
ABSTRACT
Protein kinase D 3 (PKD3) is a member of the PKD family that has been linked to many intracellular signaling pathways. However, defined statements regarding isoform specificity and in vivo functions are rare. Here, we use mouse embryonic fibroblast cells that are genetically depleted of PKD3 to identify isoform-specific functions. We show that PKD3 is involved in the regulation of the cell cycle by modulating microtubule nucleation and dynamics. In addition we also show that PKD1 partially can compensate for PKD3 function. Taken together our data provide new insights of a specific PKD3 signaling pathway by identifying a new function, which has not been identified before.
Introduction
The protein kinase D (PKD) family is represented by three individual members, PKD1-3, which are transcribed from different gene loci. Initially identified as new members of the protein kinase C (PKC) family based on nucleotide sequence homology, PKD1 was also named PKCμ, whereas PKD3 was originally identified as PKCν.Citation1,2 In addition, the overall structure of PKCs and PKDs revealed certain similarities, such as the presence of an N-terminal regulatory domain and a C-terminal kinase domain in both families. In addition, similarly to PKCs, PKDs contain tandem C1A/C1B motifs within their N-terminal domains. These factors reveal that PKDs are connected to the diacylglycerol-mediated signaling pathway, which is involved in a plethora of cellular functions. This role defines PKDs as multifunctional kinases (for a review see ref.Citation3). One major structural difference between PKC and PKD enzymes is that the latter group contains an N-terminal pleckstrin homology domain, which is believed to serve an auto-inhibitory function by influencing the conformational status of the enzyme. Indeed, PKDs and PKCs do share high homology within the kinase domain, but because PKDs are more homologous to myosin light chain kinase and Ca2+/calmodulin-dependent protein kinase (CAMK) they belong to the CAMK family.Citation4
Like PKD1 and 2, PKD3 has been linked to many intercellular signaling pathways, but isoform-specific data regarding the in vivo functions of individual PKD family members remain very limited. In general, PKD activation is believed to be mediated by the trans-phosphorylation of serine residues 738 and 742 (in human PKD1), which are located in the activation loop, by novel PKC isoforms.Citation5 More recent work has also demonstrated that only Ser738 is a target of PKC, Ser742 may instead represent an auto-phosphorylation target.Citation6 PKCs also localize to lipid membranes, thus providing an additional connection between the PKC and PKD signaling cascades. Upon activation loop phosphorylation, which is mediated by PKCs, a C-terminal serine residue (Ser916 in mouse PKD1) becomes auto-phosphorylated.Citation7 Although all PKD isoforms contain an activation loop motif, only PKD1 and 2 contain the C-terminal auto-phosphorylation motif. Nevertheless, both phosphorylation events are markers for the activation status of PKDs. Thus, the PKC/PKD axis is currently recognized as an established signaling cascade in PKD-mediated signal transduction.Citation8
The cell cycle represents a highly organized series of events that result in the generation of two daughter cells. In a simplified view, the cell cycle can be subdivided into 4 different phases (G1, S, G2 and M), which can further be sub-divided into several distinguishable stages. In general, interphase (G1, S and G2) represents the time when a cell prepares for the next division, including cell growth, DNA and centrosome duplication and checkpoints, which guarantee successful entry into the M-phase. During M-phase (mitosis), the cell undergoes a coordinated set of processes that ultimately result in the generation of two daughter cells. During mitosis, microtubules, which consist of α- and β-tubulin, represent a major structural component of the spindle apparatus, which is required for the separation of sister chromatids. Although a number of inhibitors have shown the importance of this structure, the concrete mechanism that underlies its establishment remains unknown.
In contrast to the large body of published work connecting the in vivo functions of PKCs in the cell cycle either directly or indirectly, only a few reports have indicated a possible role of PKD in that process. One such study has defined a role for PKD in the context of GPCR-mediated cell proliferation,Citation9 which has further been shown to promote cell cycle progression via c-fos expression.Citation10 More recently, the direct phosphorylation of HDAC5 by PKD has been reportedCitation11; this phosphorylation could potentially influence cell proliferation. To date, PKD3 has been described as promoting proliferation and survival only in prostate cancer cell lines and human keratinocytes,Citation12,13 and the exact mechanisms remain unclear. In addition, earlier work has identified PKD3 and PKD2 as being associated with microtubule structures during mitosis.Citation14
In the present study, by analyzing genetically depleted cell lines, we identified PKD3 as a modulator of microtubule dynamics during mitosis.
Results
To analyze the consequences of the genetic depletion of PKD3, we isolated mouse embryonic fibroblasts (MEFs) from heterozygous intercrosses of PKD3+/− mice that we recently generated (details described in Materials & Methods). Whereas primary wild-type (wt) and PKD3-deficient MEFs (passage 1–5) did not differ in their proliferation performance (), immortalised PKD3-deficient MEFs exhibited a reduced proliferation rate compared to that of the corresponding wt cells (). Interestingly, the expression of an inactive PKD3.DN mutant (containing a S to A mutation of Ser738/742 within the activation loop) in wt MEFs also yielded a reduced proliferation rate but to a smaller extent than the complete knockout, most likely due to the remaining PKD3 protein in wt cells (). In addition, the transfer of wt PKD3 into PKD3−/− MEFs was able to rescue the proliferation defect (). Thus, based on this initial analysis, we predicted the active involvement of PKD3 in the cell cycle.
Figure 1. Proliferation alteration mediated by PKD3 deficiency and its sub cellular localization during mitosis: (A) MEFs were seeded out at day 0 and cell numbers were counted each day for 6–8 days after seeding. Left graph indicates proliferation rates of primary wild type (blue) and PKD3 deficient (green) MEFs. Right graph indicates proliferation rates of immortalized wild type (blue), PKD3 deficient (green), wild type + exogenously expressing PKD.DN construct (purple) and PKD3 deficient + exogenously expressing PKD3 construct (orange) MEFs. PKD3 deficiency and expression of exogenous expressed proteins was verified by western blotting (data not shown). Quantitative results (means +/− SD of triplicates) are shown. (B) PKD3/GFP localization during mitosis was analyzed by immunofluorescence. Individual stages of mitosis (indicated on the left) were estimated by DAPI staining (1st column). 2nd column shows the corresponding PKD3/GFP signal followed by α/β tubulin antibody staining for verification of mitotic structures. Merge of the PKD3/GFP and α/β tubulin signal and a complete merge are shown in the last two columns. Expression of the PKD3/GFP construct was verified by protein gel blot (data not shown), all images are representative for each stage and applied staining. Scale bars: 48 μM.
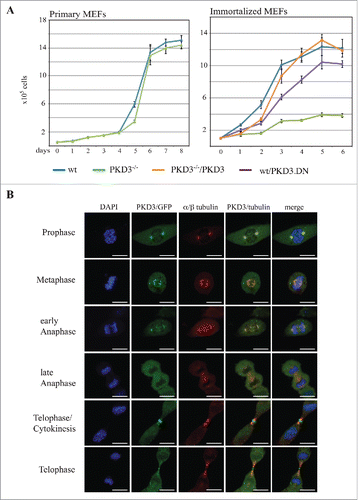
To further illuminate the role of PKD3 in that process, we characterized the sub-cellular localization of PKD3 during different stages of the cell cycle, using an exogenously expressed PKD3/GFP construct in wt MEFs. During interphase under normal growth conditions, an ubiquitous PKD3/GFP pattern was observed in wt MEFs, expression was also observed in the nucleus, with no indication of specific sub-cellular localization (data not shown). In contrast, during prophase, PKD3/GFP was specifically localized to centrosome structures that were identified via co-staining with antibodies directed against α/β-tubulin () and γ-tubulin (data not shown). During metaphase, PKD3/GFP localized to spindle structures, including the two centrosomes, which remained during early anaphase. Interestingly, the overlapping staining with α/β-tubulin disappeared during late anaphase, resulting in an ubiquitous PKD3/GFP signal, whereas α/β-tubulin staining still identified microtubule structures at that stage (). We identified two very distinct patterns for PKD3/GFP during telophase. On the one hand, the PKD3/GFP signal localized to the cleavage furrow. Thus, when the cell starts to divide, PKD3/GFP is located near structures that are actively involved in that process but are clearly separated from the previously observed overlapping α/β-tubulin staining. On the other hand, at a slightly more advanced stage, the PKD3/GFP signal again overlaps with microtubules but a separate signal in the midbody remains ().
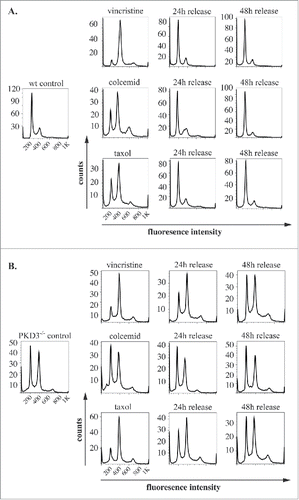
Given the reduced proliferation rate of PKD3-deficient MEFs and the specific localization of PKD3/GFP to microtubules during mitosis, we further analyzed PKD3-deficient MEFs with regard to the cell cycle. For this approach, we performed DNA staining with propidium iodide (PI), followed by a flow cytometric analysis using a set of specific cell cycle inhibitors. By comparing wt and PKD3-deficient MEFs, we intended to identify the exact time point at which PKD3 deficiency causes an alteration in the cell cycle (). Wild-type MEFs at 50% confluence were starved for 24 h in 0.2% FCS medium, followed by a 24 h incubation with aphidicolin. Starvation leads to a block at G0/G1,Citation15 whereas aphidicolin arrests cells in early S-phase;Citation16 both of these effects were represented by a diploid DNA peak after PI staining (). A 12 h release restored the original performance, whereas immediate treatment with RO-3306 for 24 h arrested the cells at G2/M by inhibiting CDK1,Citation17 as indicated by the tetraploid DNA peak after PI staining (). In addition, in this case, a 12 h release restored the original performance. Finally, RO-3306-treated MEFs were treated with nocodazole for another 24 h to achieve arrest at prometaphase/metaphase due to the blockade of microtubule polymerisation. The cells were subsequently released for 16 h. As expected, the cells exited mitosis and returned to G1 synchronously (individual PI diagrams of each step are shown in ). Interestingly, when PKD3-deficient MEFs were subjected to the same panel of treatments, it became obvious that only nocodazole treatment caused a dramatic difference in the performance of wt MEFs (). It is worth noting that the control (PI analysis of untreated MEFs at 50% confluence) had already exhibited a difference from the wt due to an obvious increase in tetraploid MEFs, which accounted for an average of approximately 50% of the counted cells. Nevertheless, neither aphidicolin treatment nor RO-3306 treatment caused any alteration of the ability to return to the original performance after release. In contrast, nocodazole treatment caused a block after a 16 h release; no clear increase in the diploid cell population was detected. Instead, it appeared that cells with a greater-than-tetraploid DNA content were generated after that treatment. To determine whether this phenomenon represents a complete block after nocodazole treatment or a delay during release, we analyzed the nocodazole effect in more detail. Therefore, wt MEFs at 80% confluence under normal growth conditions were again starved for 24 h, followed by nocodazole treatment for another 24 h. PI staining was performed before treatment, after 24 h of starvation, after 24 h of nocodazole treatment and 24, 36 and 48 h after release. Again, starvation induced a block at G0/G1 (diploid DNA peak), whereas immediate nocodazole treatment shifted this cell population to an arrest in G2/M (tetraploid DNA peak). As expected, a 24 h release reconstituted the original phenotype before treatment; there were no changes, even after 36 and 48 h (). In contrast, although PKD3-deficient MEFs exhibited a dramatic delay in reaching the initial phenotype, the PI staining pattern eventually (after 48 h) became identical to the pattern observed before starvation (). The previously described multiploid DNA peak disappeared over time, most likely due to apoptosis because these cells failed to divide. Based on these results, we assumed that PKD3 deficiency causes a mechanistic problem during microtubule polymerisation. To further evaluate the nature of the involvement of PKD3 in microtubule dynamics, we applied three additional compounds (vincristine, colcemid and taxol), which all are known to inhibit cell cycle progression via G2/M arrest due to their interference with microtubule dynamics, albeit through slightly distinct mechanisms. Following a similar regime as described above, wt MEFs at 80% confluence were starved for 24 h and then switched to complete medium containing the indicated inhibitor for another 24 h, followed by release for 24 and 48 h. The progression of the cell cycle was monitored by PI staining at the indicated time points. Vincristine caused wt MEFs to arrest at the G2/M transition () due to its ability to bind to tubulin dimers and thereby block the polymerisation of microtubule structures (e.g., spindle fibers ) (for a review see ref.Citation18). After a 24 h release, the original performance was achieved. When colcemid was applied at a range of low concentrations, the wt MEFs also arrested at G2/M, albeit not as efficiently as observed for vincristine. In addition, a third peak appeared, which we attributed to the documented ability of colcemid to generate polyploid cells (for a review see ref.Citation19). Again, after a 24 h release, a complete recovery to the original phenotype was observed (). Finally, taxol also caused wt MEFs to arrest at G2/M based on the ability of taxol to stabilize microtubules, thereby preventing depolymerisation and chromosome segregation.Citation20 Similar to colcemid, taxol was not as efficient as vincristine at the given concentration, but again, after a 24 h release, the resulting PI staining pattern revealed a complete recovery. When subjecting PKD3-deficient MEFs to treatment with the same panel of inhibitors, we observed that vincristine and taxol showed comparable results to the earlier nocodazole treatment. Thus, the recovery time upon release took up to 48 h, rather than 24 h, as observed for wt MEFs (). In addition, colcemid appeared to be unable to arrest PKD3-deficient MEFs in this assay (). In conclusion, PKD3 deficiency appeared to cause a general delay in microtubule dynamics during the cell cycle and a complete lack of response to colcemid.
Figure 2. Propidium iodide (PI) based analysis of the cell cycle of wildtype and PKD3 deficient MEFs: (A) Wild type MEFs were seeded and starved for 24 h at 50% confluence followed by an incubation with aphidicoline for 24 h and released for another 12 h. Alternatively aphidicoline treated MEFs were incubated with RO-3306 for 24 h and released for 12 h or treated with nocodazole for 24 h before the release (16 h). Individual PI stainings of each step are shown, with each labeled on top of the graph. (B) PKD3 deficient MEFs were treated the same way and analyzed accordingly. (C) Detailed analysis of wild type and PKD3 deficient MEFs after nocodazole treatment. Individual steps are indicated on top of the graphs. For all assays, multiple independent experiments were performed which all showed similar results.
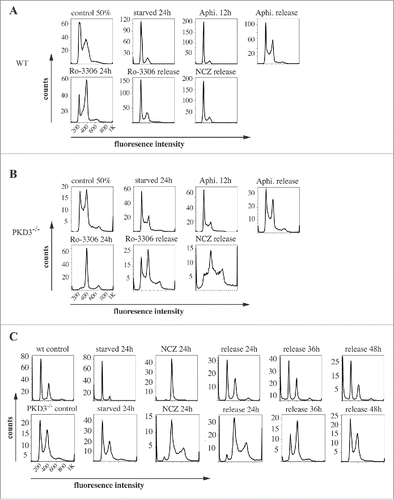
Figure 3. PKD3 deficiency induces alterations in microtubule dynamics after synchronization: Wild type and PKD3 deficient MEFs were starved for 24 h and treated with indicated inhibitors for another 24 h before release. PI stainings were performed for individual time points as indicated. (A) Wild type samples are shown. Single left graph indicates wt control before treatment followed by vincristine (top), colcemid (middle) and taxol (bottom) treated samples after starvation but before release, followed by graphs showing PI stainings after 24 h and 48 h release. (B) PKD3 deficient samples are shown. Single left graph indicates PKD3 deficient control before treatment followed by vincristine (top), colcemid (middle) and taxol (bottom) treated samples after starvation but before release, followed by graphs showing PI stainings after 24 h and 48 h release. For all assays, multiple independent experiments were performed which all showed similar results.
We next investigated the microtubule network during the cell cycle via immunofluorescence by applying a α/β-tubulin-specific antibody and comparing wt MEFs with PKD3-deficient MEFs. Individual cell cycle stages were defined by parallel DAPI staining. As shown in , wt MEFs displayed the expected α/β-tubulin pattern, starting in prophase with two centrosomes located at opposite poles and proceeding with the establishment of the spindle apparatus during prometaphase and metaphase. During early and late anaphase, the microtubules lengthened and subsequently depolymerised. Finally, during telophase, the constricted remains of microtubules were visible. In sharp contrast, a large proportion of PKD3-deficient MEFs failed to exhibit any significant α/β-tubulin staining from prophase through metaphase (e.g., no centrosomes or clear spindle structure) (). Overall, in all three stages (prophase, prometaphase and metaphase), we counted approximately 40% of cells expressing this phenotype, whereas fewer than 5% of wt MEFs showed abnormal α/β-tubulin staining. This difference between wt and PKD3-deficient MEFs disappeared when the cells were evaluated during anaphase or later stages (). We interpreted this finding as evidence that approximately 60% of PKD3-deficient MEFs are able to establish a spindle apparatus. The lack of α/β-tubulin staining in a subset of PKD3-deficient MEFs during prophase suggests that this cell population may lack centrosomes, including the microtubule-organizing center (MTOC). Therefore, we applied a γ-tubulin-specific antibody in a subsequent immunofluorescence assay to compare wt and PKD3-deficient MEFs. Indeed, we detected centrosome structures stained by γ-tubulin in all MEFs from prophase to metaphase, irrespective of genotype. Representative examples during metaphase are shown in . Whereas the wt cells contained chromosomes aligned at the equatorial plate with centrosomes at opposite poles, the PKD3-deficient cell in this figure exhibited one example of improperly aligned chromosomes (representative of 40% of PKD3-deficient MEFs); however, that sample retained two γ-tubulin-positive spots, reflecting the presence of centrosomal structures. Furthermore we investigated how much of this cell population is apoptotic by evaluating the cytoplasmic cytochrome C (Cyt C) content, which is an early indication of intrinsic induced apoptosis. Top row in indicates the control in which wt MEFs were incubated with 50μM H2O2, known to induce apoptosis, detected by the release of Cyt C from the mitochondrial inter-membrane space to the cytosol by immunofluorescence means. The middle row in shows a PKD3-deficient cell, which was able to align condensed chromosomes at the equatorial plate, thus represents one of the 60%, which are able to pass the prometaphase checkpoint. No cytosolic Cyt C was detectable while the mitochondrial Cyt C can't be detected due to the experimental procedure. In contrast, PKD3 deficient cells, which fail to align condensed chromosomes, are also positive for cytoplasmic Cyt C as shown in the bottom row (). Indeed 95% of the PKD3-deficient cell population, which failed to proceed to the metaphase, were positive for cytosolic Cyt C.
Figure 4. Failure in microtubule nucleation in PKD3 deficient MEFs: Wild type and PKD3 deficient MEFs were kept under normal cell culture conditions before fixation and incubation with indicated antibodies according to protocols described in material and methods. (A) α/β-tubulin antibody staining of wt (first three rows on the left) and PKD3−/− (last three rows on the right) MEFs at indicated mitotic phases (estimated by DAPI staining). (B) γ-tubulin antibody staining of wt (first three images on the left) and PKD3−/− (last three images on the right) MEFs at metaphase (estimated by DAPI staining). (C) First row: Wild type MEFs were treated with 50μM H2O2 for 15 min at 37°C before fixation and indicated staining or antibody incubation according to protocols described in material and methods. This sample served as control for the validation of cytoplasmic Cyt C release after apoptotic induction. Second row: Example of a PKD3 deficient MEF with appropriate aligned condensed chromosomes at prometaphase. Third row: Example of a PKD3 deficient MEF with improper aligned condensed chromosomes at prometaphase. Applied stainings and antibody are indicated above the images. Cyt C release into the cytoplasm (green color) indicates the induction of the apoptotic cascade. All images are representatives of many for the indicated staining. Scale bars: 48 μM.
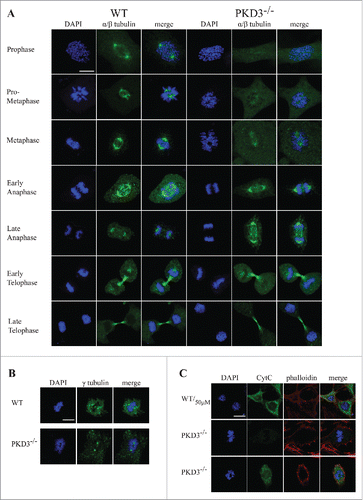
In conclusion, based on these results, we determined that PKD3-deficient MEFs do contain centrosomes but exhibit a high percentage of cells that are unable to localize α/β-tubulin to these structures and are highly prone to apoptosis.
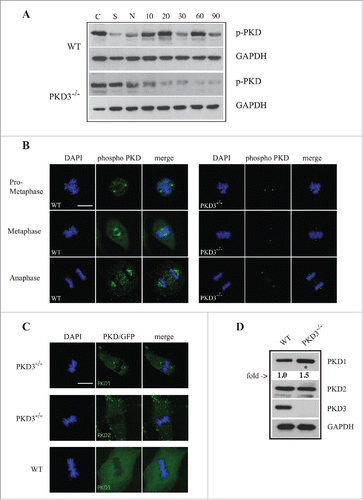
Last, we investigated whether PKD3 becomes activated in a cell cycle-specific manner, by using a phospho-specific antibody that detects activation loop phosphorylation at Ser738/742. MEFs of both genotypes were starved, treated with nocodazole and released for various time points before analysis. The western blot in clearly indicated that after starvation and nocodazole treatment, the phospho-PKD signal in wt MEFs was initially significantly diminished; increased strongly after a 10–20 min release; declined after 30 min; and increased again after 60 min. In sharp contrast, PKD3 deficiency caused a robust reduction in the phospho-PKD signal after nocodazole treatment, with no increase at all after release, suggesting that the phospho-PKD signals observed in wt extracts after release were PKD3-specific. The same antibody was then applied in an immunofluorescence assay to determine the subcellular localization of active PKD from prometaphase until anaphase. In wt MEFs, an active PKD signal was detectable first at the centrosome (prometaphase, ) and later at microtubule structures (metaphase and anaphase, ). Interestingly, PKD3-deficient MEFs still displayed a phospho-PKD signal at the centrosome but completely lacked microtubule-associated staining (). To determine which PKD isoform was responsible for the remaining phospho-PKD signal at centrosomes, we investigated PKD3-deficient MEFs expressing either PKD1 or 2 as GFP fusion proteins. In fact, only PKD1/GFP, but not PKD2/GFP, could be identified at the centrosomes in PKD3-deficient MEFs. Interestingly, both PKD/GFP constructs were unable to localize to this structure in wt MEFs (). In addition we also verified the protein amounts of individual PKDs in PKD3 deficient MEFs by western blotting. As indicated in no PKD3 protein was detectable, PKD2 protein levels were unchanged while a slight increase of 1.5 fold for PKD1 was observed when compared to wt extracts. Together, the results of this final analysis revealed that PKD activity during M-phase is represented primarily by PKD3 and that PKD3 deficiency specifically causes PKD1 to localize to centrosomes, which might constitute a redundant mechanism.
Figure 5. Analysis of PKD activity during the cell cycle: (A) Western blot analysis of wt and PKD3−/− cell extracts after starvation (S) for 24 h, nocodazole treatment (N) for 24 h and different time points after release (10, 20, 30, 60 and 90 min). Phospho-specific Ser738/742 PKD antibody was used to evaluate PKDs activity status. GAPDH antibody was applied as loading control. Shown results are representative of three independent experiments. (B) Wild type and PKD3 deficient MEFs were kept under normal cell culture conditions before fixation and incubation with the phospho-specific Ser738/742 PKD antibody according to protocols described in material and methods. Mitotic phases evaluated by DAPI staining are indicated on the left. First three rows on the left represent wt MEFs whereas last three rows on the right represent PKD3−/− MEFs. (C) Confocal images of PKD1/GFP expression (top lane) and PKD2/GFP expression (middle lane) in PKD3 deficient MEFs and PKD1/GFP expression in wt MEFs (bottom lane) at metaphase. Expression of individual PKD/GFP construct was verified by western blot (data not shown). All images are representatives of many for the indicated staining. Scale bars: 48 μM. (D) Western blot analysis of wt and PKD3−/− cell extracts. Applied antibodies are indicated on the right side. PKD1 showed a 1.5 fold increase in the PKD3 deficient extract while PKD2 showed no difference. No PKD3 was detectable in the deficient extract. GAPDH antibody was applied as loading control. For the image analysis ImageJ 1.47 v software was applied.
Discussion
Here we report that the genetic depletion of PKD3 in immortalised mouse embryonic fibroblasts leads to a severe phenotype during the cell cycle. Strikingly, this phenotype first manifested after immortalisation, which might be a reasonable explanation for why PKD3-deficient mice do not exhibit any obvious phenotype (data not shown). Immediately after the senescent crisis, PKD3-deficient MEFs significantly reduced their proliferation rate, whereas corresponding wt MEFs increased their proliferation rate (). The exogenous expression of a dominant-negative construct (PKD3.DN) in wt MEFs and of a wt PKD3 construct in PKD3-deficient MEFs supports the notion that this effect reflects a direct PKD3-specific function. A variety of reports have attempted to explain the general mechanism that underlies the immortalisation of murine fibroblasts (for a review see ref.Citation21), but there have been no findings clearly illuminating how PKD3 is involved in that process. One possibility might be that in primary cells, cell cycle regulation is mediated by a redundant system consisting of more than one regulatory pathway; however, this changes during immortalisation, resulting in a single PKD3-dependent pathway. To date, very few reports have postulated a PKD- or PKD3-specific in vivo function in the context of the cell cycle. Whereas Chen et al.Citation12 have shown that PKD3 modulated Akt and Erk1/2 activities in various prostate cancer cell lines, thereby promoting cell growth, Papazyan et al.Citation14 have reported that PKD3 and PKD2 are specifically localized to microtubule structures during mitosis. In addition, our analysis revealed that in wt MEFs, PKD3 co-localized with microtubules during mitosis (). In contrast, we did not observe any localization of PKD1 or PKD2 to microtubule structures during mitosis (data not shown), which might be explained by the different cell types used for our analysis or the rather low expression of our PKD/GFP fusion proteins, which never exceeded the endogenous PKD levels (data not shown). Notably, in addition to microtubule-associated staining, we detected PKD3/GFP at the cleavage furrow, which has not been previously reported () and will be further described in a separate report.
The decrease in proliferation and the microtubule-associated localization during mitosis led us to analyze the cell cycle in more detail by applying different inhibitors with different mechanisms of action (). Indeed, only nocodazole initially showed a clear effect in PKD3-deficient MEFs compared to wt MEFs. Whereas wt MEFs fully recovered after a 24 h release, PKD3-deficient MEFs required up to 48 h to return to their original phenotype (). Because nocodazole interferes with the polymerisation dynamics of microtubules,Citation22 we assumed that PKD3 is involved in α/β-tubulin polymerisation. An extension of this analysis using related inhibitors revealed similar defects in PKD3-deficient MEFs (). Whereas three of four tested compounds (nocodazole, vincristine and taxol) clearly indicated that PKD3 deficiency caused a significant delay in the return to original performance after release, colcemid treatment appeared to be ineffective in PKD3-deficient MEFs. Thus, PKD3 deficiency mediates resistance against colcemid treatment in MEFs. Based on the described mechanism of colcemid action (for a review see ref. Citation19), one could speculate that the colcemid-specific binding site in tubulin is altered or missing or that the functional consequence of colcemid binding is interrupted. Nevertheless, neither of these explanations provides a clear understanding of how PKD3 is involved in that mechanism owing to the imprecise nature of the actions of colcemid. All the other microtubule-interacting compounds resulted in a similar robust deceleration of the reestablishment of the microtubule network, suggesting a general impact of PKD3 on microtubule dynamics. Indeed, when we investigated the microtubule network at different stages during mitosis (), we identified a population (approximately 40%) of PKD3-deficient MEFs that exhibited no α/β-tubulin staining at the centrosomes and no spindle-like structure until metaphase. As a consequence, condensed chromosomes did not align at the mitotic plate, and the cells became arrested at the metaphase checkpoint and eventually become apoptotic, which we verified by the increased cytosolic Cyt C content in these cells. As shown by γ-tubulin, this population still contained centrosome structures but failed to establish the molecular basis for microtubule nucleation, which represents the initiation of the spindle Citation23 and is regulated by the MTOC.Citation24 The pericentriolar material (PCM), which is described as an amorphous mass of proteins surrounding the centrioles, is believed to contain more than 100 different proteins.Citation25 Some of these proteins have been identified as kinase substrates (for example, Nlp and C-Nap1), and a phosphorylation-dependent cell cycle-specific function has been described.Citation26,27 The likelihood that one or more proteins within the PCM represent possible PKD3 substrates is high, but no such candidates have been identified. Thus, we predict that future attempts will identify PKD3-specific substrates in the PCM, which will help further elucidate the physiological role of PKD3 in that context.
Nonetheless, approximately 60% of PKD3-deficient MEFs were able to establish a functional spindle apparatus and pass the metaphase checkpoint. One plausible explanation for this observation might be the finding that PKD1 (but not PKD2), which appears slightly up-regulated in PKD3 deficient MEFs (), translocates to centrosomes and becomes activated during M-phase in a PKD3-deficient background, events that were not detectable in wt MEFs (). This effect represents a classical redundant mechanism in a kinase family, in which one isoform is able to compensate for an in vivo function of another family member. In our case, the ability of PKD1 to compensate for PKD3 appeared to be incomplete; 40% of PKD3-deficient MEFs failed to establish a spindle apparatus. Because the establishment of the spindle is a tightly regulated process,Citation28 this finding might reflect (i) an inability of PKD1 to phosphorylate a predicted PKD3 substrate to the same extent in order to provide an activation or conformational change of a postulated substrate always at the right time within the PCM or (ii) it may also be possible that an insufficient kinetic of such a reaction results in a “go” or “no-go” response thus if certain amounts of signals are not present at a given time point microtubule nucleation won't take place. (iii) Alternatively it also might be possible that PKDs can act as scaffolding proteins as it has been described for PKC as well.Citation29 In this case PKD1 at the centrosome might not be able to provide equal scaffolding properties as PKD3 does and as a consequence a postulated protein complex might not work as efficient as in the wt. (iv) Lastly we also can not exclude that an alternative pathway exist which either works in parallel to the PKD3 mediated one or becomes activated when PKD3 is missing but is working less efficient. All this remains purely speculative so far since we need more information about PKD3 substrates in that context to further elaborate on the signaling pathway, which leads to microtubule nucleation and involves PKD3. Nevertheless once the PKD3-deficient MEFs succeeded in establishing a spindle apparatus, they proceeded with the cell cycle but exhibited a dramatic reduction in speed. Using specific inhibitors, we showed that this deceleration was caused by alterations in microtubule dynamics. The overlap of PKD3/GFP and the activation-specific phospho-PKD signal (demonstrated to be PKD3-specific in that context, ) with microtubule structures during mitosis suggested an additional function of PKD3 beyond its predicted role in microtubule nucleation. In addition, our western blot analysis () supported this notion; the activation-specific phospho-PKD signal in PKD3-deficient extracts decreased to background levels (also indicating that the centrosome-localized signal in these extracts was below the detection limit). Similarly to the PCM, many microtubule-associated proteins have been identified and characterized (for a review see refs.Citation28,30-32), and at least some of those proteins are also regulated by phosphorylation events.Citation32 Even β-tubulin has been reported to be phosphorylated by CDK1 at Ser172, which impaired the ability of β-tubulin to bind and interact with GTP.Citation33 Thus, it is very likely that other as-yet undefined kinases, including PKDs, can participate in the regulation of microtubule dynamics. Nonetheless, no reports have identified such a role for PKDs, particularly PKD3, except for the analysis performed by Papazyan et al.Citation14 That report has revealed a microtubule-associated localization of PKD3 in HEK293 cells but included no functional data. Therefore, future research is needed to identify and characterize PKD3-specific substrates among the large number of microtubule-associated proteins to shed light on the molecular mechanism regulated by PKD3.
The activation loop phosphorylation of PKDs (Ser738/742 in human PKD1), which is used as a marker of active PKD, has been predicted to be mediated primarily by novel PKCs.Citation34 In another context, we have been able to verify this connection using PKCδ- and PKD1-deficient MEFs.Citation35 However, with regard to the described phenotype, neither PKCδ- nor PKCε-deficient MEFs showed a related phenotype (data not shown). In addition, all other PKC-deficient MEF cell lines available to us (PKCα, β, θ, ζ and λ/ι) failed to express a comparable mitotic phenotype (data not shown). We cannot rule out the possibility that PKCη represents the activating kinase in this context, but we believe that this possibility is unlikely because the intracellular localization of PKCη has been linked to perinuclear structures, rather than spindle structures.Citation36 Instead, we believe that an undefined kinase is responsible for the activation loop phosphorylation of PKD3 (and PKD1 in PKD3-deficient MEFs) detected during mitosis or that individual PKCs can compensate for this particular function, because we have shown in other contexts that the PKC system can also be redundant.Citation37-40 Alternatively, because the antibody used in this study cannot distinguish between serine 738 and 742, it is also possible that Ser742 auto-phosphorylation occurred, which has been described to exist only under certain circumstances.Citation6 If that possibility holds true, the predicted PKD3 function during M-phase would be a PKC-independent mechanism. Together, our data show that PKD3 performs an important task during the cell cycle after immortalisation and that its deficiency leads to a dramatic decrease in proliferation rate in immortalised MEFs. Overall, the function of PKD3 was shown to be associated with microtubule dynamics, but interestingly, at least two different tasks appear to be mediated by PKD3. PKD3 is involved in microtubule nucleation, but it also participates in the dynamic regulation of microtubule polymerisation/depolymerisation. The presence of this defect only after immortalisation implies that PKD3 might represent a promising new target for interfering with the cell cycle (e.g., microtubule dynamics) specifically in cells exhibiting the cellular changes that drive tumor development.
Material & methods: Shead1
Isolation of MEFs and cell culture
For the isolation of mouse embryonic fibroblasts (MEFs) expressing the PKD3 mutant allele, we used heterozygous matings of PKD3Δ/+ animals. The generation and initial characterization of the PKD3 targeted allele will be described elsewhere. Embryos that were 12.5 to 13.5 post coitum were used for the preparation, according to standard protocols.Citation21 To immortalize MEFs, we used a modified NIH 3T3 protocol. In short, primary MEFs were cultured for 5–6 passages till senescence occurs. Prolonged cell culture gave rise to immortalized cells which were expanded. All MEFs were cultured in Gibco DMEM+ GlutaMAXTM-I (Invitrogen) medium with 10% Gibco fetal calf serum (Invitrogen), Gibco non-essential amino acids (Invitrogen), penicillin (50 units/ml) and streptomycin (100 μg/ml) (Invitrogen).
Generation of exogenous expressing PKD3/GFP and PKD3
DN/GFP MEF lines: To express a PKD3.DN/GFP construct in wild type and a PKD3/GFP construct in PKD3−/− MEFs, we used the pLenti6/TR vector system according to manufacturer's instructions obtained from Invitrogen (catalog no. V480-20). In order to use the Gateway system (Invitrogen) for sub-cloning, a full length mouse cDNA of PKD3 was sub-cloned into the pENTRTM/D-TOPO vector (Invitrogen, Cat.-No. K2400-20). To introduce the indicated dominate negative mutation (PKD3.DN) the QuickChange II XL site-directed Mutagenesis Kit (Agilent Technologies, Cat. 200521) was used. The following mutagenesis primer were used to introduce the mutation (PKD3.DN) into the wt mouse PKD3 cDNA sequence; PKD3.DN; forward: CATCATTGGTGAGAAGGCATTCCGGAGGGCAGTGGTAGGAACTCC, reverse: GGAGTTCCTACCACTGCCCTCCGGAATGCCTTCTCACCAATGATG. The introduced mutations have been verified by sequence analysis.
Proliferation and cell cycle analysis
To measure proliferation 5 × 104 primary and immortalized mouse embryonic fibroblasts (MEFs) were plated in triplicates in a series of 6-well dishes (TC-plate, 6 well, Sarstedt #83.3920) and counted for up to 8 days with a Neubauer counting chamber to perform growth curves. Graphs and statistics were generated with Microsoft Office Excel. The values represent means +/− SEM.
To analyze the cell cycle MEFs were cultured in 6 cm cell culture dishes (Sarstedt #83.3902) till 50–80% confluence, starved for 24 hours (DMEM, 0.2% FCS medium) and applied to further synchronization. Individual compounds were used as follows: Aphidicholine (Sigma-Aldrich, #38966-21-1) for 24 hours in a final concentration of 2μg/ml; Ro-3306 (Sigma-Aldrich, #872573-93-8) for 24 hour in a final concentration of 5μM; Vincristine Sulfate (Sigma-Aldrich, #2068-78-2) for 24 hours in a final concentration of 50ng/ml; Nocodazole (Sigma-Aldrich, #31430-18-9) for 16 hours in a final concentration of 100ng/ml; Colcemid (Roche, #10295892001) for 24 hours in a final concentration of 10ng/ml; Paclitaxel/Taxol (Sigma-Aldrich, #33069-62-4) for 24 hours in a final concentration of 10nM. For release MEFs were washed 3 times in PBS followed by incubation in DMEM medium supplemented with 10% FCS for indicated time points. Propidium iodide (PIP) staining: MEFs were collected and re-suspended in 200μl PBS, fixed by adding 700μl ice cold ethanol for 1 hour at 4°C before spinning down and re-suspension in 200μl PI staining solution (Sigma-Aldrich, #00M1494). After incubation for 45 min at 4°C MEFs were analyzed by the BD FACSCaliburTM flow cytometer. For data analysis FlowJo7.6.5 software was used.
Protein extraction and protein gel blotting
For whole cell protein extraction the following buffer was used: 20 mM HEPES (pH 7.9), 20% (v/v) Glycerol, 1% (v/v) Nonidet P-40, 1 mM MgCl2, 0.5 mM EDTA, 0.1 mM EGTA, 1 mM PMSF (phenylmethylsulfonyl fluoride), 10 mM DTT and 1X protease inhibitor cocktail (Sigma, NO.P-27147). MEFs from a confluent 10 cm dish were collected, centrifuged at 5000 rpm for 5 min and the pellet was re-suspended in 100µl whole cell extraction buffer. Lysates were kept on ice for 30 min and centrifuged at 14,000x rpm for 10 min. The supernatant was collected. 20μl of 4x sample buffer was added followed by an incubation at 99°C for 5min. Samples were either directly applied to SDS-PAGE or store at −20°C. After SDS-polyacrylamide gel electrophoresis (8%) wet blotting onto Bio-Rad nitrocellulose membranes using a Hoefer apparatus was applied. The protein amounts used for each electrophoresis were calculated by the Bradford method (BioRad Protein Assay, Cat. 500–0006). Membranes were blocked with 1% Tween-PBS containing 5% (w/v) skim-milk (PBST) for 1 h at RT, followed by an incubation with primary antibodies in 5% (w/v) skim-milk PBST overnight at 4°C. The secondary antibodies were applied in 2.5% (w/v) skim-milk PBST for various time, depending on the epitope (2 h RT to overnight at 4°C). SuperSignal West Pico from Pierce (Cat. 34078) was used for the detection.
Confocal imaging
For confocal imaging, MEFs were grown on gelatin coated cover slips and fixed with 4% PFA in PBS at room temperature for 15 min prior further processing. Plasma membrane permeabilization was obtained by incubation with 0.1% Saponin-PBS (Sigma-Aldrich, #BCBN3022V) at room temperature for 15 min, followed by blocking with 0.1% Saponin-PBS/3%BSA; 15mg/ml glycine for another 15 min. After washing 3 times in 0.1% Saponin-PBS/3%BSA and incubation in the staining buffer (0.1%Saponin-PBS/3%BSA including 1% sheep serum), primary antibodies were applied and incubated at 4°C overnight subsequently followed by secondary antibody incubation at room temperature for 2 hours in the dark. After antibody incubation coverslips were mounted onto microscopic glass slides using mounting medium including DAPI (Invitrogen, ProLong®Gold antifade reagent with DAPI, P36935). Cells were visualized using a Zeiss LSM 510 Meta inverted microscope equipped with a Zeiss LSM laser module. Image analysis was performed using the ZEN2009 software from Zeiss. Individual cell cycle and mitotic phases were determined by the morphology of DAPI stained chromosomes according to Levesque.Citation41 In brief, loss of the nuclear envelope indicate the prophase, highly condensed chromosomes without an equatorial alignment were defined as prometaphase while a neat alignment of condensed chromosomes was defined as metaphase. Segregating chromosomes were taken as characteristic for the anaphase while de-condensed chromosomes mark the telophase.
Used antibodies
PKD/PKCμ Rabbit (Cell signaling, #2052S, 1:1000 dilution), PKD3/PKCν Rabbit (D57E6) (Cell signaling, #5655LS, 1:1000 dilution), p-PKD rabbit (Ser744/748) (Cell signaling, #2054L, 1:1000 dilution), GAPDH (14C10) (Cell signaling, #2118L, 1:5000 dilution), α/β tubulin (Cell signaling, #2148S, 1:500 dilution), γ-Tubulin (Cell signaling, #5886S, 1:500 dilution); anti-Cytochrome c (Cell signaling, #4272, 1:500), PKD2 (E-20) (Santa Cruz, sc-74839, 1:500 dilutuion), Goat anti Rabbit HRPO (Jackson ImmunoResearch Lab, 1:10000 dilution), Donkey anti Rabbit AlexaFluor® 568 (Life Technology, #1476640, 1:1000 dilution), Phalloidin AlexaFluor® 647 (Life Technology, #1731699, 1:1000 dilution)
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We would like to thank lab members for continuous discussion of the project, in particular Philipp Sell for his initial help in generating the PKD3 knock out line. We thanks the Norwegian Transgene unit (particular Mikael Vestberg) for blastocyst injections. Also we would like to thank Sandra Lopez-Aviles for critical reading the manuscript and helpful suggestions.
Funding
This work was supported by the Norwegian Research Council (grant: 197261) and the Norwegian Cancer Society (grant: 6856910).
References
- Johannes FJ, Prestle J, Eis S, Oberhagemann P, Pfizenmaier K. PKCu is a novel, atypical member of the protein kinase C family. J Biol Chem 1994; 269, 6140-8; PMID:8119958
- Hayashi A, Seki N, Hattori A, Kozuma S, Saito T. PKCnu, a new member of the protein kinase C family, composes a fourth subfamily with PKCmu. Biochim Biophys Acta 1999; 1450(1):99-106; PMID:10231560; http://dx.doi.org/10.1016/S0167-4889(99)00040-3
- Steinberg SF. Regulation of protein kinase D1 activity. Mol Pharmacol 2012 Mar; 81(3):284-91; PMID:22188925; http://dx.doi.org/10.1124/mol.111.075986
- Rozengurt E, Rey O, Waldron RT. Protein kinase D signaling. J Biol Chem 2005; 280(14): p:13205-8; PMID:15701647; http://dx.doi.org/10.1074/jbc.R500002200
- Waldron RT, Iglesias T, Rozengurt E.. Phosphorylation-dependent protein kinase D activation. Electrophoresis 1999; 20(2):382-90; PMID:10197446; http://dx.doi.org/10.1002/(SICI)1522-2683(19990201)20:2%3c382::AID-ELPS382%3e3.0.CO;2-N
- Rybin VO, Guo J, Steinberg SF. Protein kinase D1 autophosphorylation via distinct mechanisms at Ser744/Ser748 and Ser916. J Biol Chem 2009 Jan 23; 284(4):2332-43; PMID:19029298; http://dx.doi.org/10.1074/jbc.M806381200
- Nishikawa K, Toker A, Johannes FJ, Songyang Z, Cantley LC. Determination of the specific substrate sequence motifs of protein kinase C isozymes. J Biol Chem 1997; 272(2):952-60; PMID:8995387; http://dx.doi.org/10.1074/jbc.272.2.952
- Zugaza JL, Sinnett-Smith J, Van Lint J, Rozengurt E. Protein kinase D (PKD) activation in intact cells through a protein kinase C-dependent signal transduction pathway. EMBO J 1996; 15:6220-30; PMID:8947045
- Zhukova E, Sinnett-Smith J, Rozengurt E. Protein kinase D potentiates DNA synthesis and cell proliferation induced by bombesin, vasopressin, or phorbol esters in Swiss 3T3 cells. J Biol Chem 2001; 276:40298-40305; PMID:11514571; http://dx.doi.org/10.1074/jbc.M106512200
- Sinnett-Smith J, Zhukova E, Hsieh N, Jiang X, Rozengurt E. Protein kinase D potentiates DNA synthesis induced by Gq-coupled receptors by increasing the duration of ERK signaling in swiss 3T3 cells. J Biol Chem 2004; 279:16883-16893; PMID:14963034; http://dx.doi.org/10.1074/jbc.M313225200
- Vega RB, Harrison BC, Meadows E, Roberts CR, Papst PJ, Olson EN, McKinsey TA. Protein kinases C and D mediate agonist-dependent cardiac hypertrophy through nuclear export of histone deacetylase 5. Mol Cell Biol 2004; 24:8374-8385; PMID:15367659; http://dx.doi.org/10.1128/MCB.24.19.8374-8385.2004
- Chen J, Deng F, Singh SV, Wang QJ. Protein kinase D3 (PKD3) contributes to prostate cancer cell growth and survival through a PKCepsilon/PKD3 pathway downstream of Akt and ERK 1/2. Cancer Res 2008; 68(10):3844-53; PMID:18483269; http://dx.doi.org/10.1158/0008-5472.CAN-07-5156
- Ryvkin V, Rashel M, Gaddapara T, Ghazizadeh S. Opposing growth regulatory roles of protein kinase D isoforms in human keratinocytes. J Biol Chem 2015 Apr 24; 290(17):11199-208; PMID:25802335; http://dx.doi.org/10.1074/jbc.M115.643742
- Papazyan R, Doche M, Waldron RT, Rozengurt E, Moyer MP, Rey O. Protein kinase D isozymes activation and localization during mitosis. Exp Cell Res 2008 Oct 1; 314(16):3057-68; PMID:18692497; http://dx.doi.org/10.1016/j.yexcr.2008.07.014
- Rosner M, Schipany K, Hengstschläger M. Merging high-quality biochemical fractionation with a refined flow cytometry approach to monitor nucleocytoplasmic protein expression throughout the unperturbed mammalian cell cycle. Nature Protocols 2013; 8:602-626; PMID:23449254; http://dx.doi.org/10.1038/nprot.2013.011
- Ikegami S, Taguchi T, Ohashi M, Oguro M, Nagano H, Mano Y. Aphidicolin prevents mitotic cell division by interfering with the activity of DNA polymerase-α. Nature 1978 Oct 5; 275(5679):458-60; PMID:692726; http://dx.doi.org/10.1038/275458a0
- Vassilev LT. Cell cycle synchronization at the G2/M phase border by reversible inhibition of CDK1. Cell Cycle 2006 Nov; 5(22):2555-6; http://dx.doi.org/10.4161/cc.5.22.3463
- Jordan MA. Mechanism of action of antitumor drugs that interact with microtubules and tubulin. Curr Medicinal Chem Anti-Cancer Agents 2002; 2(1):1-17; PMID:12678749; http://dx.doi.org/10.2174/1568026023394597
- Jordan MA, Wilson L. “Microtubules as a target for anticancer drugs.” Nature Rev Cancer 2004; 4(4):253-65; http://dx.doi.org/10.1038/nrc1317
- Horwitz SB. Taxol (paclitaxel): Mechanisms of action. Ann Oncol 1994; 5(Suppl 6):S3-S6; PMID:7865431
- Sharpless N. Preparation and Immortalization of Primary Murine Cells, Chapter 28, 223-228, Cell Biology A Laboratory Handbook, 2006; Third Edition (ed. Julio E. Celis) Elsevier Inc
- Vasquez RJ, Howell B, Yvon AM, Wadsworth P, Cassimeris L. Nanomolar concentrations of nocodazole alter microtubule dynamic instability in vivo and in vitro. Mol Biol Cell 1997; 8(6):973-985; PMID:9201709; http://dx.doi.org/10.1091/mbc.8.6.973
- Job D, Valiron O, Oakley B. “Microtubule nucleation.” Curr Opin Cell Biol 2003:15:111-117; PMID:12517712; http://dx.doi.org/10.1016/S0955-0674(02)00003-0
- Brinkley BR. “Microtubule Organizing Centers.” Annual Reviews: Cell Biology 1985; 1:145-172; http://dx.doi.org/10.1146/annurev.cb.01.110185.001045
- Bornens M. Centrosome composition and microtubule anchoring mechanisms. Curr Opin Cell Biol 2002; 14:25-34; PMID:11792541; http://dx.doi.org/10.1016/S0955-0674(01)00290-3
- Casenghi M, Meraldi P, Weinhart U, Duncan PI, Korner R, Nigg EA. “Polo-like kinase 1 regulates Nlp, a centrosome protein involved in microtubule nucleation.” Dev Cell 2003; 5(1):113-125; PMID:12852856; http://dx.doi.org/10.1016/S1534-5807(03)00193-X
- Fry AM, Mayor T, Meraldi P, Stierhof YD, Tanaka K, Nigg EA. “C-Nap1, a Novel Centrosomal Coiled-Coil Protein and Candidate Substrate of the Cell Cycle–regulated Protein Kinase Nek2.” J Cell Biol 1998; 141(7):1563-1574; PMID:9647649; http://dx.doi.org/10.1083/jcb.141.7.1563
- Maiato H, Sampaio P, Sunkel CE. Microtubule-associated proteins and their essential roles during mitosis. Int Rev Cytol 2004; 241:53-153; PMID:15548419; http://dx.doi.org/10.1016/S0074-7696(04)41002-X
- Cameron AJ, Procyk KJ, Leitges M, Parker PJ. PKC α protein but not kinase activity is critical for glioma cell proliferation and survival. Int J Cancer 2008 Aug 15; 123(4):769-79; PMID:18508315; http://dx.doi.org/10.1002/ijc.23560
- Mandelkow E, Mandelkow EM. Microtubules and microtubule-associated proteins. Curr Opin Cell Biol 1995 Feb; 7(1):72-81; PMID:7755992; http://dx.doi.org/10.1016/0955-0674(95)80047-6
- Fisher KH, Deane CM, Wakefield JG. The functional domain grouping of microtubule associated proteins. Commun Integr Biol 2008; 1(1):47-50; PMID:19704789; http://dx.doi.org/10.4161/cib.1.1.6795
- Drewes G, Ebneth A, Mandelkow EM. MAPs, MARKs and microtubule dynamics. Trends Biochem Sci 1998 Aug; 23(8):307-11; PMID:9757832; http://dx.doi.org/10.1016/S0968-0004(98)01245-6
- Fourest-Lieuvin A, Peris L, Gache V, Garcia-Saez I, Juillan-Binard C, Lantez V, Job D. Microtubule regulation in mitosis: tubulin phosphorylation by the cyclin-dependent kinase Cdk1. Mol Biol Cell 2006 Mar; 17(3):1041-50
- Waldron RT, Rozengurt E. Protein kinase C phosphorylates protein kinase D activation loop Ser744 and Ser748 and releases autoinhibition by the pleckstrin homology domain. J Biol Chem 2003 Jan 3; 278(1):154-63; PMID:12407104; http://dx.doi.org/10.1074/jbc.M208075200
- Zhang T, Sell P, Braun U, Leitges M. PKD1 protein is involved in reactive oxygen species-mediated mitochondrial depolarization in cooperation with protein kinase Cδ (PKCδ). J Biol Chem 2015 Apr 17; 290(16):10472-85; PMID:25759386; http://dx.doi.org/10.1074/jbc.M114.619148
- Brändlin I, Hübner S, Eiseler T, Martinez-Moya M, Horschinek A, Hausser A, Link G, Rupp S, Storz P, Pfizenmaier K, et al. Protein kinase C (PKC)eta-mediated PKC mu activation modulates ERK and JNK signal pathways. J Biol Chem 2002 Feb 22; 277(8):6490-6; http://dx.doi.org/10.1074/jbc.M106083200
- Gruber T, Hermann-Kleiter N, Pfeifhofer-Obermair C, Lutz-Nicoladoni C, Thuille N, Letschka T, Barsig J, Baudler M, Li J, Metzler B, et al. PKC theta cooperates with PKC α in alloimmune responses of T cells in vivo. Mol Immunol 2009 Jun; 46(10):2071-9; PMID:19356803; http://dx.doi.org/10.1016/j.molimm.2009.02.030
- Thuille N, Wachowicz K, Hermann-Kleiter N, Kaminski S, Fresser F, Lutz-Nicoladoni C, Leitges M, Thome M, Massoumi R, Baier G. PKCθ/â and CYLD are antagonistic partners in the NFêB and NFAT transactivation pathways in primary mouse CD3+ T lymphocytes. PLoS One 2013; 8(1):e53709; PMID:23335970; http://dx.doi.org/10.1371/journal.pone.0053709
- Lutz-Nicoladoni C, Thuille N, Wachowicz K, Gruber T, Leitges M, Baier G. PKCα and PKCβ cooperate functionally in CD3-induced de novo IL-2 mRNA transcription. Immunol Lett 2013 Mar; 151(1–2):31-8; PMID:23439007; http://dx.doi.org/10.1016/j.imlet.2013.02.002
- Carracedo S, Sacher F, Brandes G, Braun U, Leitges M. Redundant role of protein kinase C delta and epsilon during mouse embryonic development. PLoS One 2014 Aug 1; 9(8):e103686; PMID:25084151; http://dx.doi.org/10.1371/journal.pone.0103686
- Levesque AA, Compton DA. The chromokinesin Kid is necessary for chromosome arm orientation and oscillation, but not congression, on mitotic spindles. J Cell Biol 2001 Sep 17; 154(6):1135-46; PMID:11564754; http://dx.doi.org/10.1083/jcb.200106093