jain M. NGS QC Toolkit: a toolkit for quality control of next generation sequencing data. Plos One 2012; 7:e30619; PMID:22312429; http://dx.doi.org/10.1371/journal.pone.0030619
- Jung LY KP, Wold B, Sidow A, Batzoglou S, Park P. Assessment of ChIP-seq data quality using cross-correlation analysis. (Submitted).
- Diaz A, Nellore A, Song JS. CHANCE: comprehensive software for quality control and validation of ChIP-seq data. Genome Biol 2012; 13:R98; PMID:23068444; http://dx.doi.org/10.1186/gb-2012-13-10-r98
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009; 25:1754-60; PMID:19451168; http://dx.doi.org/10.1093/bioinformatics/btp324
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol 2008; 9:R137; PMID:18798982; http://dx.doi.org/10.1186/gb-2008-9-9-r137
- Zhu LJ, Gazin C, Lawson ND, Pages H, Lin SM, Lapointe DS, Green MR. ChIPpeakAnno: a Bioconductor package to annotate ChIP-seq and ChIP-chip data. BMC Bioinformatics 2010; 11:237; PMID:20459804; http://dx.doi.org/10.1186/1471-2105-11-237